
Structural Studies of Klebsiella pneumoniae Fosfomycin-Resistance Protein and Its Application for the Development of an Optical Biosensor for Fosfomycin Determination



Abstract
:1. Introduction
2. Results and Discussion
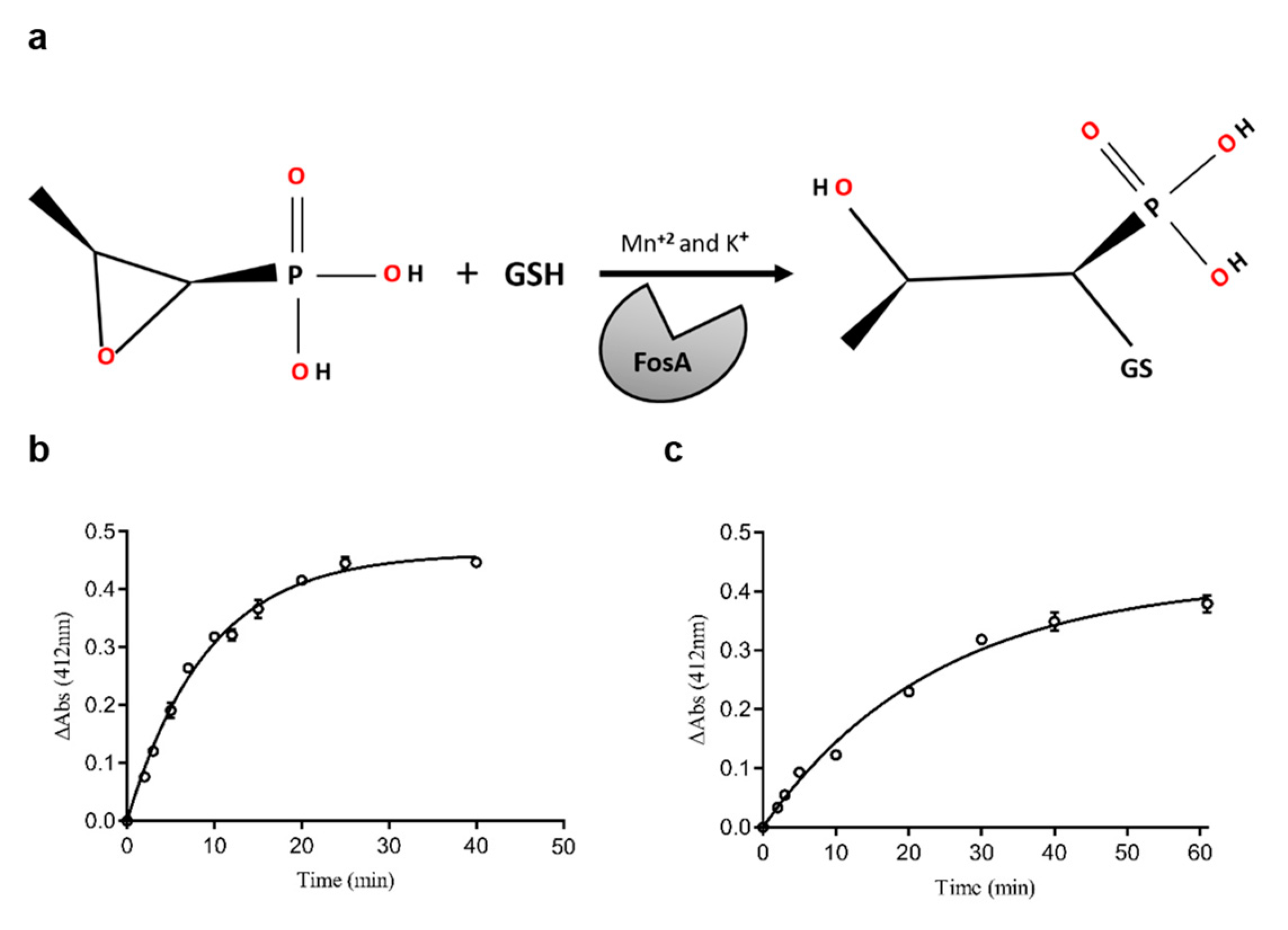
2.1. The Expression, Purification, and Substrate Specificity of FosAPA and FosAKP
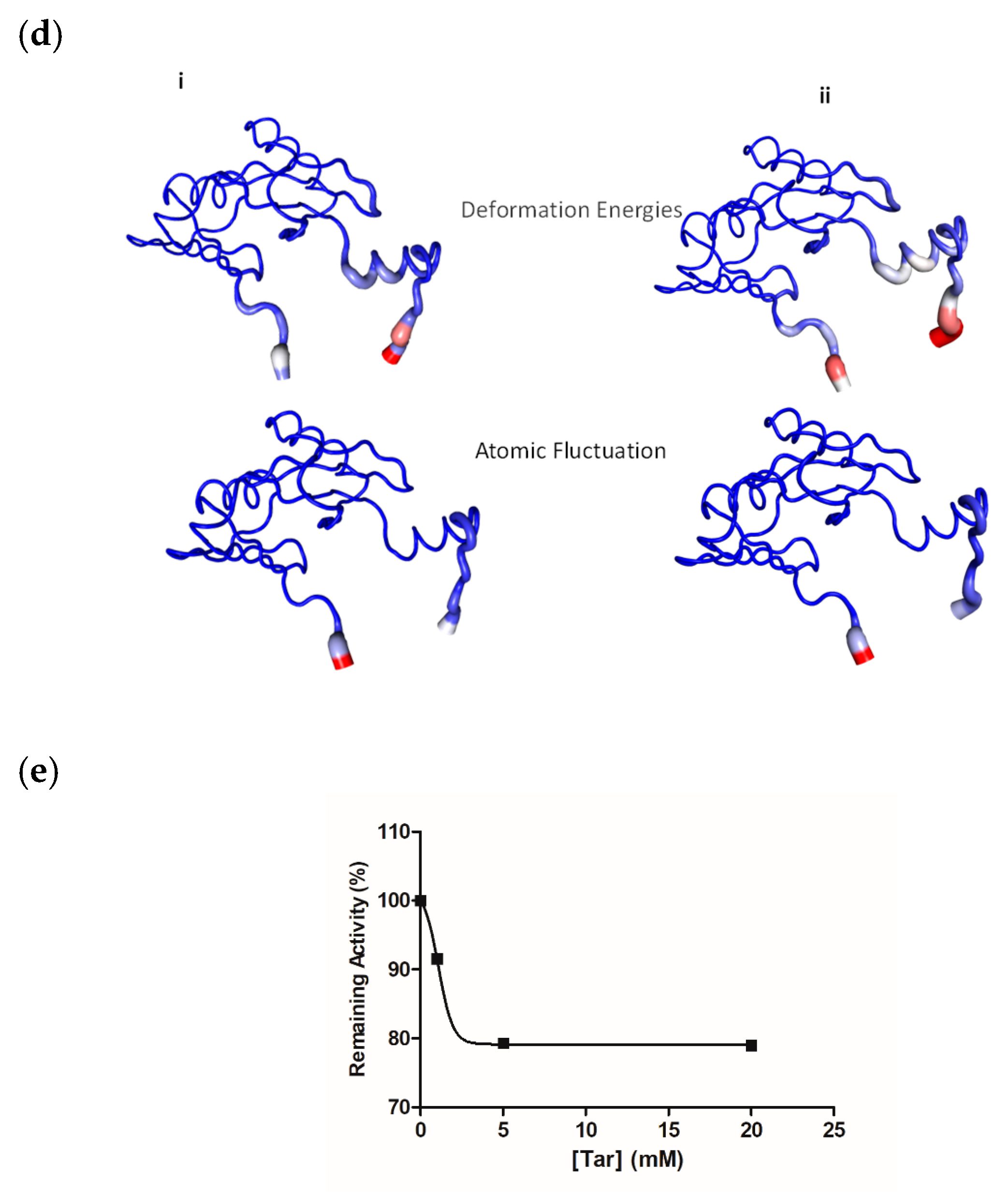
2.2. Structural Analysis
2.3. Thermal Stability of FosAKP
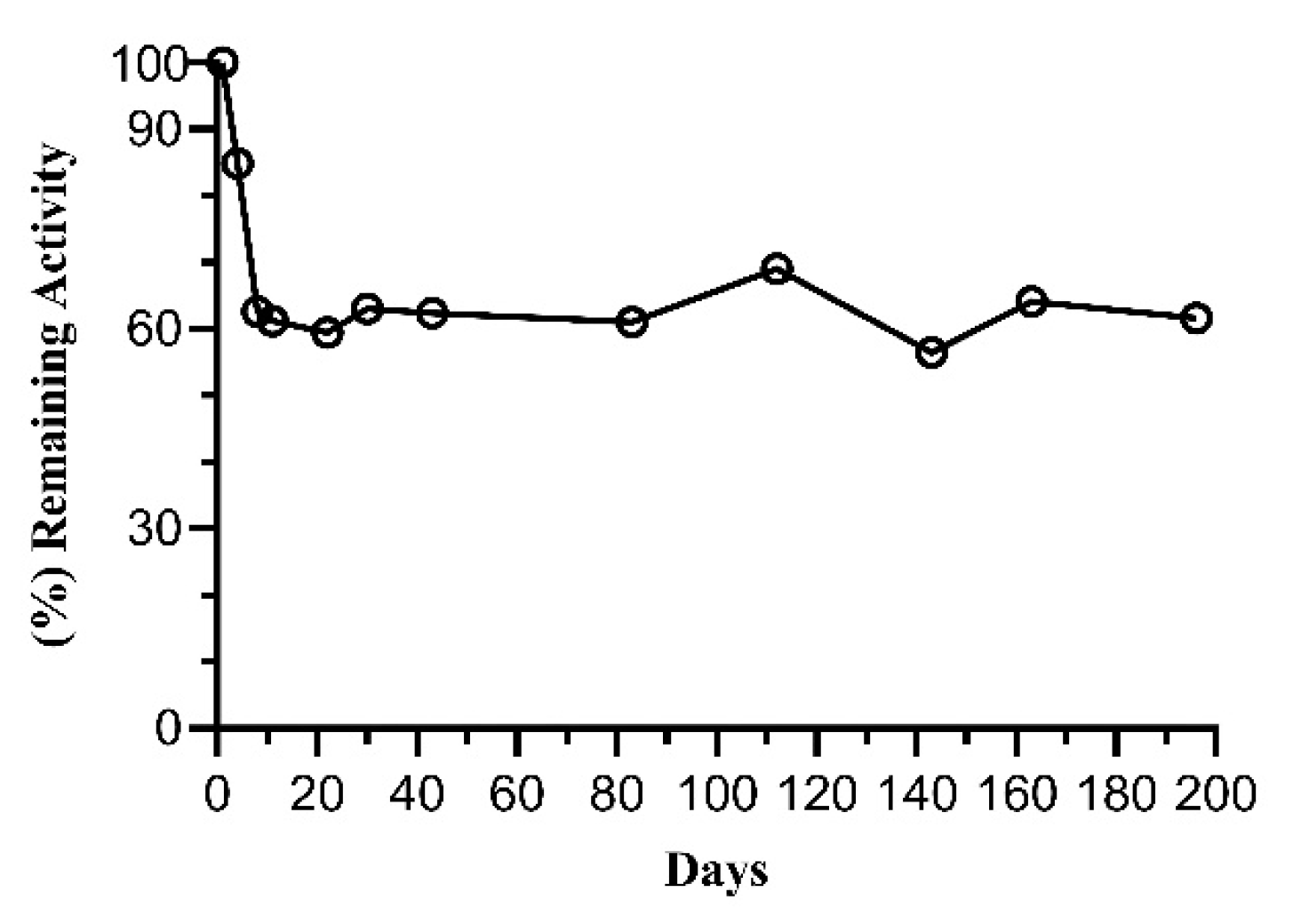
2.4. Tethering of FosAKP
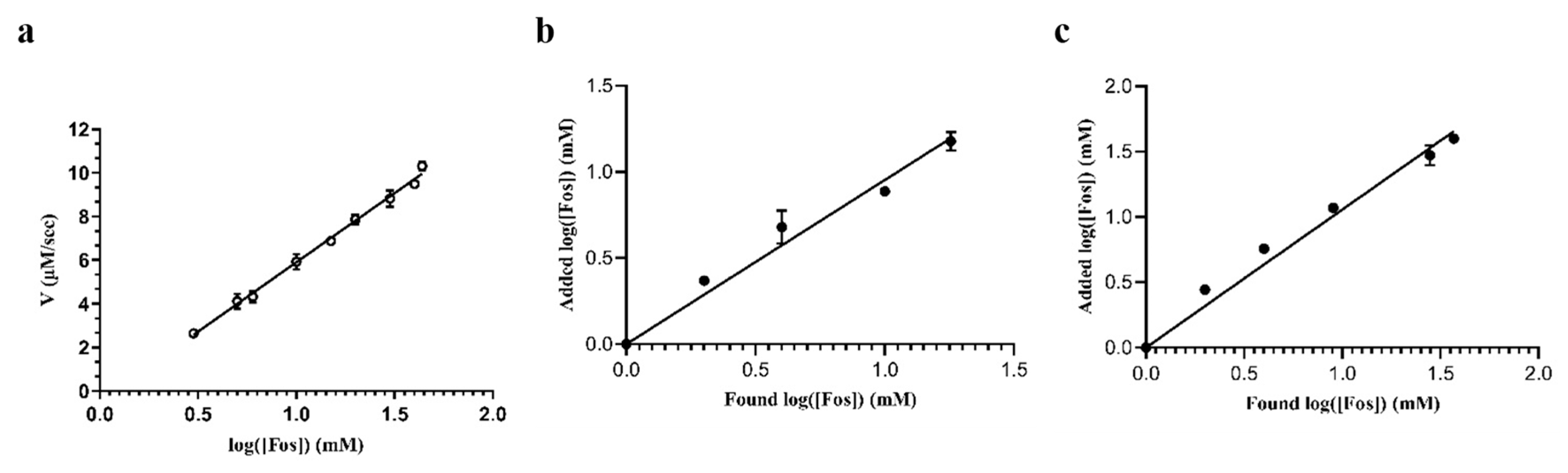
2.5. Determination of Fos in Tap Water and Sheep Plasma Samples
3. Materials and Methods
3.1. Materials
3.2. Methods
3.2.1. Molecular Cloning
3.2.2. Expression and Purification FosAPA and FosAKP
3.2.3. Assay of the Enzyme Activity and Protein Concentration Determination
3.2.4. Thermal Stability
3.2.5. Crystallization
3.2.6. Structure Determination
3.2.7. FosAKP Tethering via Covalent Cross-Linking
3.2.8. Kinetic Analysis of the Free and Tethered FosAKP
3.2.9. Biosensor Performance and Recovery Tests
3.2.10. Stability Analysis of Free and Immobilized FosAKP
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zayyad, H.; Eliakim-Raz, N.; Leibovici, L.; Paul, M. Revival of Old Antibiotics: Needs, the State of Evidence and Expectations. Int. J. Antimicrob. Agents 2017, 49, 536–541. [Google Scholar] [CrossRef] [PubMed]
- Klontz, E.H.; Tomich, A.D.; Günther, S.; Lemkul, J.A.; Deredge, D.; Silverstein, Z.; Shaw, J.F.; McElheny, C.; Doi, Y.; Wintrode, P.L.; et al. Structure and Dynamics of FosA-Mediated Fosfomycin Resistance in Klebsiella Pneumoniae and Escherichia Coli. Antimicrob. Agents Chemother. 2017, 61, e01572-17. [Google Scholar] [CrossRef]
- Dijkmans, A.C.; Ortiz Zacarías, N.V.; Burggraaf, J.; Mouton, J.W.; Wilms, E.B.; van Nieuwkoop, C.; Touw, D.J.; Stevens, J.; Kamerling, I.M.C. Fosfomycin: Pharmacological, Clinical and Future Perspectives. Antibiotics 2017, 6, 24. [Google Scholar] [CrossRef] [PubMed]
