
Structure-Based De Novo Design for the Discovery of Miniprotein Inhibitors Targeting Oncogenic Mutant BRAF
Abstract
:1. Introduction
2. Results and Discussion
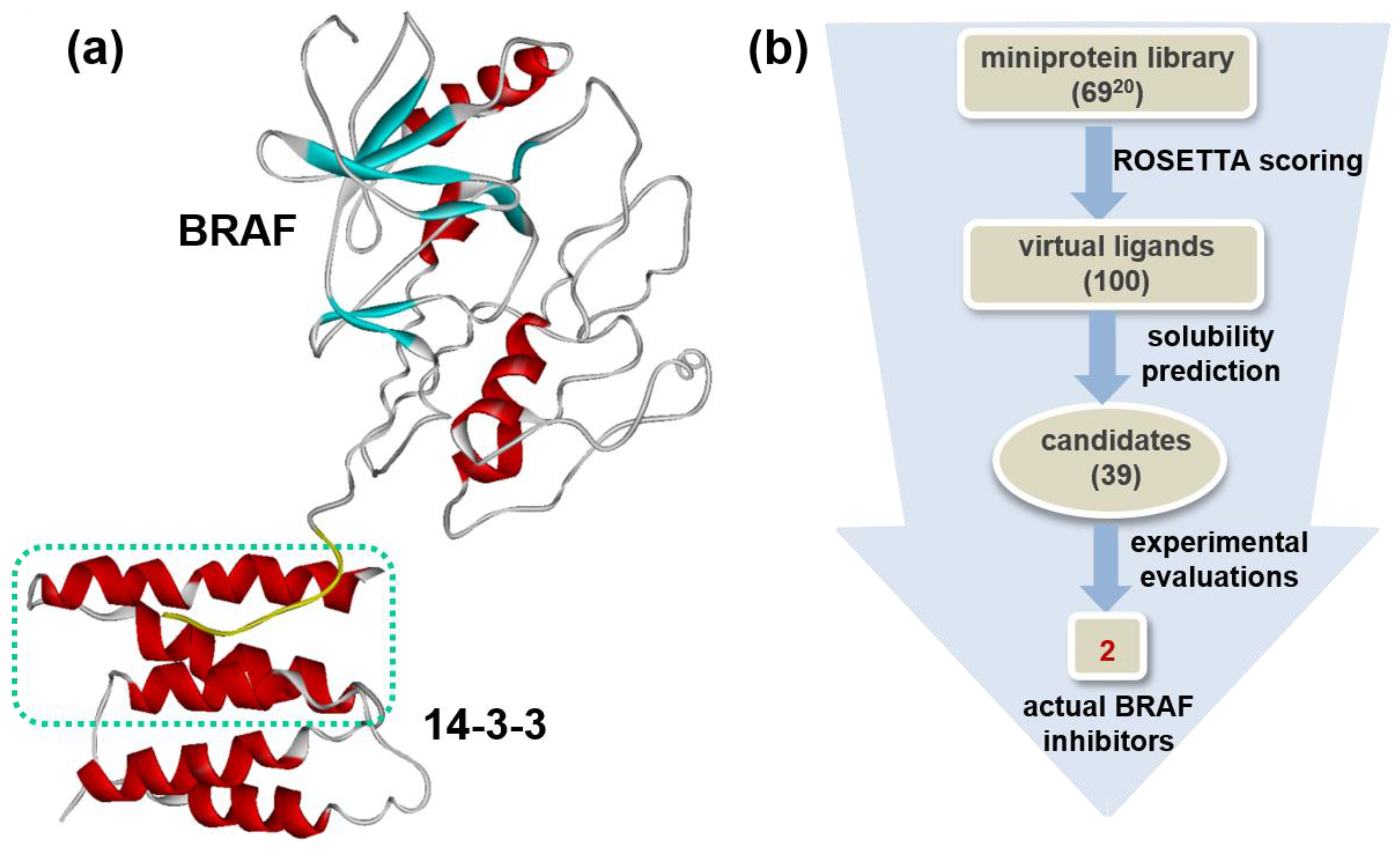
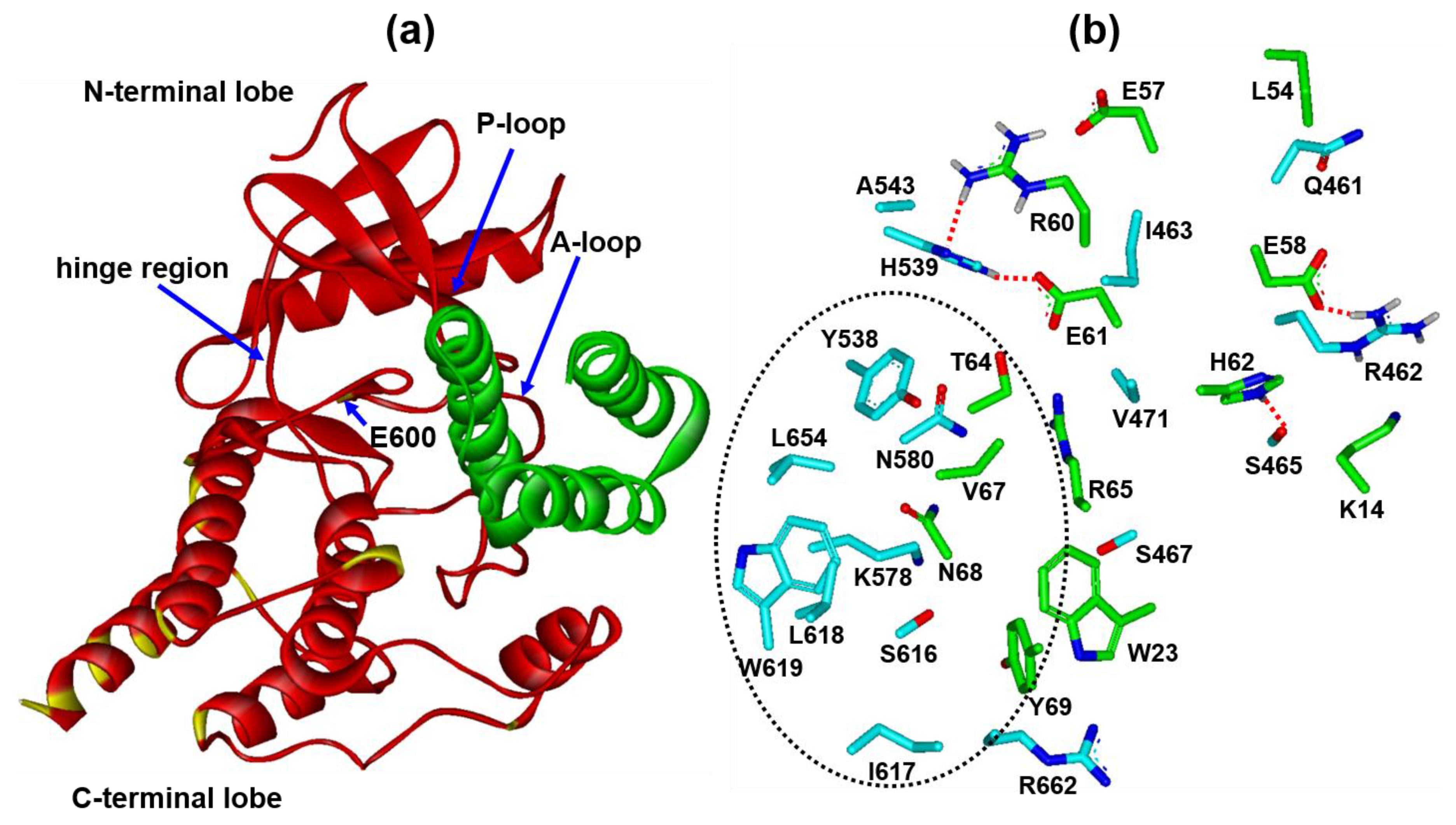
2.1. De Novo Design of Candidate Miniprotein Inhibitors of V600E Mutant BRAF15mut
2.2. Expression and Purification of Candidate Miniprotein Inhibitors
2.3. Expression and Purification of BRAF Kinase Domain and MEK1
2.4. BRAF-MEK1 Binding Assays
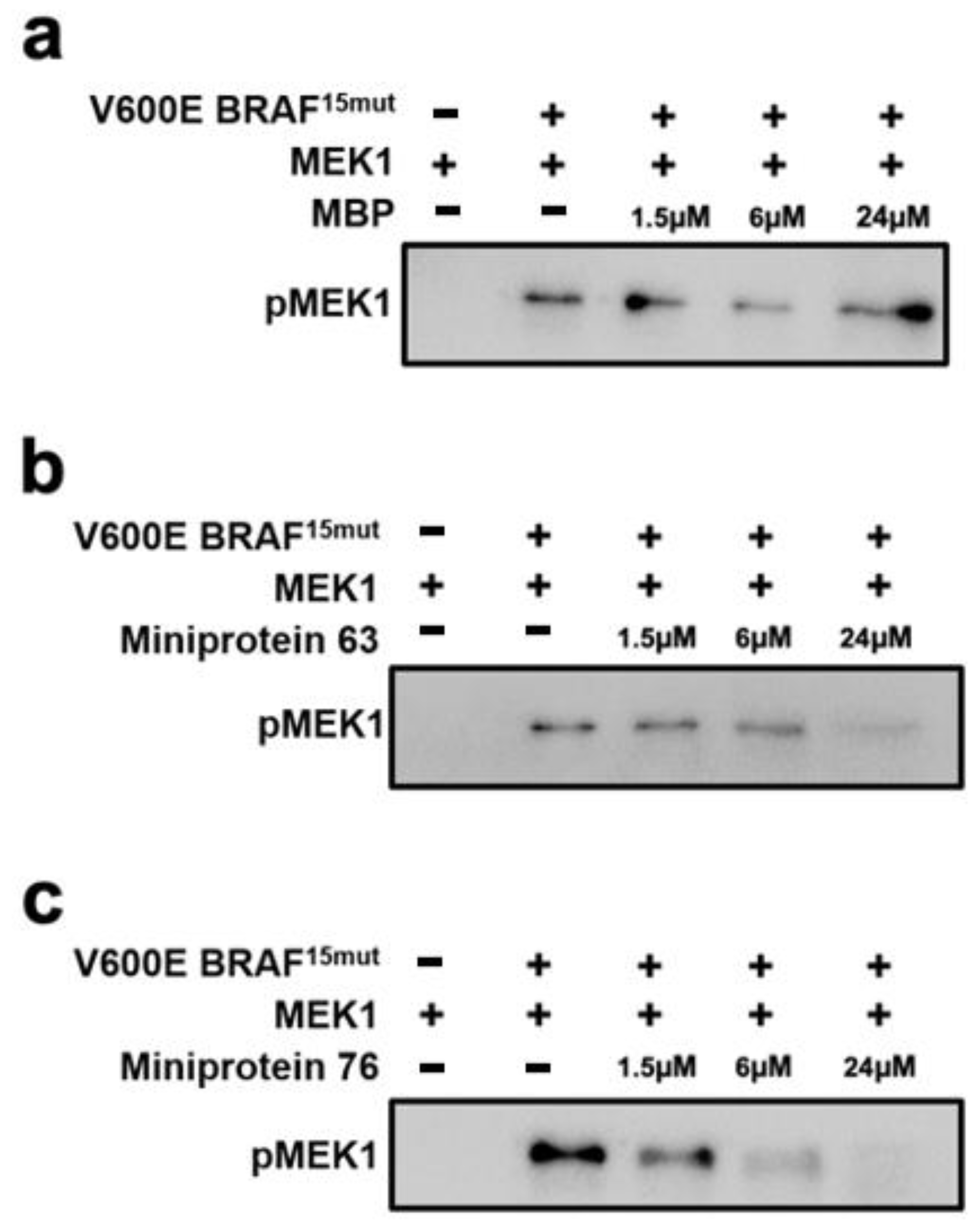
2.5. In Vitro Kinase Inhibition Assays
2.6. Cell-Based Assays
3. Materials and Methods
3.1. Preparation of All-Atom Receptor Model for V600E Mutant BRAF
3.2. Preparation of the Miniprotein Scaffold for De Novo Design
3.3. Docking Simulations to Find the Most Probable Binding Configuration
3.4. De Novo Design of Miniprotein Inhibitors of V600E Mutant BRAF
3.5. Rescoring the Putative Miniprotein Binders with the Hydration Free Energy
3.6. Molecular Constructs
3.7. Expression and Purification of Proteins
3.8. BRAF Binding Assays
3.9. In Vitro Kinase Inhibition Assays
3.10. Cell Culture and DNA Transfections
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Guo, Y.J.; Pan, W.W.; Liu, S.B.; Shen, Z.F.; Xu, Y.; Hu, L.L. ERK/MAPK signaling pathway and tumorigenesis. Exp. Ther. Med. 2020, 19, 1997–2007. [Google Scholar]
- Johansson, C.H.; Brage, S.E. BRAF inhibitors in cancer therapy. Pharmacol. Ther. 2014, 142, 176–182. [Google Scholar] [CrossRef] [PubMed]
- Wellbrock, C.; Hurlstone, A. BRAF as therapeutic target in melanoma. Biochem. Pharmacol. 2010, 80, 561–567. [Google Scholar] [CrossRef] [PubMed]
- Flaherty, K.T.; Puzanov, I.; Kim, K.B.; Ribas, A.; McArthur, G.A.; Sosman, J.A.; O’Dwyer, P.J.; Lee, R.J.; Grippo, J.F.; Nolop, K.; et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N. Engl. J. Med. 2010, 363, 809–819. [Google Scholar] [CrossRef]
