

Metabolic Dysfunction–Associated Steatotic Liver Disease: From Pathogenesis to Current Therapeutic Options

 ,
,
Abstract
1. Introduction
2. Clinical Manifestations
3. Pathogenesis
3.1. Genetic Aspects
3.2. Derangement of Lipid Homeostasis
3.3. Derangement of Carbohydrate Homeostasis
3.4. Deranged Bile Acid Homeostasis
3.5. Gut Dysbiosis
4. Therapeutic Management of MASLD
4.1. Lifestyle: Diet and Physical Exercise
4.2. Bariatric Surgery
4.3. Pharmacological Therapy
4.3.1. Antidiabetic Drugs
4.3.2. Statins
4.3.3. Peroxisome Proliferator-Activator Receptor (PPAR) Agonists
4.3.4. FXR Agonists
4.3.5. Thyroid Hormone Receptor Beta (TR-β) Agonists
4.3.6. Anti-Fibrotic and Anti-Inflammatory Agents
4.3.7. Stearoyl-Coenzyme A Desaturase-1 (SCD1) Inhibitors
4.3.8. DGAT Inhibitors and FASN Inhibitors
4.3.9. MGAT2 Inhibitors
4.3.10. Dimethyl Peptidase 4 (DPP4) Inhibitors
4.3.11. Ketohexokinase (KHK) Inhibitors
4.3.12. Miscellanea
5. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- NCD Risk Factor Collaboration. Rising rural body-mass index is the main driver of the global obesity epidemic in adults. Nature 2019, 569, 260–264. [Google Scholar] [CrossRef] [PubMed]
- Vecchie, A.; Dallegri, F.; Carbone, F.; Bonaventura, A.; Liberale, L.; Portincasa, P.; Fruhbeck, G.; Montecucco, F. Obesity phenotypes and their paradoxical association with cardiovascular diseases. Eur. J. Intern. Med. 2018, 48, 6–17. [Google Scholar] [CrossRef]
- Finucane, M.M.; Stevens, G.A.; Cowan, M.J.; Danaei, G.; Lin, J.K.; Paciorek, C.J.; Singh, G.M.; Gutierrez, H.R.; Lu, Y.A.; Bahalim, A.N.; et al. National, regional, and global trends in body-mass index since 1980: Systematic analysis of health examination surveys and epidemiological studies with 960 country-years and 9.1 million participants. Lancet 2011, 377, 557–567. [Google Scholar] [CrossRef]
- World Health Organization. Obesity and Overweight. Available online: https://www.who.int/news-room/fact-sheets/detail/obesity-and-overweight (accessed on 23 February 2023).
- Jebeile, H.; Kelly, A.S.; O’Malley, G.; Baur, L.A. Obesity in children and adolescents: Epidemiology, causes, assessment, and management. Lancet Diabetes Endocrinol. 2022, 10, 351–365. [Google Scholar] [CrossRef] [PubMed]
- Wasniewska, M.; Pepe, G.; Aversa, T.; Bellone, S.; de Sanctis, L.; Di Bonito, P.; Faienza, M.F.; Improda, N.; Licenziati, M.R.; Maffeis, C.; et al. Skeptical Look at the Clinical Implication of Metabolic Syndrome in Childhood Obesity. Children 2023, 10, 735. [Google Scholar] [CrossRef] [PubMed]
- Di Bonito, P.; Di Sessa, A.; Licenziati, M.R.; Corica, D.; Wasniewska, M.; Umano, G.R.; Morandi, A.; Maffeis, C.; Faienza, M.F.; Mozzillo, E.; et al. Is Metabolic Syndrome Useful for Identifying Youths with Obesity at Risk for NAFLD? Children 2023, 10, 233. [Google Scholar] [CrossRef]
- Portincasa, P.; Di Ciaula, A.; Bonfrate, L.; Stella, A.; Garruti, G.; Lamont, J.T. Metabolic dysfunction-associated gallstone disease: Expecting more from critical care manifestations. Intern. Emerg. Med. 2023, 18, 1897–1918. [Google Scholar] [CrossRef]