- Castañeda-García, A.; Blázquez, J.; Rodríguez-Rojas, A. Molecular Mechanisms and Clinical Impact of Acquired and Intrinsic Fosfomycin Resistance. Antibiotics 2013, 2, 217–236. [Google Scholar] [CrossRef]
- Grabein, B.; Graninger, W.; Rodríguez Baño, J.; Dinh, A.; Liesenfeld, D.B. Intravenous Fosfomycin—Back to the Future. Systematic Review and Meta-Analysis of the Clinical Literature. Clin. Microbiol. Infect. 2017, 23, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Karageorgopoulos, D.E.; Wang, R.; Yu, X.-H.; Falagas, M.E. Fosfomycin: Evaluation of the Published Evidence on the Emergence of Antimicrobial Resistance in Gram-Negative Pathogens. J. Antimicrob. Chemother. 2012, 67, 255–268. [Google Scholar] [CrossRef] [PubMed]
- Silver, L.L. Fosfomycin: Mechanism and Resistance. Cold Spring Harb. Perspect. Med. 2017, 7, a025262. [Google Scholar] [CrossRef] [PubMed]
- Benzerara, Y.; Gallah, S.; Hommeril, B.; Genel, N.; Decré, D.; Rottman, M.; Arlet, G. Emergence of Plasmid-Mediated Fosfomycin-Resistance Genes among Escherichia coli Isolates, France. Emerg. Infect. Dis. 2017, 23, 1564–1567. [Google Scholar] [CrossRef]
- Beharry, Z.; Palzkill, T. Functional Analysis of Active Site Residues of the Fosfomycin Resistance Enzyme FosA from Pseudomonas Aeruginosa. J. Biol. Chem. 2005, 280, 17786–17791. [Google Scholar] [CrossRef]
- Zurfluh, K.; Treier, A.; Schmitt, K.; Stephan, R. Mobile Fosfomycin Resistance Genes in Enterobacteriaceae—An Increasing Threat. MicrobiologyOpen 2020, 9, e1135. [Google Scholar] [CrossRef]
- Guo, Q.; Tomich, A.D.; McElheny, C.L.; Cooper, V.S.; Stoesser, N.; Wang, M.; Sluis-Cremer, N.; Doi, Y. Glutathione-S-Transferase FosA6 of Klebsiella Pneumoniae Origin Conferring Fosfomycin Resistance in ESBL-Producing Escherichia coli. J. Antimicrob. Chemother. 2016, 71, 2460–2465. [Google Scholar] [CrossRef] [PubMed]
