
1,25-Dihydroxyvitamin D3 Suppresses UV-Induced Poly(ADP-Ribose) Levels in Primary Human Keratinocytes, as Detected by a Novel Whole-Cell ELISA
 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. UV Dose–Response Study to Detect Phosphorylated PARP Product, pADPr with a Novel Whole-Cell ELISA
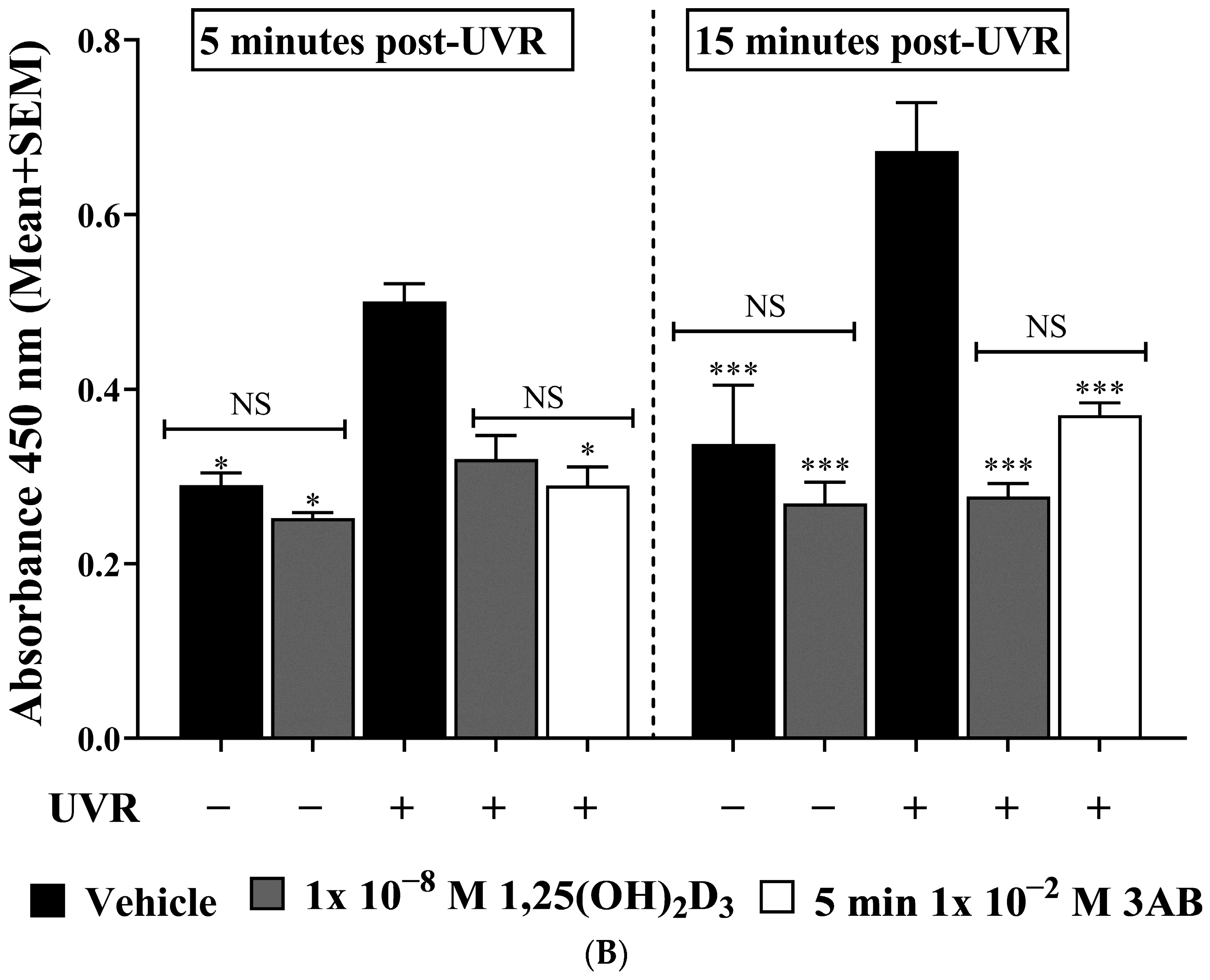
2.2. UV-Induced pADPr Levels Were Significantly Reduced by Treatment with 1,25(OH)2D3 or 3AB in Human Primary Keratinocytes
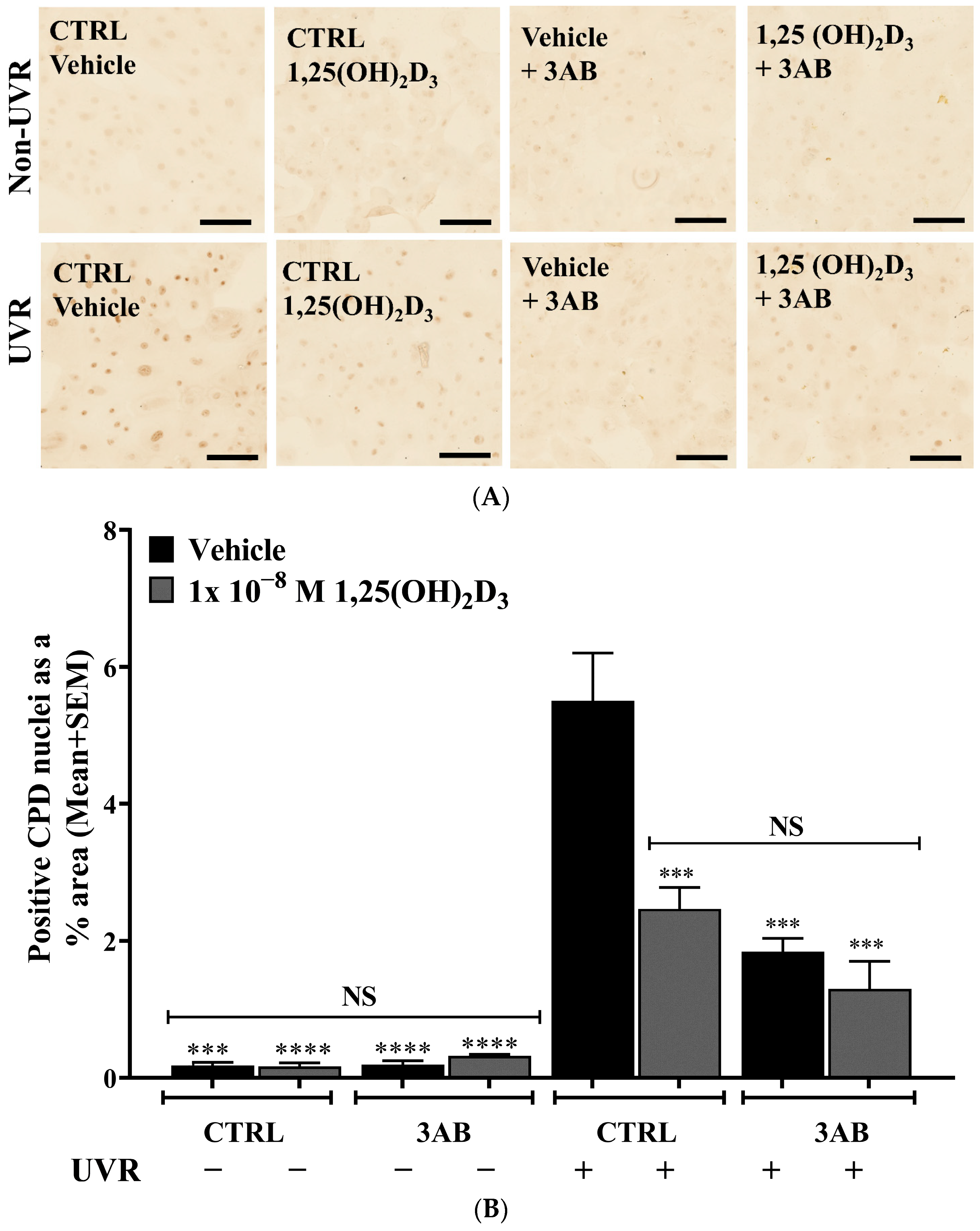
2.3. Treatment of UV-Irradiated Keratinocytes with 1,25(OH)2D3 or 3AB Resulted in a Significant Reduction in CPD Levels
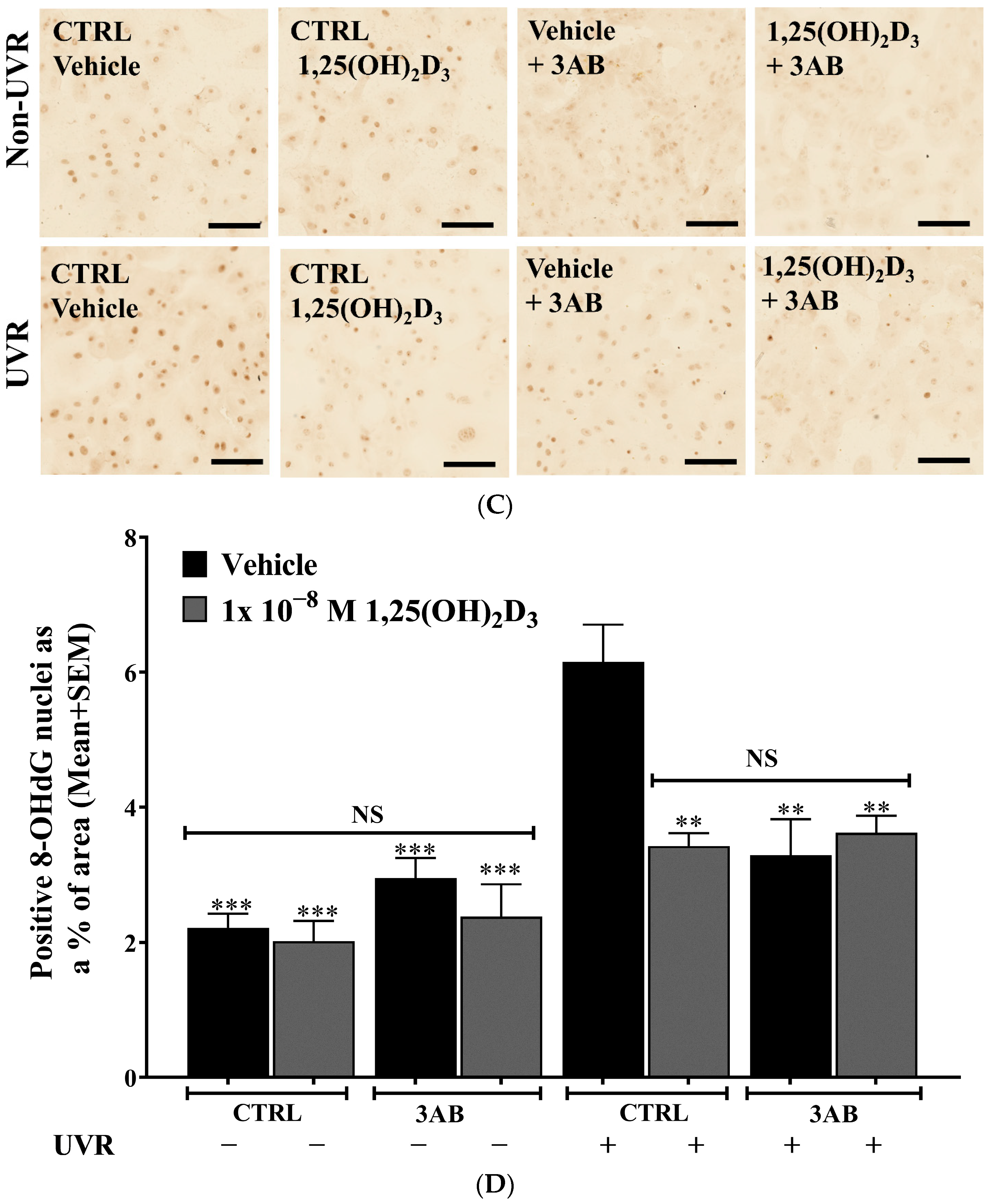
2.4. Treatment of UV-Irradiated Keratinocytes with 1,25(OH)2D3 or 3AB Resulted in a Significant Reduction in 8-OHdG Levels
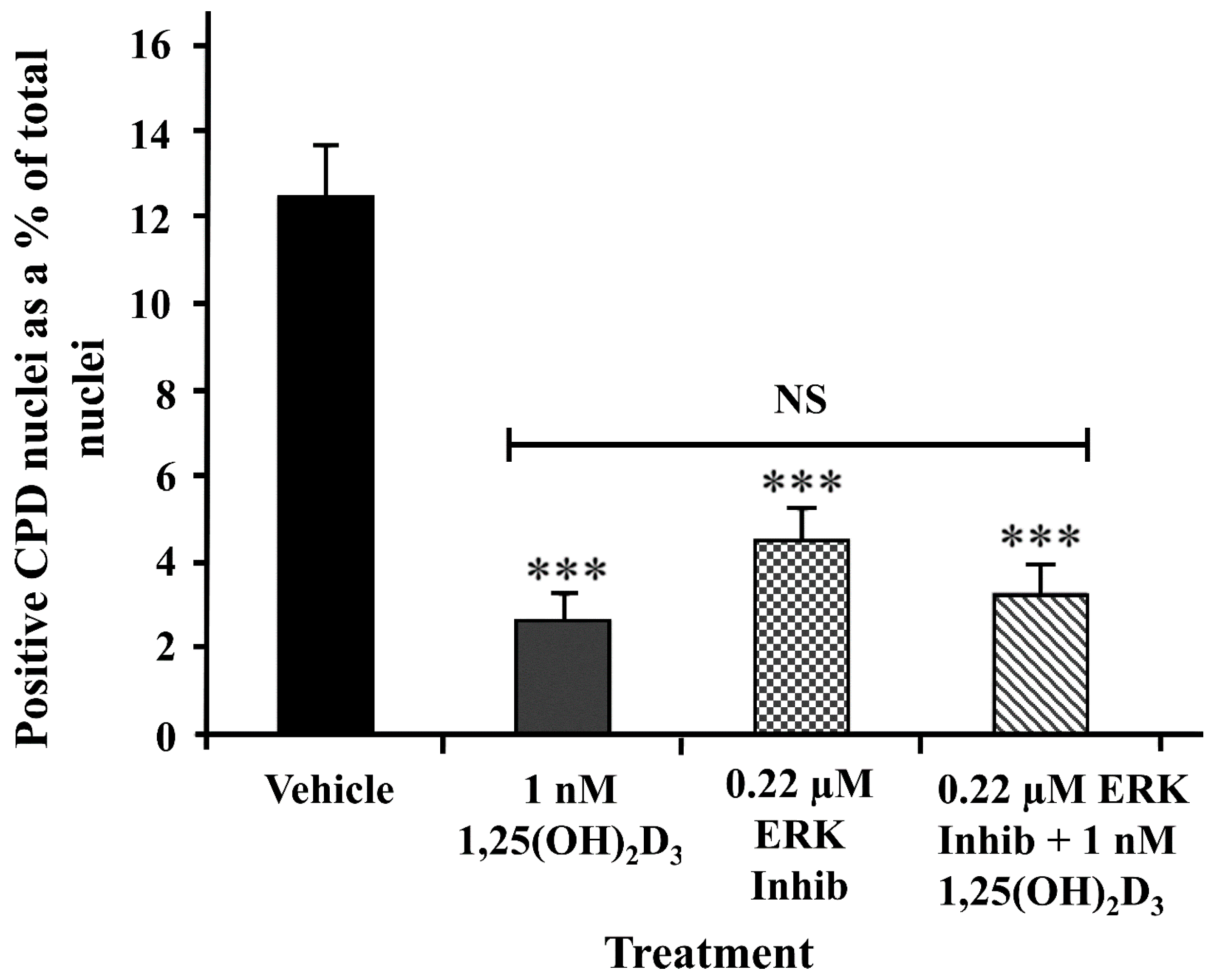
2.5. A Peptide ERK Inhibitor Reduced UV-Induced CPD Levels to a Similar Extent as 1,25(OH)2D3
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Treatments
4.3. UV Irradiation
4.4. PARP Measurements by Novel Whole-Cell ELISA
4.5. CPD and 8-OHdG Immunohistochemistry
4.6. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chambon, P.; Weill, J.D.; Mandel, P. Nicotinamide mononucleotide activation of a new DNA-dependent polyadenylic acid synthesizing nuclear enzyme. Biochem. Biophys. Res. Commun. 1963, 11, 39–43. [Google Scholar] [CrossRef] [PubMed]
- Alkhatib, H.M.; Chen, D.F.; Cherney, B.; Bhatia, K.; Notario, V.; Giri, C.; Stein, G.; Slattery, E.; Roeder, R.G.; Smulson, M.E. Cloning and expression of cDNA for human poly(ADP-ribose) polymerase. Proc. Natl. Acad. Sci. USA 1987, 84, 1224–1228. [Google Scholar] [CrossRef]
- Uchida, K.; Morita, T.; Sato, T.; Ogura, T.; Yamashita, R.; Noguchi, S.; Suzuki, H.; Nyunoya, H.; Miwa, M.; Sugimura, T. Nucleotide sequence of a full-length cDNA for human fibroblast poly(ADP-ribose) polymerase. Biochem. Biophys. Res. Commun. 1987, 148, 617–622. [Google Scholar] [CrossRef]
- Ludwig, A.; Behnke, B.; Holtlund, J.; Hilz, H. Immunoquantitation and size determination of intrinsic poly(ADP-ribose) polymerase from acid precipitates. An analysis of the in vivo status in mammalian species and in lower eukaryotes. J. Biol. Chem. 1988, 263, 6993–6999. [Google Scholar] [CrossRef]
- Dantzer, F.; Giraud-Panis, M.-J.; Jaco, I.; Amé, J.-C.; Schultz, I.; Blasco, M.; Koering, C.-E.; Gilson, E.; Ménissier-de Murcia, J.; de Murcia, G.; et al. Functional interaction between poly(ADP-Ribose) polymerase 2 (PARP-2) and TRF2: PARP activity negatively regulates TRF2. Mol. Cell. Biol. 2004, 24, 1595–1607. [Google Scholar] [CrossRef] [PubMed]
- Javle, M.; Curtin, N.J. The role of PARP in DNA repair and its therapeutic exploitation. Br. J. Cancer 2011, 105, 1114–1122. [Google Scholar] [CrossRef]
- de Murcia, G.; de Murcia, J.M. Poly(ADP-ribose) polymerase: A molecular nick-sensor. Trends Biochem. Sci. 1994, 19, 172–176. [Google Scholar] [CrossRef]
- Kauppinen, T.M.; Chan, W.Y.; Suh, S.W.; Wiggins, A.K.; Huang, E.J.; Swanson, R.A. Direct phosphorylation and regulation of poly(ADP-ribose) polymerase-1 by extracellular signal-regulated kinases 1/2. Proc. Natl. Acad. Sci. USA 2006, 103, 7136–7141. [Google Scholar] [CrossRef] [PubMed]