- White, R.; Otaibi, Z.; Rao, R.; Finley, G. BRAF V600E mutation in multiple primary malignancies: A hairy affair. Cureus 2018, 10, e3600. [Google Scholar] [CrossRef] [PubMed]
- Hoeflich, K.P.; Gray, D.C.; Eby, M.T.; Tien, J.Y.; Wong, L.; Bower, J.; Gogineni, A.; Zha, J.; Cole, M.J.; Stern, H.M.; et al. Oncogenic BRAF is required for tumor growth and maintenance in melanoma models. Cancer Res. 2006, 66, 999–1006. [Google Scholar] [CrossRef]
- Berger, D.M.; Torres, N.; Dutia, M.; Powell, D.; Ciszewski, G.; Gopalsamy, A.; Levin, J.I.; Kim, K.H.; Xu, W.; Wilhelm, J.; et al. Non-hinge-binding pyrazolo[1,5-a]pyrimidines as potent B-Raf kinase inhibitors. Bioorg. Med. Chem. Lett. 2009, 19, 6519–6523. [Google Scholar] [CrossRef]
- Qin, J.; Xie, P.; Ventocilla, C.; Zhou, G.; Vultur, A.; Chen, Q.; Liu, Q.; Herlyn, M.; Winkler, J.; Marmorstein, R. Identification of a novel family of BRAF V600E inhibitors. J. Med. Chem. 2012, 55, 5220–5230. [Google Scholar] [CrossRef]
- Pan, J.H.; Zhou, H.; Zhu, S.B.; Huang, J.L.; Zhao, X.X.; Ding, H.; Pan, Y.L. Development of small-molecule therapeutics and strategies for targeting RAF kinase in BRAF-mutant colorectal cancer. Cancer Manag. Res. 2018, 10, 2289–2301. [Google Scholar] [CrossRef]
- Karoulia, Z.; Gavathiotis, E.; Poulikakos, P.I. New perspectives for targeting RAF kinase in human cancer. Nat. Rev. Cancer 2017, 17, 676–691. [Google Scholar] [CrossRef]
- Gunderwala, A.Y.; Nimbvikar, A.A.; Cope, N.J.; Li, Z.; Wang, Z. Development of allosteric BRAF peptide inhibitors targeting the dimer interface of BRAF. ACS Chem. Biol. 2019, 14, 1471–1480. [Google Scholar] [CrossRef] [PubMed]