- Pakhomova, S.; Rife, C.L.; Armstrong, R.N.; Newcomer, M.E. Structure of Fosfomycin Resistance Protein FosA from Transposon Tn2921. Protein Sci. 2004, 13, 1260–1265. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Wan, N.; Cheng, Z.; Mo, Y.; Liu, B.; Liu, H.; Raboanatahiry, N.; Yin, Y.; Li, M. Whole-Genome Identification and Expression Pattern of the Vicinal Oxygen Chelate Family in Rapeseed (Brassica napus L.). Front. Plant Sci. 2017, 8, 745. [Google Scholar] [CrossRef] [PubMed]
- Thornalley, P.J. Glyoxalase I—Structure, Function and a Critical Role in the Enzymatic Defence against Glycation. Biochem. Soc. Trans. 2003, 31, 1343–1348. [Google Scholar] [CrossRef] [PubMed]
- Brennerova, M.V.; Zavala-Meneses, S.G.; Josefiova, J.; Branny, P.; Buriankova, K.; Vetrovsky, T.; Junca, H. A Global Survey Reveals a Divergent Extradiol Dioxygenase Clade as a Widespread Complementary Contributor to the Biodegradation of Mono- and Polycyclic Aromatic Hydrocarbons. Environ. Res. 2022, 204, 111954. [Google Scholar] [CrossRef] [PubMed]
- Thompson, M.K.; Keithly, M.E.; Sulikowski, G.A.; Armstrong, R.N. Diversity in Fosfomycin Resistance Proteins. Perspect. Sci. 2015, 4, 17–23. [Google Scholar] [CrossRef]
- Bernat, B.A.; Armstrong, R.N. Elementary Steps in the Acquisition of Mn2+ by the Fosfomycin Resistance Protein (FosA). Biochemistry 2001, 40, 12712–12718. [Google Scholar] [CrossRef]
- Rife, C.L.; Pharris, R.E.; Newcomer, M.E.; Armstrong, R.N. Crystal Structure of a Genomically Encoded Fosfomycin Resistance Protein (FosA) at 1.19 Å Resolution by MAD Phasing off the L-III Edge of Tl+. J. Am. Chem. Soc. 2002, 124, 11001–11003. [Google Scholar] [CrossRef]
- Ortiz Zacarías, N.V.; Dijkmans, A.C.; Burggraaf, J.; Mouton, J.W.; Wilms, E.B.; van Nieuwkoop, C.; Touw, D.J.; Kamerling, I.M.C.; Stevens, J. Fosfomycin as a Potential Therapy for the Treatment of Systemic Infections: A Population Pharmacokinetic Model to Simulate Multiple Dosing Regimens. Pharmacol. Res. Perspect. 2018, 6, e00378. [Google Scholar] [CrossRef]
