
Unveiling the Nature and Strength of Selenium-Centered Chalcogen Bonds in Binary Complexes of SeO2 with Oxygen-/Sulfur-Containing Lewis Bases: Insights from Theoretical Calculations


Abstract
:1. Introduction
2. Results and Discussion
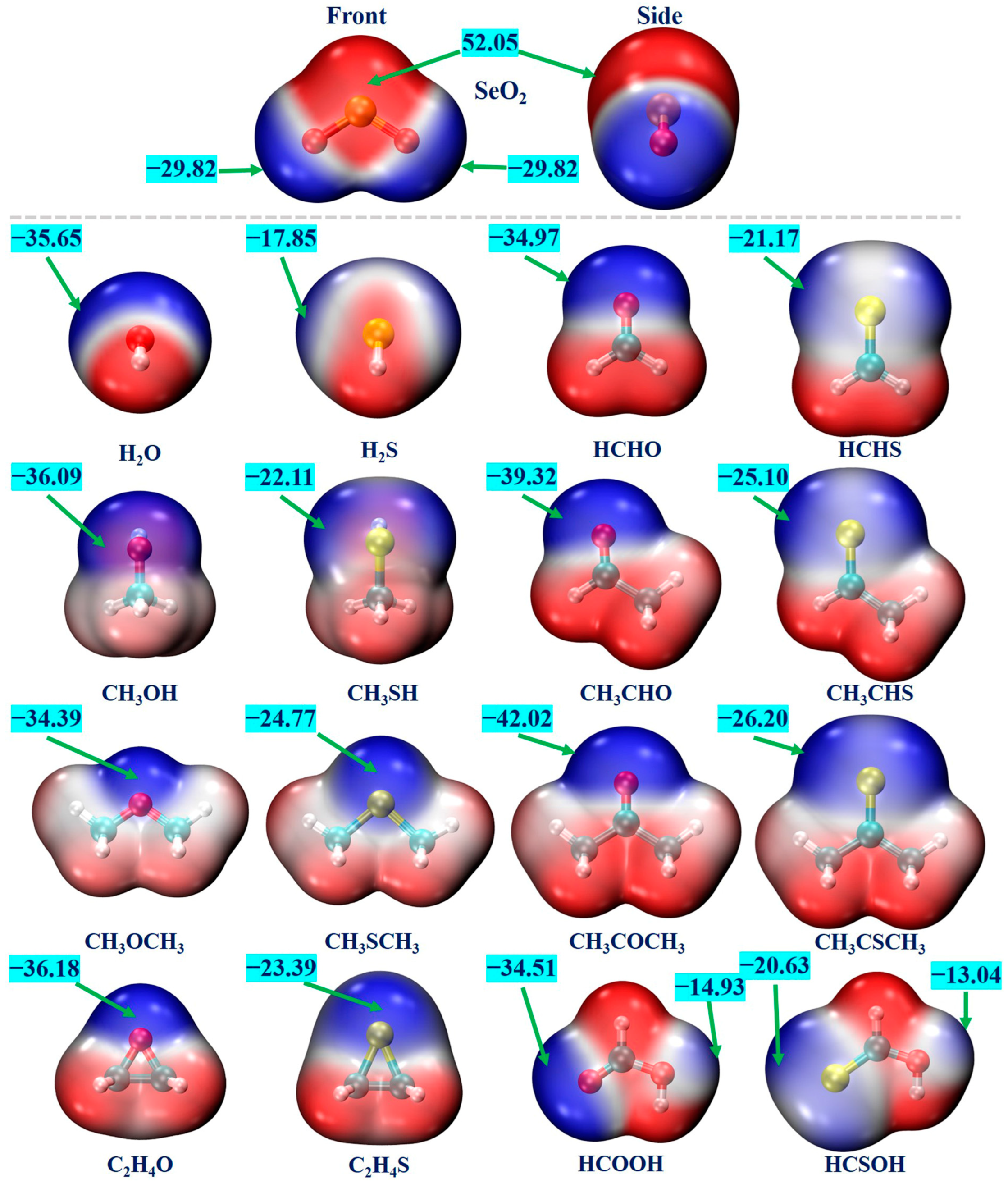
2.1. MEP Analysis of Monomers
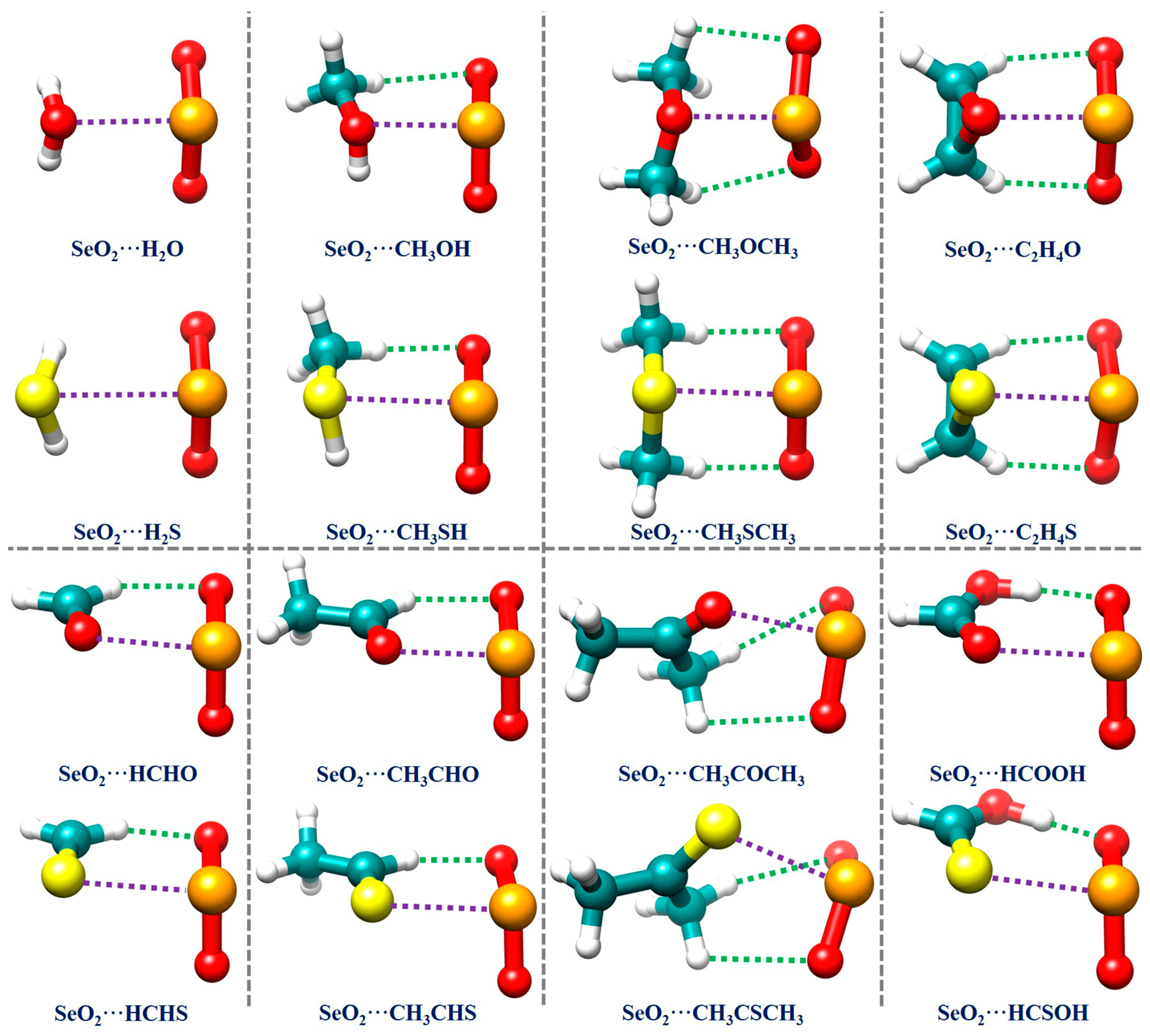
2.2. Geometrical Structures and Binding Energies of the Binary Complexes
2.3. QTAIM Analysis
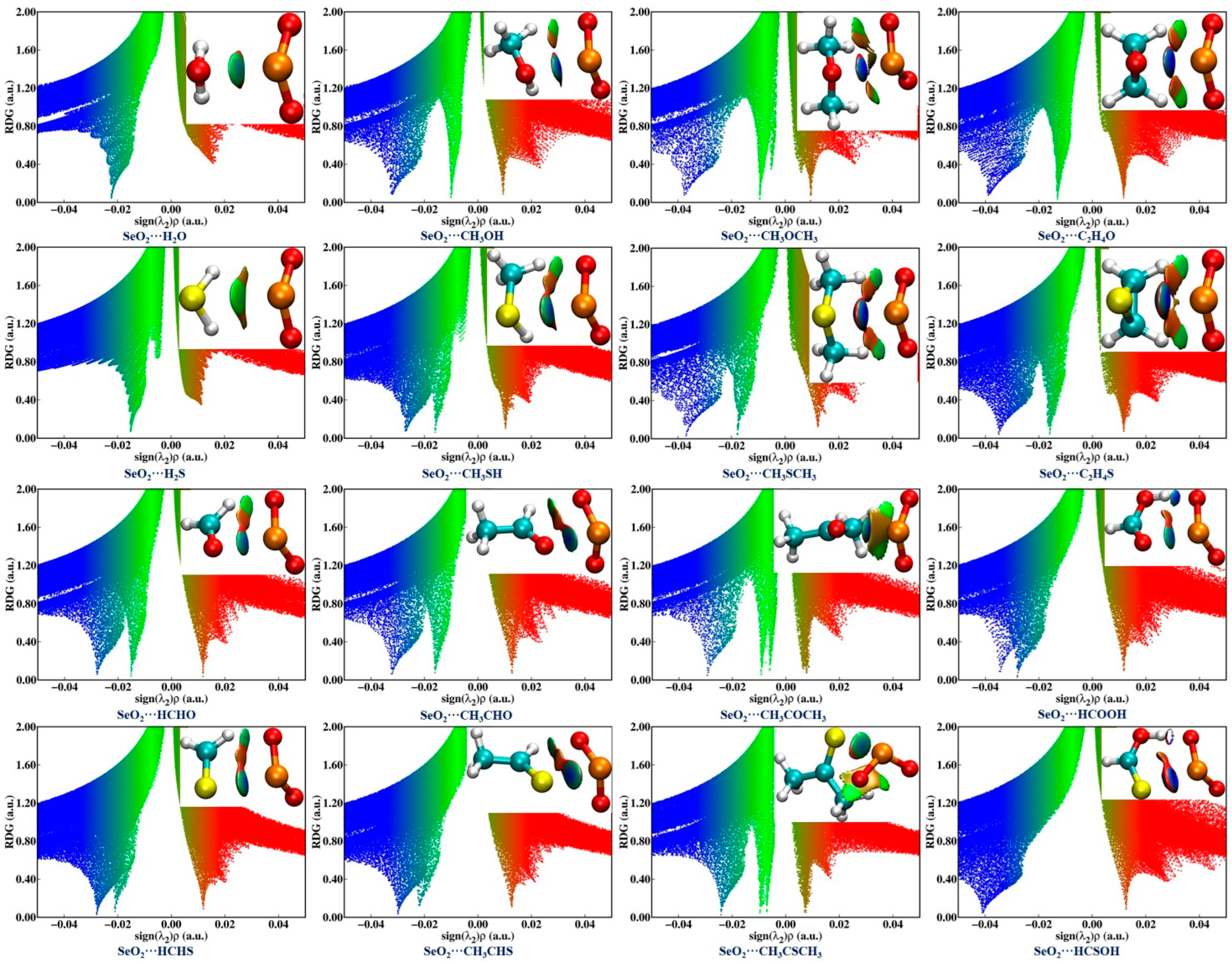
2.4. NCIplot Analysis
2.5. NBO Analysis
2.6. SAPT Analysis
3. Computational Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Moroder, L. Isosteric replacement of sulfur with other chalcogens in peptides and proteins. J. Pep. Sci. 2005, 11, 187–214. [Google Scholar] [CrossRef] [PubMed]
- Reich, H.J.; Hondal, R.J. Why nature chose selenium. ACS Chem. Biol. 2016, 11, 821–841. [Google Scholar] [CrossRef]
- Chuai, H.; Zhang, S.Q.; Bai, H.; Li, J.; Wang, Y.; Sun, J.; Wen, E.; Zhang, J.; Xin, M. Small molecule selenium-containing compounds: Recent development and therapeutic applications. Eur. J. Med. Chem. 2021, 223, 113621. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, A.J.; Zade, S.S.; Singh, H.B.; Sunoj, R.B. Organoselenium chemistry: Role of intramolecular interactions. Chem. Rev. 2010, 110, 4357–4416. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Pang, Y.; Zhao, Z.; Zhou, P.P.; Wang, Y. Supramolecular catalysis with ethers enabled by dual chalcogen bonding activation. Nat. Commun. 2023, 14, 6347. [Google Scholar] [CrossRef] [PubMed]
- Fourmigué, M.; Dhaka, A. Chalcogen bonding in crystalline diselenides and selenocyanates: From molecules of pharmaceutical interest to conducting materials. Coordin. Chem. Rev. 2020, 403, 213084. [Google Scholar] [CrossRef]
- Fan, B.; Lin, F.; Wu, X.; Zhu, Z.; Jen, A.K.Y. Selenium-containing organic photovoltaic materials. Acc. Chem. Res. 2021, 54, 3906–3916. [Google Scholar] [CrossRef]
- Birringer, M.; Pilawa, S.; Flohé, L. Trends in selenium biochemistry. Nat. Prod. Rep. 2002, 19, 693–718. [Google Scholar] [CrossRef] [PubMed]
- Wessjohann, L.A.; Schneider, A.; Abbas, M.; Brandt, W. Selenium in chemistry and biochemistry in comparison to sulfur. Biol. Chem. 2007, 388, 997–1006. [Google Scholar] [CrossRef]
- Iwaoka, M.; Komatsu, H.; Katsuda, T.; Tomada, S. Nature of nonbonded Se∙∙∙O interactions characterized by 17O NMR spectroscopy and NBO and AIM analyses. J. Am. Chem. Soc. 2004, 126, 5309–5317. [Google Scholar] [CrossRef]
- Behera, R.N.; Panda, A. Effect of chelate ring and rigidity on Se∙∙∙N interactions: A computational study. RSC Adv. 2012, 2, 6948–6956. [Google Scholar] [CrossRef]
- Tripathi, A.; Daolio, A.; Pizzi, A.; Guo, Z.; Turner, D.R.; Baggioli, A.; Famulari, A.; Deacon, G.B.; Resnati, G.; Singh, H.B. Chalcogen bonds in selenocysteine seleninic acid, a functional GPx constituent, and in other seleninic or sulfinic acid derivatives. Chem. Asian J. 2021, 16, 2351–2360. [Google Scholar] [CrossRef]
- Carugo, O.; Resnati, G.; Metrangolo, P. Chalcogen bonds involving selenium in protein structures. ACS Chem. Biol. 2021, 16, 1622–1627. [Google Scholar] [CrossRef] [PubMed]
- Biswal, H.S.; Sahu, A.K.; Galmés, B.; Frontera, A.; Chopra, D. Se∙∙∙O/S and S∙∙∙O chalcogen bonds in small molecules and proteins: A combined CSD and PDB study. ChemBioChem 2022, 23, e202100498. [Google Scholar] [CrossRef]