- Yao, Z.; Torres, N.M.; Tao, A.; Gao, Y.; Luo, L.; Li, Q.; de Stanchina, E.; Abdel-Wahab, O.; Solit, D.B.; Poulikakos, P.I.; et al. BRAF mutants evade ERK-dependent feedback by different mechanisms that determine their sensitivity to pharmacologic inhibition. Cancer Cell 2015, 28, 370–383. [Google Scholar] [CrossRef] [PubMed]
- Agianian, B.; Gavathiotis, E. Current insights of BRAF inhibitors in cancer. J. Med. Chem. 2018, 61, 5775–5793. [Google Scholar] [CrossRef] [PubMed]
- Crook, Z.R.; Nairn, N.W.; Olson, J.M. Miniproteins as a powerful modality in drug development. Trends Biochem. Sci. 2020, 45, 332–346. [Google Scholar] [CrossRef] [PubMed]
- Marchand, A.; Van Hall-Beauvais, A.K.; Correia, B.E. Computational design of novel protein–protein interactions—An overview on methodological approaches and applications. Curr. Opin. Struct. Biol. 2022, 74, 102370. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Goreshnik, I.; Coventry, B.; Case, J.B.; Miller, L.; Kozodoy, L.; Chen, R.E.; Carter, L.; Walls, A.C.; Park, Y.J.; et al. De novo design of picomolar SARS-CoV-2 miniprotein inhibitors. Science 2020, 370, 426–431. [Google Scholar] [CrossRef] [PubMed]
- Chevalier, A.; Silva, D.A.; Rocklin, G.J.; Hicks, D.R.; Vergara, R.; Murapa, P.; Bernard, S.M.; Zhang, L.; Lam, K.H.; Yao, G.; et al. Massively parallel de novo protein design for targeted therapeutics. Nature 2017, 550, 74–79. [Google Scholar] [CrossRef] [PubMed]
- Ozga, K.; Berlicki, L. Design and engineering of miniproteins. ACS Bio Med Chem Au 2022, 2, 316–327. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.; Kang, H.; Park, H. Extended solvent-contact model for protein solvation: Test cases for dipeptides. J. Mol. Graph. Model. 2013, 42, 50–59. [Google Scholar] [CrossRef]
- Smialowski, P.; Martin-Galiano, A.J.; Mikolajka, A.; Girschick, T.; Holak, T.A.; Frishman, D. Protein solubility: Sequence-based prediction and experimental verification. Bioinformatics 2007, 23, 2536–2542. [Google Scholar] [CrossRef]