- Cohen-Armon, M.; Visochek, L.; Rozensal, D.; Kalal, A.; Geistrikh, I.; Klein, R.; Bendetz-Nezer, S.; Yao, Z.; Seger, R. DNA-independent PARP-1 activation by phosphorylated ERK2 increases Elk1 activity: A link to histone acetylation. Mol. Cell 2007, 25, 297–308. [Google Scholar] [CrossRef]
- Cohen-Armon, M. PARP-1 activation in the ERK signaling pathway. Trends Pharmacol. Sci. 2007, 28, 556–560. [Google Scholar] [CrossRef]
- Boehi, F.; Manetsch, P.; Hottiger, M.O. Interplay between ADP-ribosyltransferases and essential cell signaling pathways controls cellular responses. Cell Discov. 2021, 7, 104. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, D.W.; Ali, A.; Thornberry, N.A.; Vaillancourt, J.P.; Ding, C.K.; Gallant, M.; Gareau, Y.; Griffin, P.R.; Labelle, M.; Lazebnik, Y.A.; et al. Identification and inhibition of the ICE/CED-3 protease necessary for mammalian apoptosis. Nature 1995, 376, 37–43. [Google Scholar] [CrossRef]
- Gradwohl, G.; Ménissier de Murcia, J.M.; Molinete, M.; Simonin, F.; Koken, M.; Hoeijmakers, J.H.; de Murcia, G. The second zinc-finger domain of poly(ADP-ribose) polymerase determines specificity for single-stranded breaks in DNA. Proc. Natl. Acad. Sci. USA 1990, 87, 2990–2994. [Google Scholar] [CrossRef] [PubMed]
- Langelier, M.F.; Servent, K.M.; Rogers, E.E.; Pascal, J.M. A third zinc-binding domain of human poly(ADP-ribose) polymerase-1 coordinates DNA-dependent enzyme activation. J. Biol. Chem. 2008, 283, 4105–4114. [Google Scholar] [CrossRef] [PubMed]
- Langelier, M.F.; Planck, J.L.; Roy, S.; Pascal, J.M. Structural basis for DNA damage-dependent poly(ADP-ribosyl)ation by human PARP-1. Science 2012, 336, 728–732. [Google Scholar] [CrossRef] [PubMed]
- Loeffler, P.A.; Cuneo, M.J.; Mueller, G.A.; DeRose, E.F.; Gabel, S.A.; London, R.E. Structural studies of the PARP-1 BRCT domain. BMC Struct. Biol. 2011, 11, 37. [Google Scholar] [CrossRef] [PubMed]
- Langelier, M.F.; Eisemann, T.; Riccio, A.A.; Pascal, J.M. PARP family enzymes: Regulation and catalysis of the poly(ADP-ribose) posttranslational modification. Curr. Opin. Struct. Biol. 2018, 53, 187–198. [Google Scholar] [CrossRef] [PubMed]
- Simonin, F.; Ménissier-de Murcia, J.; Poch, O.; Muller, S.; Gradwohl, G.; Molinete, M.; Penning, C.; Keith, G.; de Murcia, G. Expression and site-directed mutagenesis of the catalytic domain of human poly(ADP-ribose)polymerase in Escherichia coli. Lysine 893 is critical for activity. J. Biol. Chem. 1990, 265, 19249–19256. [Google Scholar] [CrossRef]
- Ruf, A.; de Murcia, G.; Schulz, G.E. Inhibitor and NAD+ binding to poly(ADP-ribose) polymerase as derived from crystal structures and homology modeling. Biochemistry 1998, 37, 3893–3900. [Google Scholar] [CrossRef]
- Castri, P.; Lee, Y.J.; Ponzio, T.; Maric, D.; Spatz, M.; Bembry, J.; Hallenbeck, J. Poly(ADP-ribose) polymerase-1 and its cleavage products differentially modulate cellular protection through NF-kappaB-dependent signaling. Biochim. Biophys. Acta 2014, 1843, 640–651. [Google Scholar] [CrossRef]
- Lazebnik, Y.A.; Kaufmann, S.H.; Desnoyers, S.; Poirier, G.G.; Earnshaw, W.C. Cleavage of poly(ADP-ribose) polymerase by a proteinase with properties like ICE. Nature 1994, 371, 346–347. [Google Scholar] [CrossRef]