- Scheiner, S. Participation of S and Se in hydrogen and chalcogen bonds. CrystEngComm 2021, 23, 6821–6837. [Google Scholar] [CrossRef]
- Chand, A.; Biswal, H.S. Hydrogen bonds with chalcogens: Looking beyond the second row of the periodic table. J. Indian Inst. Sci. 2020, 100, 77–100. [Google Scholar] [CrossRef]
- Biot, N.; Bonifazi, D. Chalcogen-bond driven molecular recognition at work. Coordin. Chem. Rev. 2020, 413, 213243. [Google Scholar] [CrossRef]
- Mundlapati, V.R.; Sahoo, D.K.; Ghosh, S.; Purame, U.K.; Pandey, S.; Acharya, R.; Pal, N.; Tiwari, P.; Biswal, H.S. Spectroscopic evidences for strong hydrogen bonds with selenomethionine in proteins. J. Phys. Chem. Lett. 2017, 8, 794–800. [Google Scholar] [CrossRef]
- Mishra, K.K.; Singh, S.K.; Ghosh, P.; Ghosh, D.; Das, A. The nature of selenium hydrogen bonding: Gas phase spectroscopy and quantum chemistry calculations. Phys. Chem. Chem. Phys. 2017, 19, 24179–24187. [Google Scholar] [CrossRef]
- Mishra, K.K.; Singh, S.K.; Kumar, S.; Singh, G.; Sarkar, B.; Madhusudhan, M.S.; Das, A. Water-mediated selenium hydrogen-bonding in proteins: PDB analysis and gas-phase spectroscopy of model complexes. J. Phys. Chem. A 2019, 123, 5995–6002. [Google Scholar] [CrossRef]
- Chand, A.; Sahoo, D.K.; Rana, A.; Jena, S.; Biswal, H.S. The prodigious hydrogen bonds with sulfur and selenium in molecular assemblies, structural biology, and functional materials. Acc. Chem. Res. 2020, 53, 1580–1592. [Google Scholar] [CrossRef] [PubMed]
- Pal, D.; Charaya, H.; Chakraborty, S. An experimental exploration of C−H∙∙∙X hydrogen bond in [CHCl3−X(CH3)2] complexes (X = O, S, and Se). ChemPhysChem 2023, 24, e202300124. [Google Scholar] [CrossRef] [PubMed]
- Pal, D.; Agrawal, S.K.; Chakraborty, A.; Chakraborty, S. Hydrogen bond properties of Se in [ROH–Se (CH3)2] complexes (R = H, CH3, C2H5): Matrix-isolation infrared spectroscopy and theoretical calculations. Phys. Chem. Chem. Phys. 2023, 25, 11286–11300. [Google Scholar] [CrossRef] [PubMed]
- Jaju, K.; Pal, D.; Chakraborty, A.; Chakraborty, S. Electronic substituent effect on Se−H∙∙∙N hydrogen bond: A computational study of para-substituted pyridine-SeH2 complexes. Chem. Phys. Lett. 2019, 737, 100031. [Google Scholar] [CrossRef]
- Rafat, R.; Nowroozi, A. A comprehensive theoretical study of conformational analysis, intramolecular hydrogen bond, π-electron delocalization, and tautomeric preferences in 2-selenoformyl-3-thioxo-propionaldehyde. Struct. Chem. 2018, 29, 1057–1065. [Google Scholar] [CrossRef]
- Gómez Castaño, J.A.; Romano, R.M.; Beckers, H.; Willner, H.; Boese, R.; Della Védova, C.O. Selenoacetic acid, CH3C(O)SeH: Preparation, characterization, and conformational properties. Angew. Chem. Int. Ed. 2008, 47, 10114–10118. [Google Scholar] [CrossRef] [PubMed]
- Senćanski, M.; Djordjević, I.; Grubišić, S. Assessing the dispersive and electrostatic components of the selenium–aromatic interaction energy by DFT. J. Mol. Model. 2017, 23, 162. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.P.; Sathishkumar, R.; Row, T.N.G. Organic alloys of room temperature liquids thiophenol and selenophenol. Chem. Commun. 2015, 51, 14255–14258. [Google Scholar] [CrossRef]