- Ptak-Kaczor, M.; Banach, M.; Stapor, K.; Fabian, P.; Konieczny, L.; Roterman, I. Solubility and aggregation of selected proteins interpreted on the basis of hydrophobicity distribution. Int. J. Mol. Sci. 2021, 22, 5002. [Google Scholar] [CrossRef] [PubMed]
- Qing, R.; Hao, S.; Smorodina, E.; Jin, D.; Zalevsky, A.; Zhang, S. Protein design: From the aspect of water solubility and stability. Chem. Rev. 2022, 122, 14085–14179. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.E.; Wilsbacher, J.L.; Collisson, T.; Cobb, M.H. The N-terminal ERK-binding site of MEK1 is required for efficient feedback phosphorylation by ERK2 in vitro and ERK activation in vivo. J. Biol. Chem. 1999, 274, 34029–34035. [Google Scholar] [CrossRef] [PubMed]
- Wan, P.T.C.; Garnett, M.J.; Roe, S.M.; Lee, S.; Niculescu-Duvaz, D.; Good, V.M.; Jones, C.M.; Marshall, C.J.; Springer, C.J.; Barford, D.; et al. Mechanism of activation of the Raf-Erk signaling pathway by oncogenic mutations of B-Raf. Cell 2004, 116, 855–867. [Google Scholar] [CrossRef] [PubMed]
- Tsai, J.; Lee, J.T.; Wang, W.; Zhang, J.; Cho, H.; Mamo, S.; Bremer, R.; Gillette, S.; Kong, J.; Haass, N.K.; et al. Discovery of a selective inhibitor of oncogenic B-Raf kinase with potent antimelanoma activity. Proc. Natl. Acad. Sci. USA 2008, 105, 3041–3046. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.; Lin, L.Y.; Zhou, J.O.; Kibby, E.; Sia, T.W.; Tillis, T.D.; Vapuryan, N.; Xu, M.R.; Potluri, R.; Shin, Y.; et al. Identification and characterization of a B-Raf kinase α-helix critical for the activity of MEK kinase in MAPK signaling. Biochemistry 2020, 59, 4755–4765. [Google Scholar] [CrossRef] [PubMed]