- Wiltsie, V.; Travis, S.; Shay, M.R.; Simmons, Z.; Frantom, P.; Thompson, M.K. Structural and Functional Characterization of Fosfomycin Resistance Conferred by FosB from Enterococcus Faecium. Protein Sci. 2022, 31, 580–590. [Google Scholar] [CrossRef]
- Sheldon, R.A.; Van Pelt, S. Enzyme Immobilisation in Biocatalysis: Why, What and How. Chem. Soc. Rev. 2013, 42, 6223–6235. [Google Scholar] [CrossRef] [PubMed]
- Chapman, J.; Ismail, A.; Dinu, C. Industrial Applications of Enzymes: Recent Advances, Techniques, and Outlooks. Catalysts 2018, 8, 238. [Google Scholar] [CrossRef]
- Chaniotakis, N.A. Enzyme Stabilization Strategies Based on Electrolytes and Polyelectrolytes for Biosensor Applications. Anal. Bioanal. Chem. 2004, 378, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Perperopoulou, F.; Fragoulaki, M.; Papageorgiou, A.C.; Labrou, N.E. Directed Evolution of a Glutathione Transferase for the Development of a Biosensor for Alachlor Determination. Symmetry 2021, 13, 461. [Google Scholar] [CrossRef]
- Hassan, M.E.; Yang, Q.; Xiao, Z.; Liu, L.; Wang, N.; Cui, X.; Yang, L. Impact of Immobilization Technology in Industrial and Pharmaceutical Applications. 3 Biotech. 2019, 9, 440. [Google Scholar] [CrossRef]
- Wijma, R.A.; Bahmany, S.; Wilms, E.B.; van Gelder, T.; Mouton, J.W.; Koch, B.C.P. A Fast and Sensitive LC–MS/MS Method for the Quantification of Fosfomycin in Human Urine and Plasma Using One Sample Preparation Method and HILIC Chromatography. J. Chromatogr. B 2017, 1061–1062, 263–269. [Google Scholar] [CrossRef]
- Parker, S.L.; Lipman, J.; Dimopoulos, G.; Roberts, J.A.; Wallis, S.C. A Validated Method for the Quantification of Fosfomycin on Dried Plasma Spots by HPLC–MS/MS: Application to a Pilot Pharmacokinetic Study in Humans. J. Pharm. Biomed. Anal. 2015, 115, 509–514. [Google Scholar] [CrossRef]