- Aakeroy, C.B.; Bryce, D.I.; Desiraju, G.R.; Frontera, A.; Legon, A.C.; Nicotra, F.; Rissanen, K.K.; Scheiner, S.; Terraneo, G.; Metrangolo, P.; et al. Definition of the chalcogen bond (IUPAC Recommendations 2019). Pure Appl. Chem. 2019, 91, 1889–1892. [Google Scholar] [CrossRef]
- Dhaka, A.; Jeon, I.R.; Fourmigué, M. Selective activation of chalcogen bonding: An efficient structuring tool toward crystal engineering strategies. Acc. Chem. Res. 2024, 57, 362–374. [Google Scholar] [CrossRef]
- Scilabra, P.; Terraneo, G.; Resnati, G. The chalcogen bond in crystalline solids: A world parallel to halogen bond. Acc. Chem. Res. 2019, 52, 1313–1324. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.S. Anion recognition based on halogen, chalcogen, pnictogen and tetrel bonding. Coordin. Chem. Rev. 2020, 413, 213270. [Google Scholar] [CrossRef]
- Sekar, G.; Nair, V.V.; Zhu, J. Chalcogen bonding catalysis. Chem. Soc. Rev. 2024, 53, 586–605. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Wang, Y. Chalcogen bonding catalysis with phosphonium chalcogenide (PCH). Acc. Chem. Res. 2023, 56, 608–621. [Google Scholar] [CrossRef] [PubMed]
- Beno, B.R.; Yeung, K.-S.; Bartberger, M.D.; Pennington, L.D.; Meanwell, N.A. A Survey of the role of noncovalent sulfur interactions in drug design. J. Med. Chem. 2015, 58, 4383–4438. [Google Scholar] [CrossRef] [PubMed]
- Ho, P.C.; Wang, J.Z.; Meloni, F.; Vargas-Baca, I. Chalcogen bonding in materials chemistry. Coordin. Chem. Rev. 2020, 422, 213464. [Google Scholar] [CrossRef]
- Eckstein, B.J.; Brown, L.C.; Noll, B.C.; Moghadasnia, M.P.; Balaich, G.J.; McGuirk, C.M. A porous chalcogen-bonded organic framework. J. Am. Chem. Soc. 2021, 143, 20207–20215. [Google Scholar] [CrossRef] [PubMed]
- Tupikina, E.Y. Non-covalent interactions in the glutathione peroxidase active center and their influence on the enzyme activity. Org. Biomol. Chem. 2022, 20, 5551–5557. [Google Scholar] [CrossRef]
- Piña, M.N.; Bauzá, A. On the importance of halogen and chalcogen bonds in the solid state of nucleic acids: A combined crystallographic and theoretical perspective. Int. J. Mol. Sci. 2023, 24, 13035. [Google Scholar] [CrossRef]
- Wang, H.; Wang, W.; Jin, W.J. σ-Hole bond vs π-hole bond: A comparison based on halogen bond. Chem. Rev. 2016, 116, 5072–5104. [Google Scholar] [CrossRef]
- Bauzá, A.; Mooibroek, T.J.; Frontera, A. The bright future of unconventional σ/π-hole interactions. ChemPhysChem 2015, 16, 2496–2517. [Google Scholar] [CrossRef] [PubMed]
- Gilli, G.; Gilli, P. The Nature of the Hydrogen Bond; Oxford University Press: Oxford, UK, 2009. [Google Scholar]
- Gleiter, R.; Haberhauer, G.; Werz, D.B.; Rominger, F.; Bleiholder, C. From noncovalent chalcogen–chalcogen interactions to supramolecular aggregates: Experiments and calculations. Chem. Rev. 2018, 118, 2010–2041. [Google Scholar] [CrossRef] [PubMed]
- Frontera, A.; Bauza, A. Metal Coordination enhances chalcogen bonds: CSD survey and theoretical calculations. Int. J. Mol. Sci. 2022, 23, 4188. [Google Scholar] [CrossRef] [PubMed]
- Lei, F.; Liu, Q.; Zhong, Y.; Cui, X.; Yu, J.; Hu, Z.; Feng, G.; Zeng, Z.; Lu, T. Computational insight into the nature and strength of the π-hole type chalcogen∙∙∙chalcogen interactions in the XO2∙∙∙CH3YCH3 Complexes (X = S, Se, Te; Y = O, S, Se, Te). Int. J. Mol. Sci. 2023, 24, 16193. [Google Scholar] [CrossRef] [PubMed]