- Schneider, M.; Delfosse, V.; Gelin, M.; Grimaldi, M.; Granell, M.; Heriaud, L.; Pons, J.; Gonsaud, M.; Balaguer, B.; Bourguet, W.; et al. Structure-based and knowledge-informed design of B-Raf inhibitors devoid of deleterious PXR binding. J. Med. Chem. 2021, 65, 1552–1566. [Google Scholar] [CrossRef] [PubMed]
- Aoki, K.; Yamada, M.; Kunida, K.; Yasuda, S.; Matsuda, M. Processive phosphorylation of ERK MAP kinase in mammalian cells. Proc. Natl. Acad. Sci. USA 2011, 108, 12675–12680. [Google Scholar] [CrossRef] [PubMed]
- Whitehurst, A.; Cobb, M.H.; White, M.A. Stimulus-coupled spatial restriction of extracellular signal-regulated kinase 1/2 activity contributes to the specificity of signal-response pathways. Mol. Cell. Biol. 2004, 24, 10145–10150. [Google Scholar] [CrossRef]
- Tang, H.C.; Chen, Y.C. Insight into molecular dynamics simulation of BRAF(V600E) and potent novel inhibitors for malignant melanoma. Int. J. Nanomed. 2015, 10, 3131–3146. [Google Scholar]
- Muley, L.; Baum, B.; Smolinski, M.; Freindorf, M.; Heine, A.; Klebe, G.; Hangauer, D.G. Enhancement of hydrophobic interactions and hydrogen bond strength by cooperativity: Synthesis, modeling, and molecular dynamics simulations of a congeneric series of thrombin Inhibitors. J. Med. Chem. 2010, 53, 2126–2135. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.H.; Ueng, S.H.; Tseng, C.T.; Hung, M.S.; Song, J.S.; Wu, J.S.; Liao, F.Y.; Fan, Y.S.; Wu, M.H.; Hsiao, W.C.; et al. Important hydrogen bond net-works in indoleamine 2,3-dioxygenase 1 (IDO1) inhibitor design revealed by crystal structures of imidazoleisoindole derivatives with IDO1. J. Med. Chem. 2016, 59, 282–293. [Google Scholar] [CrossRef] [PubMed]
- Arozarena, I.; Wellbrock, C. Overcoming resistance to BRAF inhibitors. Ann. Transl. Med. 2017, 5, 387. [Google Scholar] [CrossRef] [PubMed]