- Dieguez, S.; Soraci, A.; Tapia, O.; Carciochi, R.; Pérez, D.; Harkes, R.; Romano, O. Determination of Antibiotic Fosfomycin in Chicken Serum by Liquid Chromatography-Tandem Mass Spectrometry. J. Liq. Chromatogr. Relat. Technol. 2011, 34, 116–128. [Google Scholar] [CrossRef]
- Robert, X.; Gouet, P. Deciphering Key Features in Protein Structures with the New ENDscript Server. Nucleic Acids Res. 2014, 42, W320–W324. [Google Scholar] [CrossRef]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, Scalable Generation of High-quality Protein Multiple Sequence Alignments Using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef]
- Rodrigues, C.H.; Pires, D.E.; Ascher, D.B. DynaMut: Predicting the Impact of Mutations on Protein Conformation, Flexibility and Stability. Nucleic Acids Res. 2018, 46, W350–W355. [Google Scholar] [CrossRef] [PubMed]
- Rigsby, R.E.; Rife, C.L.; Fillgrove, K.L.; Newcomer, M.E.; Armstrong, R.N. Phosphonoformate: A Minimal Transition State Analogue Inhibitor of the Fosfomycin Resistance Protein, FosA. Biochemistry 2004, 43, 13666–13673. [Google Scholar] [CrossRef] [PubMed]
- Voběrková, S.; Solčány, V.; Vršanská, M.; Adam, V. Immobilization of Ligninolytic Enzymes from White-Rot Fungi in Cross-Linked Aggregates. Chemosphere 2018, 202, 694–707. [Google Scholar] [CrossRef] [PubMed]
- López-Gallego, F.; Guisán, J.M.; Betancor, L. Glutaraldehyde-Mediated Protein Immobilization. Methods Mol. Biol. 2013, 1051, 33–41. [Google Scholar] [CrossRef]
- Melo, R.R.D.; Alnoch, R.C.; Vilela, A.F.L.; Souza, E.M.D.; Krieger, N.; Ruller, R.; Sato, H.H.; Mateo, C. New Heterofunctional Supports Based on Glutaraldehyde-Activation: A Tool for Enzyme Immobilization at Neutral pH. Molecules 2017, 22, 1088. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.H.; Lee, S.H.; Lee, U.J.; Fermin, C.D.; Kim, M. Immobilized Enzymes in Biosensor Applications. Materials 2019, 12, 121. [Google Scholar] [CrossRef] [PubMed]