- Pascoe, D.J.; Ling, K.B.; Cockroft, S.L. The origin of chalcogen-bonding interactions. J. Am. Chem. Soc. 2017, 139, 15160–15167. [Google Scholar] [CrossRef] [PubMed]
- Kříž, K.; Fanfrlík, J.; Lepšík, M. Chalcogen bonding in protein−ligand complexes: PDB survey and quantum mechanical calculation. ChemPhysChem 2018, 19, 2540–2548. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Liu, R.; Tang, J.; Li, P.; Cui, Y.; Zhang, H. On the properties of Se∙∙∙N interaction: The analysis of substituent effects by energy decomposition and orbital interaction. J. Mol. Model. 2016, 22, 29. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; An, X.; Li, Q. Se∙∙∙N chalcogen bond and Se∙∙∙X halogen bond Involving F2C=Se: Influence of hybridization, substitution, and cooperativity. J. Phys. Chem. A 2015, 119, 3518–3527. [Google Scholar] [CrossRef] [PubMed]
- Afonin, A.V.; Pavlov, D.V.; Albanov, A.I.; Levanova, E.P.; Levkovskaya, G.G. Study of stereospecificity of 1H, 13C, 15N and 77Se shielding constants in the configurational isomers of the selenophene-2-carbaldehyde azine by NMR spectroscopy and MP2-GIAO calculations. Magn. Reson. Chem. 2011, 49, 740–748. [Google Scholar] [CrossRef]
- Wang, H.; Liu, J.; Wang, W. Intermolecular and very strong intramolecular C–Se∙∙∙O/N chalcogen bonds in nitrophenyl selenocyanate crystals. Phys. Chem. Chem. Phys. 2018, 20, 5227–5234. [Google Scholar] [CrossRef]
- Aliyeva, V.A.; Gurbanov, A.V.; Guedes da Silva, M.F.C.; Gomila, R.M.; Frontera, A.; Mahmudov, K.T.; Pombeiro, A.J.L. Substituent effect on chalcogen bonding in 5-Substituted Benzo[c][1,2,5]selenadiazoles and their copper (II) complexes: Experimental and theoretical study. Cryst. Growth Des. 2024, 24, 781–791. [Google Scholar] [CrossRef]
- Thomas, S.P.; Satheeshkumar, K.; Mugesh, G.; Guru Row, T.N. Unusually short chalcogen bonds involving organoselenium: Insights into the Se–N bond cleavage mechanism of the antioxidant ebselen and analogues. Chem. Eur. J. 2015, 21, 6793–6800. [Google Scholar] [CrossRef] [PubMed]
- Rusakov, Y.Y.; Krivdin, L.B.; Istomina, N.V.; Levanova, E.P.; Levkovskaya, G.G. Conformational analysis of 2-formylselenophene by means of 13C–1H, 13C–13C, and 77Se–1H spin–spin coupling constants. Aust. J. Chem. 2009, 62, 734–738. [Google Scholar] [CrossRef]
- Riveras, J.A.F.; Frontera, A.; Bauzá, A. Selenium chalcogen bonds are involved in protein–carbohydrate recognition: A combined PDB and theoretical study. Phys. Chem. Chem. Phys. 2021, 23, 17656–17662. [Google Scholar] [CrossRef] [PubMed]
- Bauzá, A.; Frontera, A. Chalcogen ‘like-like’interactions involving trisulphide and triselenide compounds: A combined CSD and ab initio study. Molecules 2018, 23, 699. [Google Scholar] [CrossRef] [PubMed]
- Veljković, I.S.; Kretić, D.S.; Veljković, D.Ž. Geometrical and energetic characteristics of Se∙∙∙Se interactions in crystal structures of organoselenium molecules. CrystEngComm 2021, 23, 3383–3390. [Google Scholar] [CrossRef]
- Esrafili, M.D.; Mohammadian-Sabet, F. Prediction and characterisation of a chalcogen∙∙∙π interaction with acetylene as a potential electron donor in XHS∙∙∙HCCH and XHSe∙∙∙HCCH (X= F, Cl, Br, CN, OH, OCH3, NH2, CH3) σ-hole complexes. Mol. Phys. 2015, 113, 3559–3566. [Google Scholar] [CrossRef]