- Haling, J.R.; Sudhamsu, J.; Yen, I.; Sideris, S.; Sandoval, W.; Phung, W.; Bravo, B.J.; Giannetti, A.M.; Peck, A.; Masselot, A.; et al. Structure of the BRAF-MEK complex reveals a kinase activity independent role for BRAF in MAPK signaling. Cancer Cell 2014, 26, 402–413. [Google Scholar] [CrossRef] [PubMed]
- Sali, A.; Blundell, T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef] [PubMed]
- Kondo, Y.; Ognjenovic, J.; Banerjee, S.; Karandur, D.; Merk, A.; Kulhanek, K.; Wong, K.; Roose, J.P.; Subramaniam, S.; Kuriyan, J. Cryo-EM structure of a dimeric B-Raf:14-3-3 complex reveals asymmetry in the active sites of B-Raf kinases. Science 2019, 366, 109–115. [Google Scholar] [CrossRef]
- Case, D.A.; Cheatham, T.E., III; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M., Jr.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The AMBER biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [PubMed]
- Gray, J.J.; Moughon, S.; Wang, C.; Schueler-Furman, O.; Kuhlman, B.; Rohl, C.A.; Baker, D. Protein-protein docking with simultaneous optimization of rigid-body displacement and side-chain conformations. J. Mol. Biol. 2003, 331, 281–299. [Google Scholar] [CrossRef] [PubMed]
- Alford, R.F.; Leaver-Fay, A.; Jeliazkov, J.R.; O’Meara, M.J.; DiMaio, F.P.; Park, H.; Shapovalov, M.V.; Renfrew, P.D.; Mulligan, V.K.; Kappel, K.; et al. The Rosetta all-atom energy function for macromolecular modeling and design. J. Chem. Theory Comput. 2017, 13, 3031–3048. [Google Scholar] [CrossRef]
- Huang, P.S.; Ban, Y.E.A.; Richter, F.; Andre, I.; Vernon, R.; Schief, W.R.; Baker, D. RosettaRemodel: A generalized framework for flexible backbone protein design. PLoS ONE 2011, 6, e24109. [Google Scholar] [CrossRef]