- Cohen, G.M. Caspases: The executioners of apoptosis. Biochem. J. 1997, 326 Pt 1, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Tewari, M.; Quan, L.T.; O’Rourke, K.; Desnoyers, S.; Zeng, Z.; Beidler, D.R.; Poirier, G.G.; Salvesen, G.S.; Dixit, V.M. Yama/CPP32 beta, a mammalian homolog of CED-3, is a CrmA-inhibitable protease that cleaves the death substrate poly(ADP-ribose) polymerase. Cell 1995, 81, 801–809. [Google Scholar] [CrossRef] [PubMed]
- Mashimo, M.; Onishi, M.; Uno, A.; Tanimichi, A.; Nobeyama, A.; Mori, M.; Yamada, S.; Negi, S.; Bu, X.; Kato, J.; et al. The 89-kDa PARP1 cleavage fragment serves as a cytoplasmic PAR carrier to induce AIF-mediated apoptosis. J. Biol. Chem. 2021, 296, 100046. [Google Scholar] [CrossRef]
- Kaufmann, S.H.; Desnoyers, S.; Ottaviano, Y.; Davidson, N.E.; Poirier, G.G. Specific Proteolytic Cleavage of Poly(ADP-ribose) Polymerase: An Early Marker of Chemotherapy-induced Apoptosis. Cancer Res. 1993, 53, 3976–3985. [Google Scholar]
- Yung, T.M.; Satoh, M.S. Functional competition between poly(ADP-ribose) polymerase and its 24-kDa apoptotic fragment in DNA repair and transcription. J. Biol. Chem. 2001, 276, 11279–11286. [Google Scholar] [CrossRef]
- Nie, L.; Wang, C.; Huang, M.; Liu, X.; Feng, X.; Tang, M.; Li, S.; Hang, Q.; Teng, H.; Shen, X.; et al. DePARylation is critical for S phase progression and cell survival. bioRxiv 2024. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, P.M.; Lewis, P.J.; Davies, M.I.; Skidmore, C.J.; Shall, S. The effect of gamma radiation and neocarzinostatin of NAD and ATP levels in mouse leukaemia cells. Biochim. Biophys. Acta (BBA)-Gen. Subj. 1978, 543, 576–582. [Google Scholar] [CrossRef]
- Skidmore, C.J.; Davis, M.I.; Goodwin, P.M.; Halldorsson, H.; Lewis, P.J.; Shall, S.; Zia’ee, A. The Involvement of Poly(ADP-ribose) Polymerase in the Degradation of NAD Caused by γ-Radiation and N-Methyl-N-Nitrosourea. Eur. J. Biochem. 1979, 101, 135–142. [Google Scholar] [CrossRef]
- Scaife, J.F. Effect of Ionizing Radiation on the Pyridine Nucleotides of Thymocytes. Can. J. Biochem. Physiol. 1963, 41, 1469–1481. [Google Scholar] [CrossRef]
- Jacobson, E.L.; Antol, K.M.; Juarez-Salinas, H.; Jacobson, M.K. Poly(ADP-ribose) metabolism in ultraviolet irradiated human fibroblasts. J. Biol. Chem. 1983, 258, 103–107. [Google Scholar] [CrossRef]
- Jacobson, E.L.; Giacomoni, P.U.; Roberts, M.J.; Wondrak, G.T.; Jacobson, M.K. Optimizing the energy status of skin cells during solar radiation. J. Photochem. Photobiol. B Biol. 2001, 63, 141–147. [Google Scholar] [CrossRef]
- Park, J.; Halliday, G.M.; Surjana, D.; Damian, D.L. Nicotinamide prevents ultraviolet radiation-induced cellular energy loss. Photochem. Photobiol. 2010, 86, 942–948. [Google Scholar] [CrossRef]
- Andrabi, S.A.; Umanah, G.K.E.; Chang, C.; Stevens, D.A.; Karuppagounder, S.S.; Gagné, J.-P.; Poirier, G.G.; Dawson, V.L.; Dawson, T.M. Poly(ADP-ribose) polymerase-dependent energy depletion occurs through inhibition of glycolysis. Proc. Natl. Acad. Sci. USA 2014, 111, 10209–10214. [Google Scholar] [CrossRef] [PubMed]
- Fouquerel, E.; Sobol, R.W. ARTD1 (PARP1) activation and NAD(+) in DNA repair and cell death. DNA Repair 2014, 23, 27–32. [Google Scholar] [CrossRef]
- Harrision, D.; Gravells, P.; Thompson, R.; Bryant, H.E. Poly(ADP-Ribose) Glycohydrolase (PARG) vs. Poly(ADP-Ribose) Polymerase (PARP)-Function in Genome Maintenance and Relevance of Inhibitors for Anti-cancer Therapy. Front. Mol. Biosci. 2020, 7, 191. [Google Scholar] [CrossRef]