- Sapronov, A.A.; Artemjev, A.A.; Burkin, G.M.; Khrustalev, V.N.; Kubasov, A.S.; Nenajdenko, V.G.; Gomila, R.M.; Frontera, A.; Kritchenkov, A.S.; Tskhovrebov, A.G. Robust supramolecular dimers derived from benzylic-substituted 1,2,4-selenodiazolium salts featuring selenium∙∙∙π chalcogen bonding. Int. J. Mol. Sci. 2022, 23, 14973. [Google Scholar] [CrossRef] [PubMed]
- Sapronov, A.A.; Kubasov, A.S.; Khrustalev, V.N.; Artemjev, A.A.; Burkin, G.M.; Dukhnovsky, E.A.; Chizhov, A.O.; Kritchenkov, A.S.; Gomila, R.M.; Frontera, A.; et al. Se∙∙∙chalcogen bonding in 1,2,4-selenodiazolium tetraphenylborate complexes. Symmetry 2023, 15, 212. [Google Scholar] [CrossRef]
- Sarma, B.K.; Mugesh, G. Theoretical investigation on the effect of different nitrogen donors on intramolecular Se∙∙∙N interactions. ChemPhysChem 2009, 10, 3013–3020. [Google Scholar] [CrossRef]
- Esrafili, M.D.; Vakili, M.; Solimannejad, M. Cooperative interaction between π-hole and single-electron σ-hole interactions in O2S∙∙∙NCX∙∙∙CH3 and O2Se∙∙∙NCX∙∙∙CH3 complexes (X = F, Cl, Br and I). Mol. Phys. 2014, 112, 2078–2084. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S.; Concha, M.C. σ-hole bonding between like atoms; a fallacy of atomic charges. J. Mol. Model. 2008, 14, 659–665. [Google Scholar] [CrossRef] [PubMed]
- Mantina, M.; Chamberlin, A.C.; Valero, R.; Cramer, C.J.; Truhlar, D.G. Consistent van der Waals radii for the whole main group. J. Phys. Chem. A 2009, 113, 5806–5812. [Google Scholar] [CrossRef] [PubMed]
- Bleiholder, C.; Gleiter, R.; Werz, D.B.; Köppel, H. Theoretical investigations on heteronuclear chalcogen—Chalcogen interactions: On the nature of weak bonds between chalcogen centers. Inorg. Chem. 2007, 46, 2249–2260. [Google Scholar] [CrossRef] [PubMed]
- Bauzá, A.; Frontera, A. Halogen and chalcogen bond energies evaluated using electron density properties. ChemPhysChem 2020, 21, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Emamian, S.; Lu, T.; Kruse, H.; Emamian, H. Exploring nature and predicting strength of hydrogen bonds: A correlation analysis between atoms-in-molecules descriptors, binding energies, and energy components of symmetry-adapted perturbation theory. J. Comput. Chem. 2019, 40, 2868–2881. [Google Scholar] [CrossRef] [PubMed]
- Møller, C.; Plesset, M.S. Note on an approximation treatment for many-electron systems. Phys. Rev. B 1934, 46, 618–622. [Google Scholar] [CrossRef]
- Woon, D.E.; Dunning, T.H., Jr. Gaussian basis sets for use in correlated molecular calculations. III. The atoms aluminum through argon. J. Chem. Phys. 1993, 98, 1358–1371. [Google Scholar] [CrossRef]
- Yan, N.; Huo, S.; Li, X.; Zeng, Y.; Meng, L. The chalcogen bond in F2P(S)N∙∙∙SX2, F2PNS∙∙∙SX2, F2PSN∙∙∙SX2 (X = F, Cl, Br, OH, CH3, NH2) complexes. J. Mol. Model. 2019, 25, 19. [Google Scholar] [CrossRef]
- Ibrahim, M.A.A.; Saeed, R.R.A.; Shehata, M.N.I.; Moussa, N.A.M.; Tawfeek, A.M.; Ahmed, M.N.; El-Rahman, M.K.A.; Shoeib, T. Sigma-hole and lone-pair-hole site-based interactions of seesaw tetravalent chalcogen-bearing molecules with Lewis bases. ACS Omega 2023, 8, 32828–32837. [Google Scholar] [CrossRef]
- Boys, S.F.; Bernardi, F.D. The Calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision A.03; Gaussian, Inc.: Wallingford, CT, USA, 2016.