- Koga, N.; Tatsumi-Koga, R.; Liu, G.; Xiao, R.; Acton, T.B.; Montelione, G.T.; Baker, D. Principles for designing ideal protein structures. Nature 2012, 491, 222–227. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.; Choi, H.; Park, H. Prediction of molecular solvation free energy based on the optimization of atomic solvation parameters with genetic algorithm. J. Chem. Inf. Model. 2007, 47, 509–514. [Google Scholar] [CrossRef] [PubMed]
- Chung, K.C.; Park, H. Accuracy enhancement in the estimation of molecular hydration free energies by implementing the intramolecular hydrogen bond effects. J. Cheminform. 2015, 7, 57. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No | Amino Acid Sequence |
|---|---|
| 21 | AVIKNKALQMCWNRTCVWAANEQLQASLDLIVFYMSGLRNTCLMLMLRLMFVSELLVLDRELHKDMVAV |
| 33 | APKREVAFMELLTRCLVTVLNNQAQTKLQLIDCYLADLLMRVLNRLAMMMLMAHLNLLMQLAYQGMHAQ |
| 45 | ADIMMCALGWEISVVKVFGYNNPSMDQLLNNKWFLGNGWNYKFMRMEVMLGMRAWSLTTVENLLIELIG |
| 51 | LVSCQFNSMWKFTSMEVWNYNTMLASQMWGHVYTNATLLANLLMGMMIMCKKLRLTSWMATDVVLGGDW |
| 59 | GHRCNMARANLLTSMASIGLYTMYDGIMYTECILIHDNQIRVKMWLAQMFKMEVSAFDLRDDSSLYHLG |
| 60 | QANGMTTLMFLNILNWVWHYQTMPRAAEDGYVHWLNIGVKMELMRMCWCMWKARACAHMLQDDTQFSSH |
| 63 | SIVGQGAQQMLVSKIKCTVANEWLDRSMLTSKFAMFVLLNEIDMLDIMLEAMGISLLEDREHQTRRVNY |
| 64 | PDKCAMASEWLLGTNWCWMYYGRQASIYQTIHFYLAMKAGYQFVYMFVMMAMRHWLIAMMRKDYRMLAK |
| 70 | AVINQVAWDQWNTLMTPDVYACCLIALRLGIFTALACGTARLDMRYTIRQEDKQLVMQMNLLDKIVFEV |
| 71 | AVIRDVAKWWLLHNMWSIVQTNQQIGNTHDQVCNSYDTLVRVLMSPEMIFDATRLDNFEVLHCYLMMQV |
| 75 | CNSLILKWLLFGGDHWHLRGVTQILTREIACTAWMEQWLSVDSSLMSNNRASNYMNVERCWLWKETDAA |
| 76 | SIQGQGARQCLVSKLKCTVANEWLDRWMLTSKFAMFVLLNEIDMLDIMLEAMGISIEEDREHQTRRVNY |
| 77 | GTIDQRWWNHFDSTTERTVHEMTSDCHMRMQKSMTQNRCSMVVVPHRMRYVGYKGMRNINNMGYCGDAA |
| 78 | LELLRAEETAHTSTLHIDVEVISTTAFAYCRMDQFTQISNDALETMNQLESGLAWMAVRGCYMMTCVGE |
| 79 | VTMQFSFNSLLEGRAEAHMGTILITAMAITIKKLKGRGAFGKKLLEWNMAMACSHNAQLGGMDFQHKEG |