- Wei, L.; Nakajima, S.; Hsieh, C.-L.; Kanno, S.; Masutani, M.; Levine, A.S.; Yasui, A.; Lan, L. Damage response of XRCC1 at sites of DNA single strand breaks is regulated by phosphorylation and ubiquitylation after degradation of poly(ADP-ribose). J. Cell Sci. 2013, 126, 4414–4423. [Google Scholar] [CrossRef]
- Gogola, E.; Duarte, A.A.; de Ruiter, J.R.; Wiegant, W.W.; Schmid, J.A.; de Bruijn, R.; James, D.I.; Guerrero Llobet, S.; Vis, D.J.; Annunziato, S.; et al. Selective Loss of PARG Restores PARylation and Counteracts PARP Inhibitor-Mediated Synthetic Lethality. Cancer Cell 2018, 33, 1078–1093.e1012. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, B.; Sauter, W.; Knuschke, P.; Dreßler, S.; Meurer, M. Demonstration of UVB-induced synthesis of 1α,25-dihydroxyvitamin D3 (calcitriol) in human skin by microdialysis. Arch. Dermatol. Res. 2003, 295, 24–28. [Google Scholar] [CrossRef] [PubMed]
- Slominski, A.T.; Kim, T.-K.; Li, W.; Yi, A.-K.; Postlethwaite, A.; Tuckey, R.C. The role of CYP11A1 in the production of vitamin D metabolites and their role in the regulation of epidermal functions. J. Steroid Biochem. Mol. Biol. 2014, 144 Pt A, 28–39. [Google Scholar] [CrossRef]
- Bikle, D.D.; Nemanic, M.K.; Whitney, J.O.; Elias, P.W. Neonatal human foreskin keratinocytes produce 1,25-dihydroxyvitamin D3. Biochemistry 1986, 25, 1545–1548. [Google Scholar] [CrossRef]
- Bikle, D.D.; Nemanic, M.K.; Gee, E.; Elias, P. 1,25-Dihydroxyvitamin D3 production by human keratinocytes. Kinetics and regulation. J. Clin. Investig. 1986, 78, 557–566. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.; Dixon, K.M.; Deo, S.S.; Holliday, C.J.; Slater, M.; Halliday, G.M.; Reeve, V.E.; Mason, R.S. Photoprotection by 1,25 dihydroxyvitamin D3 is associated with an increase in p53 and a decrease in nitric oxide products. J. Investig. Dermatol. 2007, 127, 707–715. [Google Scholar] [CrossRef]
- Carter, S.E. Mechanisms of Photoprotection by 20-Hydroxyvitamin D3, a Naturally Occurring 1a25-Dihydroxyvitamin D3 Analogue. Master’s Thesis, University of Sydney, Sydney, Australia, 2014. [Google Scholar]
- Rybchyn, M.S.; De Silva, W.G.M.; Sequeira, V.B.; McCarthy, B.Y.; Dilley, A.V.; Dixon, K.M.; Halliday, G.M.; Mason, R.S. Enhanced repair of UV-induced DNA damage by 1,25-Dihydroxyvitamin D3 in skin is linked to pathways that control cellular energy. J. Investig. Dermatol. 2018, 138, 1146–1156. [Google Scholar] [CrossRef] [PubMed]
- Song, E.J.; Gordon-Thomson, C.; Cole, L.; Stern, H.; Halliday, G.M.; Damian, D.L.; Reeve, V.E.; Mason, R.S. 1α,25-Dihydroxyvitamin D3 reduces several types of UV-induced DNA damage and contributes to photoprotection. J. Steroid Biochem. Mol. Biol. 2013, 136, 131–138. [Google Scholar] [CrossRef]
- Gordon-Thomson, C.; Gupta, R.; Tongkao-on, W.; Ryan, A.; Halliday, G.M.; Mason, R.S. 1α,25 Dihydroxyvitamin D3 enhances cellular defences against UV-induced oxidative and other forms of DNA damage in skin. Photochem. Photobiol. Sci. 2012, 11, 1837–1847. [Google Scholar] [CrossRef] [PubMed]
- Sequeira, V.B.; Rybchyn, M.S.; Gordon-Thomson, C.; Tongkao-on, W.; Mizwicki, M.T.; Norman, A.W.; Reeve, V.E.; Halliday, G.M.; Mason, R.S. Opening of Chloride Channels by 1α,25-Dihydroxyvitamin D3 Contributes to Photoprotection against UVR-Induced Thymine Dimers in Keratinocytes. J. Investig. Dermatol. 2013, 133, 776–782. [Google Scholar] [CrossRef]