- Migneault, I.; Dartiguenave, C.; Bertrand, M.J.; Waldron, K.C. Glutaraldehyde: Behavior in Aqueous Solution, Reaction with Proteins, and Application to Enzyme Crosslinking. Biotechniques 2004, 37, 790–796, 798–802. [Google Scholar] [CrossRef] [PubMed]
- Pérez, D.S.; Tapia, M.O.; Soraci, A.L. Fosfomycin: Uses and Potentialities in Veterinary Medicine. Open Vet. J. 2014, 4, 26–43. [Google Scholar]
- Goto, M.; Sugiyama, M.; Nakajima, S.; Yamashina, H. Fosfomycin Kinetics after Intravenous and Oral Administration to Human Volunteers. Antimicrob. Agents Chemother. 1981, 20, 393–397. [Google Scholar] [CrossRef]
- Labrou, N.E.; Mello, L.V.; Clonis, Y.D. Functional and Structural Roles of the Glutathione-Binding Residues in Maize (Zea Mays) Glutathione S-Transferase I. Biochem. J. 2001, 358, 101–110. [Google Scholar] [CrossRef]
- Georgakis, N.; Poudel, N.; Papageorgiou, A.C.; Labrou, N.E. Comparative Structural and Functional Analysis of Phi Class Glutathione Transferases Involved in Multiple-Herbicide Resistance of Grass Weeds and Crops. Plant Physiol. Biochem. 2020, 149, 266–276. [Google Scholar] [CrossRef] [PubMed]
- Laemmli, U.K. Cleavage of Structural Proteins during the Assembly of the Head of Bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.M. A Rapid and Sensitive Method for the Quantitation of Microgram Quantities of Protein Utilizing the Principle of Protein-Dye Binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Huynh, K.; Partch, C.L. Analysis of Protein Stability and Ligand Interactions by Thermal Shift Assay. CP Protein Sci. 2015, 79, 28.9.1–28.9.14. [Google Scholar] [CrossRef]
- Adams, P.D.; Afonine, P.V.; Bunkóczi, G.; Chen, V.B.; Davis, I.W.; Echols, N.; Headd, J.J.; Hung, L.-W.; Kapral, G.J.; Grosse-Kunstleve, R.W.; et al. PHENIX: A Comprehensive Python-Based System for Macromolecular Structure Solution. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 213–221. [Google Scholar] [CrossRef]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and Development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Data Processing | FosAKP |