| 83 | SEGAAKASELLQSMNLCWMYIGCQNSIQQTIAVYLAMQAGMQFVYMFVCSAAQHWLKTMDRKNYLMLVK |
| 84 | ADDQNTEIVWLLVSQWWWLYNFVEIANLLLVSNMNTIWNNRMKGRMNMNCLMYRMLGNRGMLCLQDGQA |
| 86 | TAVLIVFLEGLSAATGRHVGARMPDAAYSKNYHMMFAAAQACDYRGNMKSLARGFAAAQWMMTWVSFIS |
| 91 | NACEFKALDVDRGNLHDAHRNCGGRKNNAMQVEAMIVLSNGFGAGPNVWMLKAALLEVMTLLTCDTLKR |
| 92 | GVIRGHARVRALAGRMYALLDVTWCADSKSRRIMQGGFLVHIFNQEWTLYGELAKTTWVWRHFFQIMDF |
| 93 | CKQMQALHAFSSEADRQHLQACAWKGMNVGCVLEVVVLDAIMGNQNNDQYFSTNIMMQMSTVFWNLQGM |
| 94 | PCVWTGFREFKGGQRGKERLAMCEDLMCEADWWGGNQNRDRGSIFTRVKGSGALGKISIAERGARNNAV |
| 98 | TMNTIMMKVSFCAGARIVHLAVLVIGLDVDRSELMDTAIDTMVHWNFAISEVYMVLWGSSQSKSDCQEV |
| 100 | ACTETIWTVTCNGHLEMRTYWAYCAMFTFMESHITDNLHWTAAMFTQEVRTRTFYAGVQGNSMWWTVRN |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ham, J.M.; Kim, M.; Kim, T.; Ryu, S.E.; Park, H. Structure-Based De Novo Design for the Discovery of Miniprotein Inhibitors Targeting Oncogenic Mutant BRAF. Int. J. Mol. Sci. 2024, 25, 5535. https://doi.org/10.3390/ijms25105535
Ham JM, Kim M, Kim T, Ryu SE, Park H. Structure-Based De Novo Design for the Discovery of Miniprotein Inhibitors Targeting Oncogenic Mutant BRAF. International Journal of Molecular Sciences. 2024; 25(10):5535. https://doi.org/10.3390/ijms25105535
Chicago/Turabian StyleHam, Jae Min, Myeongbin Kim, Taeho Kim, Seong Eon Ryu, and Hwangseo Park. 2024. "Structure-Based De Novo Design for the Discovery of Miniprotein Inhibitors Targeting Oncogenic Mutant BRAF" International Journal of Molecular Sciences 25, no. 10: 5535. https://doi.org/10.3390/ijms25105535
APA StyleHam, J. M., Kim, M., Kim, T., Ryu, S. E., & Park, H. (2024). Structure-Based De Novo Design for the Discovery of Miniprotein Inhibitors Targeting Oncogenic Mutant BRAF. International Journal of Molecular Sciences, 25(10), 5535. https://doi.org/10.3390/ijms25105535