- Lee, J.-h.; Youn, J.I. The photoprotective effect of 1,25-dihydroxyvitamin D3 on ultraviolet light B-induced damage in keratinocyte and its mechanism of action. J. Dermatol. Sci. 1998, 18, 11–18. [Google Scholar] [CrossRef]
- De Haes, P.; Garmyn, M.; Verstuyf, A.; De Clercq, P.; Vandewalle, M.; Degreef, H.; Vantieghem, K.; Bouillon, R.; Segaert, S. 1,25-Dihydroxyvitamin D3 and analogues protect primary human keratinocytes against UVB-induced DNA damage. J. Photochem. Photobiol. B Biol. 2005, 78, 141–148. [Google Scholar] [CrossRef]
- Dixon, K.M.; Norman, A.W.; Sequeira, V.B.; Mohan, R.; Rybchyn, M.S.; Reeve, V.E.; Halliday, G.M.; Mason, R.S. 1α,25(OH)2-Vitamin D and a Nongenomic Vitamin D Analogue Inhibit Ultraviolet Radiation–Induced Skin Carcinogenesis. Cancer Prev. Res. 2011, 4, 1485–1494. [Google Scholar] [CrossRef]
- Dixon, K.M.; Deo, S.S.; Wong, G.; Slater, M.; Norman, A.W.; Bishop, J.E.; Posner, G.H.; Ishizuka, S.; Halliday, G.M.; Reeve, V.E.; et al. Skin cancer prevention: A possible role of 1,25dihydroxyvitamin D3 and its analogs. J. Steroid Biochem. Mol. Biol. 2005, 97, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S.; Jung, M.; Yoo, J.; Choi, E.H.; Park, B.C.; Kim, M.H.; Hong, S.P. Protective Effect of Topical Vitamin D(3) against Photocarcinogenesis in a Murine Model. Ann. Dermatol. 2016, 28, 304–313. [Google Scholar] [CrossRef] [PubMed]
- Damian, D.L.; Kim, Y.J.; Dixon, K.M.; Halliday, G.M.; Javeri, A.; Mason, R.S. Topical calcitriol protects from UV-induced genetic damage but suppresses cutaneous immunity in humans. Exp. Dermatol. 2010, 19, e23–e30. [Google Scholar] [CrossRef] [PubMed]
- Ming, M.; Han, W.; Maddox, J.; Soltani, K.; Shea, C.R.; Freeman, D.M.; He, Y.Y. UVB-induced ERK/AKT-dependent PTEN suppression promotes survival of epidermal keratinocytes. Oncogene 2010, 29, 492–502. [Google Scholar] [CrossRef]
- Molnár, E. Cell-Based Enzyme-Linked Immunosorbent Assay (Cell-ELISA) Analysis of Native and Recombinant Glutamate Receptors. Methods Mol. Biol. 2019, 1941, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Zingarelli, B.; Cuzzocrea, S.; Zsengellér, Z.; Salzman, A.L.; Szabó, C. Protection against myocardial ischemia and reperfusion injury by 3-aminobenzamide, an inhibitor of poly (ADP-ribose) synthetase. Cardiovasc. Res. 1997, 36, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Douki, T.; Court, M.; Sauvaigo, S.; Odin, F.; Cadet, J. Formation of the Main UV-induced Thymine Dimeric Lesions within Isolated and Cellular DNA as Measured by High Performance Liquid Chromatography-Tandem Mass Spectrometry. J. Biol. Chem. 2000, 275, 11678–11685. [Google Scholar] [CrossRef] [PubMed]
- Agnez-Lima, L.F.; Melo, J.T.A.; Silva, A.E.; Oliveira, A.H.S.; Timoteo, A.R.S.; Lima-Bessa, K.M.; Martinez, G.R.; Medeiros, M.H.G.; Di Mascio, P.; Galhardo, R.S.; et al. DNA damage by singlet oxygen and cellular protective mechanisms. Mutat. Res./Rev. Mutat. Res. 2012, 751, 15–28. [Google Scholar] [CrossRef] [PubMed]
- Cadet, J.; Treoule, R. Comparative study of oxidation of nucleic acid components by hydroxyl radicals, singlet oxygen and superoxide anion radicals. Photochem. Photobiol. 1978, 28, 661–667. [Google Scholar] [CrossRef]