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Bader, R.F.W. A quantum theory of molecular structure and its applications. Chem. Rev. 1991, 91, 893–928. [Google Scholar] [CrossRef]
- Johnson, E.R.; Keinan, S.; Mori-Sánchez, P.; Contreras-García, J.; Cohen, A.J.; Yang, W. Revealing Noncovalent Interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef] [PubMed]
- Williams, T.; Kelley, C. Gnuplot 5.2.2: An interactive Plotting Program. 2017. Available online: http://www.gnuplot.info (accessed on 30 December 2023).
- Glendening, E.D.; Landis, C.R.; Weinhold, F. Natural bond orbital methods. WIREs Comput. Mol. Sci. 2011, 2, 1–42. [Google Scholar] [CrossRef]
- Parker, T.M.; Burns, L.A.; Parrish, R.M.; Ryno, A.G.; Sherrill, C.D. Levels of symmetry adapted perturbation theory (SAPT). I. Efficiency and performance for interaction energies. J. Chem. Phys. 2014, 140, 094106. [Google Scholar] [CrossRef]
- Parrish, R.M.; Burns, L.A.; Smith, D.G.A.; Simmonett, A.C.; DePrince, A.E., III; Hohenstein, E.G.; Bozkaya, U.; Sokolov, A.Y.; Di Remigio, R.; Richard, R.M.; et al. Psi4 1.1: An open-source electronic structure program emphasizing automation, advanced libraries, and interoperability. J. Chem. Theory Comput. 2017, 13, 3185–3197. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex | Symmetry | Eint a (kcal/mol) | EB b (kcal/mol) | Edef (kcal/mol) | NCIs | RNCI c (Å) | Rsum,1 d (Å) |
|---|---|---|---|---|---|---|---|
| SeO2∙∙∙H2O | Cs | −4.68 | −3.39 | 1.28 | Se∙∙∙O | 2.765 | 3.42 (19.2%) e |
| SeO2∙∙∙CH3OH | C1 | −7.86 | −5.37 | 2.49 | Se∙∙∙O | 2.598 | 3.42 (24.0%) |
| C–H∙∙∙O | 2.490 (116.9°) f | 2.62 (5.0%) | |||||
| SeO2∙∙∙CH3OCH3 | C1 | −10.12 | −6.73 | 3.39 | Se∙∙∙O | 2.535 | 3.42 (25.9%) |
| C–H∙∙∙OU g | 2.598 (106.0°) | 2.62 (0.8%) | |||||
| C–H∙∙∙OL g | 2.550 (114.3°) | 2.62 (2.7%) | |||||
| SeO2∙∙∙C2H4O | Cs | −10.83 | −7.72 | 3.11 | Se∙∙∙O | 2.512 | 3.42 (26.5%) |
| C–H∙∙∙O h | 2.340 (124.7°) | 2.62 (10.7%) | |||||
| SeO2∙∙∙HCHO | C1 | −6.81 | −4.70 | 2.11 | Se∙∙∙O | 2.672 | 3.42 (21.9%) |
| C–H∙∙∙O | 2.261 (123.0°) | 2.62 (13.7%) | |||||
| SeO2∙∙∙CH3CHO | C1 | −8.50 | −6.00 | 2.50 | Se∙∙∙O | 2.602 | 3.42 (23.9%) |
| C–H∙∙∙O | 2.237 (124.3°) | 2.62 (14.6%) | |||||
| SeO2∙∙∙CH3COCH3 | C1 | −9.08 | −5.83 | 3.25 | Se∙∙∙O | 2.631 | 3.42 (23.1%) |
| C–H∙∙∙OU | 2.512 (144.4°) | 2.62 (4.1%) | |||||
| C–H∙∙∙OL | 2.884 (101.0°) | 2.62 (10.1%) | |||||
| SeO2∙∙∙HCOOH | C1 | −9.88 | −6.57 | 3.31 | Se∙∙∙O | 2.642 | 3.42 (22.7%) |
| O–H∙∙∙O | 1.787 (166.2°) | 2.62 (31.8%) | |||||
| SeO2∙∙∙H2S | Cs | −3.53 | −2.42 | 1.11 | Se∙∙∙S | 3.309 | 3.70 (10.6%) |
| SeO2∙∙∙CH3SH | C1 | −7.80 | −5.47 | 2.33 | Se∙∙∙S | 3.029 | 3.70 (18.1%) |
| C–H∙∙∙O | 2.206 (139.2°) | 2.62 (15.8%) | |||||
| SeO2∙∙∙CH3SCH3 | Cs | −13.42 | −9.64 | 3.78 | Se∙∙∙S | 2.884 | 3.70 (22.1%) |
| C–H∙∙∙O | 2.182 (131.1°) | 2.62 (16.7%) | |||||
| SeO2∙∙∙C2H4S | Cs | −12.21 | −8.90 | 3.31 | Se∙∙∙S | 2.906 | 3.70 (21.5%) |
| C–H∙∙∙O | 2.211 (136.9°) | 2.62 (15.6%) | |||||
| SeO2∙∙∙HCHS | C1 | −7.67 | −5.55 | 2.12 | Se∙∙∙S | 3.021 | 3.70 (18.4%) |
| C–H∙∙∙O | 2.076 (137.1°) | 2.62 (20.8%) | |||||
| SeO2∙∙∙CH3CHS | C1 | −9.14 | −6.71 | 2.43 | Se∙∙∙S | 2.982 | 3.70 (19.4%) |
| C–H∙∙∙O | 2.059 (140.1°) | 2.62 (21.4%) | |||||