|---|---|
| Beamline | BioMAX (MAX IV, Lund, Sweden) |
| Wavelength (Å) | 0.9919 |
| Resolution (Å) | 149.2–1.48 (1.51–1.48) |
| Space group | P212121 |
| Unit cell a, b, c (Å) | 40.6, 47.0, and 149.2 |
| No. of molecules/asymmetric unit | 2 |
| No. of unique reflections | 48,410 (2174) |
| Completeness (%) | 99.4 (92.2) |
| Multiplicity | 9.8 (6.3) |
| Mosaicity (°) | 0.13 |
| Rmeas | 0.089 (1.452) |
| CC1/2 | 0.999 (0.666) |
| Mean (I/σ(I)) | 11.8 (1.2) |
| Wilson B factor (Å2) | 22.2 |
| Refinement | |
| Resolution (Å) | 44.85–1.48 |
| No. of reflections used | 48,242 |
| Rcryst/Rfree | 0.173/0.204 |
| RMSD in bonds (Å) | 0.005 |
| RMSD in angles (°) | 0.899 |
| No. of protein atoms | 2203 |
| No. of water molecules | 383 |
| Average B-factor (Å2) | 27.7 |
| Ramachandran favored/outliers (%) | 99.6/0.0 |
| Clashscore | 4.6 |
| PDB ID | 8R37 |
| Buffer System | Tm (°C) |
|---|---|
| 20 mM NaH2PO4 (pH 8.0) | 65.9 ± 0.3 |
| 20 mM NaH2PO4 (pH 8.0) and 0.05 mM MnCl2 | 67.3 ± 0.1 |
| 20 mM NaH2PO4 (pH 8.0) and 100 mM KCl | 64.7 ± 0.3 |
| 20 mM NaH2PO4 (pH 8.0) and 0.05 mM MnCl2/100 mM KCl | 67.5 ± 0.2 |
| 20 mM NaH2PO4 (pH 7.0) | 63.0 ± 0.2 |
| 20 mM HEPES (pH 8.0) | 61.9 ± 0.2 |
| 20 mM MOPS (pH 8.0) | 61.0 ± 0.2 |
| 10 mM Tris/75 mM NaCl (pH 7.5) | 59.4 ± 0.1 |
| 20 mM Tris (pH 7.8) | 58.7 ± 0.2 |
| 20 mM HEPES (pH 7.0) | 57.6 ± 0.1 |
| 20 mM L-Arg/20 mM L-Glu (pH 7.5) | 56.8 ± 0.3 |
| 10 mM acetate (pH 4.2) | 53.7 ± 0.0 |
| 10 mM acetate (pH 5.2) | 44.3 ± 0.0 |
| Run Order | Glutaraldehyde (%v/v) | BSA (mg) | Enzyme (units) |
|---|---|---|---|
| 1 | 0.5 | 16.5 | 13.5 |
| 2 | 0.5 | 16.5 | 6.8 |
| 3 | 0.5 | 33.0 | 13.5 |
| 4 | 0.5 | 33.0 | 6.8 |
| 5 | 0.7 | 16.5 | 13.5 |
| 6 | 0.7 | 16.5 | 6.8 |
| 7 | 0.7 | 33.0 | 13.5 |
| 8 | 0.7 | 33.0 | 6.8 |
| 9 | 1.0 | 16.5 | 13.5 |
| 10 | 1.0 | 16.5 | 6.8 |
| 11 | 1.0 | 33.0 | 13.5 |
| 12 | 1.0 | 33.0 | 6.8 |