- Hallett, F.R.; Hallett, B.P.; Snipes, W. Reactions between Singlet Oxygen and the Constituents of Nucleic Acids: Importance of Reactions in Photodynamic Processes. Biophys. J. 1970, 10, 305–315. [Google Scholar] [CrossRef]
- Kasai, H.; Crain, P.F.; Kuchino, Y.; Nishimura, S.; Ootsuyama, A.; Tanooka, H. Formation of 8-hydroxyguanine moiety in cellular DNA by agents producing oxygen radicals and evidence for its repair. Carcinogenesis 1986, 7, 1849–1851. [Google Scholar] [CrossRef] [PubMed]
- Kasai, H.; Nishimura, S. Hydroxylation of deoxyguanosine at the C-8 position by ascorbic acid and other reducing agents. Nucleic Acids Res. 1984, 12, 2137–2145. [Google Scholar] [CrossRef]
- Bai, P. Biology of Poly(ADP-Ribose) Polymerases: The Factotums of Cell Maintenance. Mol. Cell 2015, 58, 947–958. [Google Scholar] [CrossRef] [PubMed]
- Silveira, S.C.; Buhagiar-Labarchède, G.; Onclercq-Delic, R.; Gemble, S.; Bou Samra, E.; Mameri, H.; Duchambon, P.; Machon, C.; Guitton, J.; Amor-Guéret, M. A decrease in NAMPT activity impairs basal PARP-1 activity in cytidine deaminase deficient-cells, independently of NAD+. Sci. Rep. 2020, 10, 13907. [Google Scholar] [CrossRef]
- Aasen, T.; Izpisua Belmonte, J.C. Isolation and cultivation of human keratinocytes from skin or plucked hair for the generation of induced pluripotent stem cells. Nat. Protoc. 2010, 5, 371–382. [Google Scholar] [CrossRef] [PubMed]
- Mabley, J.G.; Wallace, R.; Pacher, P.; Murphy, K.; SzabÓ, C. Inhibition of poly(adenosine diphosphate-ribose) polymerase by the active form of vitamin D. Int. J. Mol. Med. 2007, 19, 947–952. [Google Scholar] [CrossRef] [PubMed]
- Mabley, J.G.; Szabo, C. Inflammatory disease and sunlight: The vitamin D-poly (ADP-ribose) polymerase connection. Future Rheumatol. 2008, 3, 169. [Google Scholar] [CrossRef]
- Schuhwerk, H.; Atteya, R.; Siniuk, K.; Wang, Z.Q. PARPing for balance in the homeostasis of poly(ADP-ribosyl)ation. Semin. Cell Dev. Biol. 2017, 63, 81–91. [Google Scholar] [CrossRef]
- De Silva, W.G.M.; Han, J.Z.R.; Yang, C.; Tongkao-On, W.; McCarthy, B.Y.; Ince, F.A.; Holland, A.J.A.; Tuckey, R.C.; Slominski, A.T.; Abboud, M.; et al. Evidence for Involvement of Nonclassical Pathways in the Protection From UV-Induced DNA Damage by Vitamin D-Related Compounds. J. Bone Miner. Res. Plus 2021, 5, e10555. [Google Scholar] [CrossRef]
- Karampinis, E.; Aloizou, A.M.; Zafiriou, E.; Bargiota, A.; Skaperda, Z.; Kouretas, D.; Roussaki-Schulze, A.V. Non-Melanoma Skin Cancer and Vitamin D: The “Lost Sunlight” Paradox and the Oxidative Stress Explanation. Antioxidants 2023, 12, 1107. [Google Scholar] [CrossRef]
- Zinser, G.M.; Sundberg, J.P.; Welsh, J. Vitamin D3 receptor ablation sensitizes skin to chemically induced tumorigenesis. Carcinogenesis 2002, 23, 2103–2109. [Google Scholar] [CrossRef] [PubMed]
- Ellison, T.I.; Smith, M.K.; Gilliam, A.C.; MacDonald, P.N. Inactivation of the Vitamin D Receptor Enhances Susceptibility of Murine Skin to UV-induced Tumorigenesis. J. Investig. Dermatol. 2008, 128, 2508–2517. [Google Scholar] [CrossRef] [PubMed]
- Visochek, L.; Cohen-Armon, M. PARP1-Erk synergism in proliferating cells. Oncotarget 2018, 9, 29140–29145. [Google Scholar] [CrossRef] [PubMed]