| SeO2∙∙∙CH3CSCH3 | C1 | −7.92 | −5.01 | 2.91 | Se∙∙∙S | 3.090 | 3.70 (16.5%) |
| C–H∙∙∙OU | 2.502 (150.6°) | 2.62 (4.5%) | |||||
| C–H∙∙∙OL | 2.738 (106.2°) | 2.62 (4.5%) | |||||
| SeO2∙∙∙HCSOH | C1 | −13.77 | −7.57 | 6.20 | Se∙∙∙S | 2.818 | 3.70 (23.8%) |
| O–H∙∙∙O | 1.534 (174.6°) | 2.62 (41.5%) |
| Complex | BCPs | ENCI (kcal/mol) | ρ (a.u) | ∇2ρ (a.u) | G (a.u) | V (a.u) | H (a.u) |
|---|---|---|---|---|---|---|---|
| SeO2∙∙∙H2O | Se∙∙∙O | −4.01 b | 0.0223 | 0.0666 | 0.0156 | −0.0146 | 0.0010 |
| SeO2∙∙∙CH3OH | Se∙∙∙O | −5.95 b | 0.0324 | 0.0852 | 0.0221 | −0.0229 | −0.0008 |
| C–H∙∙∙O | −1.51 c | 0.0101 | 0.0384 | 0.0080 | −0.0063 | 0.0016 | |
| SeO2∙∙∙CH3OCH3 | Se∙∙∙O | −6.99 b | 0.0377 | 0.0911 | 0.0250 | −0.0273 | −0.0023 |
| C–H∙∙∙O | −1.38 c | 0.0095 | 0.0365 | 0.0077 | −0.0062 | 0.0015 | |
| SeO2∙∙∙C2H4O | Se∙∙∙O | −7.26 b | 0.0390 | 0.0945 | 0.0260 | −0.0285 | −0.0024 |
| C–H∙∙∙OU a | −2.20 c | 0.0132 | 0.0501 | 0.0105 | −0.0084 | 0.0021 | |
| C–H∙∙∙OL a | −2.20 c | 0.0132 | 0.0501 | 0.0105 | −0.0084 | 0.0021 | |
| SeO2∙∙∙HCHO | Se∙∙∙O | −4.94 b | 0.0280 | 0.0761 | 0.0188 | −0.0186 | 0.0002 |
| C–H∙∙∙O | −2.65 c | 0.0152 | 0.0583 | 0.0123 | −0.0099 | 0.0023 | |
| SeO2∙∙∙CH3CHO | Se∙∙∙O | −5.79 b | 0.0323 | 0.0839 | 0.0216 | −0.0222 | −0.0006 |
| C–H∙∙∙O | −2.87 c | 0.0162 | 0.0613 | 0.0130 | −0.0107 | 0.0023 | |
| SeO2∙∙∙CH3COCH3 | Se∙∙∙O | −5.34 b | 0.0294 | 0.0822 | 0.0204 | −0.0203 | 0.0001 |
| C–H∙∙∙O | −1.33 c | 0.0093 | 0.0321 | 0.0068 | −0.0055 | 0.0012 | |
| SeO2∙∙∙HCOOH | Se∙∙∙O | −5.15 b | 0.0281 | 0.0807 | 0.0198 | −0.0195 | 0.0003 |
| O–H∙∙∙O | −7.00 c | 0.0347 | 0.0975 | 0.0292 | −0.0339 | −0.0048 | |
| SeO2∙∙∙H2S | Se∙∙∙S | −2.40 b | 0.0151 | 0.0322 | 0.0079 | −0.0078 | 0.0001 |
| SeO2∙∙∙CH3SH | Se∙∙∙S | −4.10 b | 0.0271 | 0.0415 | 0.0127 | −0.0150 | −0.0023 |
| C–H∙∙∙O | −2.85 c | 0.0161 | 0.0603 | 0.0128 | −0.0105 | 0.0023 | |
| SeO2∙∙∙CH3SCH3 | Se∙∙∙S | −5.55 b | 0.0371 | 0.0420 | 0.0159 | −0.0212 | −0.0053 |
| C–H∙∙∙OU a | −3.25 c | 0.0179 | 0.0668 | 0.0143 | −0.0119 | 0.0024 | |
| C–H∙∙∙OL a | −3.25 c | 0.0179 | 0.0668 | 0.0143 | −0.0119 | 0.0024 | |
| SeO2∙∙∙C2H4S | Se∙∙∙S | −5.30 b | 0.0351 | 0.0435 | 0.0155 | −0.0201 | −0.0046 |
| C–H∙∙∙OU a | −2.89 c | 0.0163 | 0.0608 | 0.0130 | −0.0107 | 0.0022 | |
| C–H∙∙∙OL a | −2.89 c | 0.0163 | 0.0608 | 0.0130 | −0.0107 | 0.0022 | |
| SeO2∙∙∙HCHS | Se∙∙∙S | −4.16 b | 0.0279 | 0.0418 | 0.0129 | −0.0153 | −0.0024 |
| C–H∙∙∙O | −3.99 c | 0.0212 | 0.0788 | 0.0173 | −0.0150 | 0.0023 | |
| SeO2∙∙∙CH3CHS | Se∙∙∙S | −4.49 b | 0.0300 | 0.0427 | 0.0137 | −0.0167 | −0.0030 |
| C–H∙∙∙O | −4.21 c | 0.0222 | 0.0808 | 0.0181 | −0.0160 | 0.0021 | |
| SeO2∙∙∙CH3CSCH3 | Se∙∙∙S | −3.63 b | 0.0242 | 0.0400 | 0.0115 | −0.0130 | −0.0015 |
| C–H∙∙∙O | −1.42 c | 0.0097 | 0.0328 | 0.0070 | −0.0058 | 0.0012 | |
| C–H∙∙∙O | −0.84 c | 0.0071 | 0.0275 | 0.0056 | −0.0043 | 0.0013 | |
| SeO2∙∙∙HCSOH | Se∙∙∙S | −6.34 b | 0.0410 | 0.0440 | 0.0178 | −0.0245 | −0.0068 |
| O–H∙∙∙O | −14.20 c | 0.0670 | 0.1100 | 0.0532 | −0.0790 | −0.0257 |
| Complex | Donor | Acceptor | E(2) | Complex | Donor | Acceptor | E(2) |
|---|---|---|---|---|---|---|---|
| SeO2∙∙∙H2O | LP(O) | BD*(OSe) | 5.95 | SeO2∙∙∙H2S | LP(S) | BD*(OSe) | 7.99 |
| SeO2∙∙∙CH3OH | LP(O) | BD*(OSe) | 11.02 | SeO2∙∙∙CH3SH | LP(S) | BD*(OSe) | 17.83 |