| 13 | 0.7 | 22.0 | 6.8 |
| 14 | 1.0 | 22.0 | 6.8 |
| Fos Concentration (mM) | Recovery Tests (%) | Reproducibility of Recovery Tests (%) |
|---|---|---|
| 2 | 122.5 ± 4.8 | 6.7 (n = 3) |
| 4 | 121.7 ± 4.6 | 6.6 (n = 4) |
| 10 | 88.7 ± 2.3 | 4.6 (n = 3) |
| 18 | 93.9 ± 4.3 | 8.0 (n = 4) |
| Fos Concentration (mM) | Recovery Tests (%) | Reproducibility of Recovery Tests (%) |
|---|---|---|
| 2 | 146.8 ± 0.6 | 0.7 (n = 3) |
| 4 | 116.8 ± 5.5 | 8.1(n = 3) |
| 9 | 112.0 ± 1.6 | 2.1 (n = 3) |
| 28 | 101.6 ± 5.2 | 7.2 (n = 3) |
| 37 | 102.0 ± 0.06 | 0.62 (n = 3) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Varotsou, C.; Ataya, F.; Papageorgiou, A.C.; Labrou, N.E. Structural Studies of Klebsiella pneumoniae Fosfomycin-Resistance Protein and Its Application for the Development of an Optical Biosensor for Fosfomycin Determination. Int. J. Mol. Sci. 2024, 25, 85. https://doi.org/10.3390/ijms25010085
Varotsou C, Ataya F, Papageorgiou AC, Labrou NE. Structural Studies of Klebsiella pneumoniae Fosfomycin-Resistance Protein and Its Application for the Development of an Optical Biosensor for Fosfomycin Determination. International Journal of Molecular Sciences. 2024; 25(1):85. https://doi.org/10.3390/ijms25010085
Chicago/Turabian StyleVarotsou, Christina, Farid Ataya, Anastassios C. Papageorgiou, and Nikolaos E. Labrou. 2024. "Structural Studies of Klebsiella pneumoniae Fosfomycin-Resistance Protein and Its Application for the Development of an Optical Biosensor for Fosfomycin Determination" International Journal of Molecular Sciences 25, no. 1: 85. https://doi.org/10.3390/ijms25010085
APA StyleVarotsou, C., Ataya, F., Papageorgiou, A. C., & Labrou, N. E. (2024). Structural Studies of Klebsiella pneumoniae Fosfomycin-Resistance Protein and Its Application for the Development of an Optical Biosensor for Fosfomycin Determination. International Journal of Molecular Sciences, 25(1), 85. https://doi.org/10.3390/ijms25010085