- Cohen-Armon, M.; Yeheskel, A.; Pascal, J.M. Signal-induced PARP1-Erk synergism mediates IEG expression. Signal Transduct. Target. Ther. 2019, 4, 8. [Google Scholar] [CrossRef] [PubMed]
- Visochek, L.; Grigoryan, G.; Kalal, A.; Milshtein-Parush, H.; Gazit, N.; Slutsky, I.; Yeheskel, A.; Shainberg, A.; Castiel, A.; Seger, R.; et al. A PARP1-ERK2 synergism is required for the induction of LTP. Sci. Rep. 2016, 6, 24950. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Cui, Z.; Meng, X. Knockdown of PARP-1 Inhibits Proliferation and ERK Signals, Increasing Drug Sensitivity in Osteosarcoma U2OS Cells. Oncol. Res. 2016, 24, 279–286. [Google Scholar] [CrossRef]
- Kuchmerovska, T.; Guzyk, M.; Tykhonenko, T.; Yanitska, L.; Pryvrotska, I.; Diakun, K. The parp-1 and bax genes as potential targets for treatment of the heart functioning impairments induced by type 1 diabetes mellitus. Endocr. Regul. 2021, 55, 61–71. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Li, Y.; Wang, N.; Luo, G.; Wang, J.; Luo, C.; Yu, W.; Hao, L. 1α,25-Dihydroxyvitamin D3 prevents renal oxidative damage via the PARP1/SIRT1/NOX4 pathway in Zucker diabetic fatty rats. Am. J. Physiol.-Endocrinol. Metab. 2020, 318, E343–E356. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Wu, J.; Wan, F.; Kou, L.; Yin, S.; Sun, Y.; Li, Y.; Zhou, Q.; Wang, T. Calcitriol Alleviates MPP(+)- and MPTP-Induced Parthanatos Through the VDR/PARP1 Pathway in the Model of Parkinson’s Disease. Front. Aging Neurosci. 2021, 13, 657095. [Google Scholar] [CrossRef]
- Rizvi, A.; Naseem, I. Causing DNA damage and stopping DNA repair–Vitamin D supplementation with Poly(ADP-ribose) polymerase 1 (PARP1) inhibitors may cause selective cell death of cancer cells: A novel therapeutic paradigm utilizing elevated copper levels within the tumour. Med. Hypotheses 2020, 144, 110278. [Google Scholar] [CrossRef]
- Lee, Y.W.; Ha, M.S.; Kim, Y.K. H2O2-Induced Cell Death in Human Glioma Cells: Role of Lipid Peroxidation and PARP Activation. Neurochem. Res. 2001, 26, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Lakatos, P.; Szabó, É.; Hegedűs, C.; Haskó, G.; Gergely, P.; Bai, P.; Virág, L. 3-Aminobenzamide protects primary human keratinocytes from UV-induced cell death by a poly(ADP-ribosyl)ation independent mechanism. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2013, 1833, 743–751. [Google Scholar] [CrossRef] [PubMed]
- Farkas, B.; Magyarlaki, M.; Csete, B.; Nemeth, J.; Rabloczky, G.; Bernath, S.; Literáti Nagy, P.; Sümegi, B. Reduction of acute photodamage in skin by topical application of a novel PARP inhibitor. Biochem. Pharmacol. 2002, 63, 921–932. [Google Scholar] [CrossRef] [PubMed]
- Robu, M.; Shah, R.G.; Petitclerc, N.; Brind’Amour, J.; Kandan-Kulangara, F.; Shah, G.M. Role of poly(ADP-ribose) polymerase-1 in the removal of UV-induced DNA lesions by nucleotide excision repair. Proc. Natl. Acad. Sci. USA 2013, 110, 1658–1663. [Google Scholar] [CrossRef] [PubMed]
- Flohr, C.; Bürkle, A.; Radicella, J.P.; Epe, B. Poly(ADP-ribosyl)ation accelerates DNA repair in a pathway dependent on Cockayne syndrome B protein. Nucleic Acids Res. 2003, 31, 5332–5337. [Google Scholar] [CrossRef] [PubMed]
- Hunting, D.J.; Gowans, B.J. Inhibition of repair patch ligation by an inhibitor of poly(ADP-ribose) synthesis in normal human fibroblasts damaged with ultraviolet radiation. Mol. Pharmacol. 1988, 33, 358–362. [Google Scholar] [PubMed]