| SeO2∙∙∙CH3OCH3 | LP(O) | BD*(OSe) | 14.87 | SeO2∙∙∙CH3SCH3 | LP(S) | BD*(OSe) | 29.00 |
| SeO2∙∙∙C2H4O | LP(O) | BD*(OSe) | 15.76 | SeO2∙∙∙C2H4S | LP(S) | BD*(OSe) | 26.00 |
| SeO2∙∙∙HCHO | LP(O) | BD*(OSe) | 9.35 | SeO2∙∙∙HCHS | LP(S) | BD*(OSe) | 16.38 |
| SeO2∙∙∙CH3CHO | LP(O) | BD*(OSe) | 12.46 | SeO2∙∙∙CH3CHS | LP(S) | BD*(OSe) | 19.27 |
| SeO2∙∙∙CH3COCH3 | LP(O) | BD*(OSe) | 13.09 | SeO2∙∙∙CH3CSCH3 | LP(S) | BD*(OSe) | 16.01 |
| SeO2∙∙∙HCOOH | LP(O) | BD*(OSe) | 15.53 | SeO2∙∙∙HCSOH | LP(S) | BD*(OSe) | 56.02 |
| Complex | Eelec | Eind | Edisp | Eex-re | Etotal |
|---|---|---|---|---|---|
| SeO2∙∙∙H2O | −12.86 (62%) a | −3.34 (16%) | −4.66 (22%) | 15.53 | −5.32 |
| SeO2∙∙∙CH3OH | −20.39 (56%) | −7.52 (21%) | −8.45 (23%) | 28.83 | −7.54 |
| SeO2∙∙∙CH3OCH3 | −23.56 (53%) | −9.88 (22%) | −11.41 (25%) | 35.29 | −9.56 |
| SeO2∙∙∙C2H4O | −25.02 (52%) | −11.75 (24%) | −11.65 (24%) | 38.55 | −9.86 |
| SeO2∙∙∙HCHO | −15.56 (52%) | −6.90 (23%) | −7.55 (25%) | 23.15 | −6.85 |
| SeO2∙∙∙CH3CHO | −19.12 (52%) | −8.96 (24%) | −8.71 (24%) | 28.76 | −8.02 |
| SeO2∙∙∙CH3COCH3 | −18.83 (54%) | −6.97 (20%) | −9.01 (26%) | 26.52 | −8.29 |
| SeO2∙∙∙HCOOH | −22.80 (51%) | −12.06 (27%) | −9.93 (22%) | 35.51 | −9.28 |
| SeO2∙∙∙H2S | −8.41 (53%) | −2.56 (16%) | −4.88 (31%) | 12.28 | −3.56 |
| SeO2∙∙∙CH3SH | −18.40 (49%) | −9.27 (25%) | −9.76 (26%) | 30.54 | −6.90 |
| SeO2∙∙∙CH3SCH3 | −28.43 (47%) | −17.59 (29%) | −14.63 (24%) | 48.28 | −12.38 |
| SeO2∙∙∙C2H4S | −26.48 (47%) | −15.55 (28%) | −13.78 (25%) | 44.24 | −11.56 |
| SeO2∙∙∙HCHS | −18.45 (47%) | −11.03 (28%) | −9.92 (25%) | 32.38 | −7.02 |
| SeO2∙∙∙CH3CHS | −20.99 (47%) | −12.71 (29%) | −10.79 (24%) | 36.28 | −8.21 |
| SeO2∙∙∙CH3CSCH3 | −16.43 (48%) | −7.75 (23%) | −9.79 (29%) | 26.28 | −7.70 |
| SeO2∙∙∙HCSOH | −43.82 (47%) | −32.00 (35%) | −16.96 (18%) | 80.24 | −12.54 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, T.; Chen, R.; Liu, Q.; Zhong, Y.; Lei, F.; Zeng, Z. Unveiling the Nature and Strength of Selenium-Centered Chalcogen Bonds in Binary Complexes of SeO2 with Oxygen-/Sulfur-Containing Lewis Bases: Insights from Theoretical Calculations. Int. J. Mol. Sci. 2024, 25, 5609. https://doi.org/10.3390/ijms25115609
Lu T, Chen R, Liu Q, Zhong Y, Lei F, Zeng Z. Unveiling the Nature and Strength of Selenium-Centered Chalcogen Bonds in Binary Complexes of SeO2 with Oxygen-/Sulfur-Containing Lewis Bases: Insights from Theoretical Calculations. International Journal of Molecular Sciences. 2024; 25(11):5609. https://doi.org/10.3390/ijms25115609
Chicago/Turabian StyleLu, Tao, Renhua Chen, Qingyu Liu, Yeshuang Zhong, Fengying Lei, and Zhu Zeng. 2024. "Unveiling the Nature and Strength of Selenium-Centered Chalcogen Bonds in Binary Complexes of SeO2 with Oxygen-/Sulfur-Containing Lewis Bases: Insights from Theoretical Calculations" International Journal of Molecular Sciences 25, no. 11: 5609. https://doi.org/10.3390/ijms25115609
APA StyleLu, T., Chen, R., Liu, Q., Zhong, Y., Lei, F., & Zeng, Z. (2024). Unveiling the Nature and Strength of Selenium-Centered Chalcogen Bonds in Binary Complexes of SeO2 with Oxygen-/Sulfur-Containing Lewis Bases: Insights from Theoretical Calculations. International Journal of Molecular Sciences, 25(11), 5609. https://doi.org/10.3390/ijms25115609