- Wang, Z.Q.; Auer, B.; Stingl, L.; Berghammer, H.; Haidacher, D.; Schweiger, M.; Wagner, E.F. Mice lacking ADPRT and poly(ADP-ribosyl)ation develop normally but are susceptible to skin disease. Genes Dev. 1995, 9, 509–520. [Google Scholar] [CrossRef]
- Satoh, M.S.; Lindahl, T. Role of poly(ADP-ribose) formation in DNA repair. Nature 1992, 356, 356–358. [Google Scholar] [CrossRef] [PubMed]
- Satoh, M.S.; Poirier, G.G.; Lindahl, T. NAD(+)-dependent repair of damaged DNA by human cell extracts. J. Biol. Chem. 1993, 268, 5480–5487. [Google Scholar] [CrossRef]
- Smulson, M.; Istock, N.; Ding, R.; Cherney, B. Deletion Mutants of Poly(ADP-Ribose) Polymerase A Support a Model of Cyclic Association and Dissociation of Enzyme from DNA Ends During DNA Repair. Biochemistry 1994, 33, 6186–6191. [Google Scholar] [CrossRef]
- Lindahl, T.; Satoh, M.S.; Poirier, G.G.; Klungland, A. Post-translational modification of poly(ADP-ribose) polymerase induced by DNA strand breaks. Trends Biochem. Sci. 1995, 20, 405–411. [Google Scholar] [CrossRef] [PubMed]
- Kelemen, B.R.; Hsiao, K.; Goueli, S.A. Selective in Vivo Inhibition of Mitogen-activated Protein Kinase Activation Using Cell-permeable Peptides*210. J. Biol. Chem. 2002, 277, 8741–8748. [Google Scholar] [CrossRef] [PubMed]
- Douki, T.; Cadet, J. Individual determination of the yield of the main UV-induced dimeric pyrimidine photoproducts in DNA suggests a high mutagenicity of CC photolesions. Biochemistry 2001, 40, 2495–2501. [Google Scholar] [CrossRef] [PubMed]
- De Silva, W.G.M.; McCarthy, B.Y.; Han, J.; Yang, C.; Holland, A.J.A.; Stern, H.; Dixon, K.M.; Tang, E.K.Y.; Tuckey, R.C.; Rybchyn, M.S.; et al. The Over-Irradiation Metabolite Derivative, 24-Hydroxylumister-ol(3), Reduces UV-Induced Damage in Skin. Metabolites 2023, 13, 775. [Google Scholar] [CrossRef]
- Wong, G.; Gupta, R.; Dixon, K.M.; Deo, S.S.; Choong, S.M.; Halliday, G.M.; Bishop, J.E.; Ishizuka, S.; Norman, A.W.; Posner, G.H.; et al. 1,25-Dihydroxyvitamin D and three low-calcemic analogs decrease UV-induced DNA damage via the rapid response pathway. J. Steroid Biochem. Mol. Biol. 2004, 89–90, 567–570. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Silva, W.G.M.; Sequeira, V.B.; Yang, C.; Dixon, K.M.; Holland, A.J.A.; Mason, R.S.; Rybchyn, M.S. 1,25-Dihydroxyvitamin D3 Suppresses UV-Induced Poly(ADP-Ribose) Levels in Primary Human Keratinocytes, as Detected by a Novel Whole-Cell ELISA. Int. J. Mol. Sci. 2024, 25, 5583. https://doi.org/10.3390/ijms25115583
De Silva WGM, Sequeira VB, Yang C, Dixon KM, Holland AJA, Mason RS, Rybchyn MS. 1,25-Dihydroxyvitamin D3 Suppresses UV-Induced Poly(ADP-Ribose) Levels in Primary Human Keratinocytes, as Detected by a Novel Whole-Cell ELISA. International Journal of Molecular Sciences. 2024; 25(11):5583. https://doi.org/10.3390/ijms25115583
Chicago/Turabian StyleDe Silva, Warusavithana Gunawardena Manori, Vanessa Bernadette Sequeira, Chen Yang, Katie Marie Dixon, Andrew J. A. Holland, Rebecca Sara Mason, and Mark Stephen Rybchyn. 2024. "1,25-Dihydroxyvitamin D3 Suppresses UV-Induced Poly(ADP-Ribose) Levels in Primary Human Keratinocytes, as Detected by a Novel Whole-Cell ELISA" International Journal of Molecular Sciences 25, no. 11: 5583. https://doi.org/10.3390/ijms25115583