- Bhaskaran, K.; Douglas, I.; Forbes, H.; dos-Santos-Silva, I.; Leon, D.A.; Smeeth, L. Body-mass index and risk of 22 specific cancers: A population-based cohort study of 5.24 million UK adults. Lancet 2014, 384, 755–765. [Google Scholar] [CrossRef]
- Dietz, W.; Santos-Burgoa, C. Obesity and its Implications for COVID-19 Mortality. Obesity 2020, 28, 1005. [Google Scholar] [CrossRef]
- Ludwig, J.; Viggiano, T.R.; McGill, D.B.; Oh, B.J. Nonalcoholic steatohepatitis: Mayo Clinic experiences with a hitherto unnamed disease. Mayo Clin. Proc. 1980, 55, 434–438. [Google Scholar]
- Schaffner, F.; Thaler, H. Nonalcoholic fatty liver disease. Prog. Liver Dis. 1986, 8, 283–298. [Google Scholar] [PubMed]
- Browning, J.D.; Szczepaniak, L.S.; Dobbins, R.; Nuremberg, P.; Horton, J.D.; Cohen, J.C.; Grundy, S.M.; Hobbs, H.H. Prevalence of hepatic steatosis in an urban population in the United States: Impact of ethnicity. Hepatology 2004, 40, 1387–1395. [Google Scholar] [CrossRef] [PubMed]
- Szczepaniak, L.S.; Nurenberg, P.; Leonard, D.; Browning, J.D.; Reingold, J.S.; Grundy, S.; Hobbs, H.H.; Dobbins, R.L. Magnetic resonance spectroscopy to measure hepatic triglyceride content: Prevalence of hepatic steatosis in the general population. Am. J. Physiol. Endocrinol. Metab. 2005, 288, E462–E468. [Google Scholar] [CrossRef] [PubMed]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Charlton, M.; Cusi, K.; Rinella, M.; Harrison, S.A.; Brunt, E.M.; Sanyal, A.J. The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American Association for the Study of Liver Diseases. Hepatology 2018, 67, 328–357. [Google Scholar] [CrossRef]
- Singh, S.; Allen, A.M.; Wang, Z.; Prokop, L.J.; Murad, M.H.; Loomba, R. Fibrosis progression in nonalcoholic fatty liver vs nonalcoholic steatohepatitis: A systematic review and meta-analysis of paired-biopsy studies. Clin. Gastroenterol. Hepatol. 2015, 13, 643–654.e9. [Google Scholar] [CrossRef] [PubMed]
- Portincasa, P. NAFLD, MAFLD, and beyond: One or several acronyms for better comprehension and patient care. Intern. Emerg. Med. 2023, 18, 993–1006. [Google Scholar] [CrossRef] [PubMed]
- Powell, E.E.; Wong, V.W.; Rinella, M. Non-alcoholic fatty liver disease. Lancet 2021, 397, 2212–2224. [Google Scholar] [CrossRef] [PubMed]
- Sanyal, A.J.; Harrison, S.A.; Ratziu, V.; Abdelmalek, M.F.; Diehl, A.M.; Caldwell, S.; Shiffman, M.L.; Aguilar Schall, R.; Jia, C.; McColgan, B.; et al. The Natural History of Advanced Fibrosis due to Nonalcoholic Steatohepatitis: Data from the Simtuzumab Trials. Hepatology 2019, 70, 1913–1927. [Google Scholar] [CrossRef] [PubMed]
- Eslam, M.; Sanyal, A.J.; George, J. MAFLD: A Consensus-Driven Proposed Nomenclature for Metabolic Associated Fatty Liver Disease. Gastroenterology 2020, 158, 1999–2014.e1. [Google Scholar] [CrossRef]
- Mendez-Sanchez, N.; Bugianesi, E.; Gish, R.G.; Lammert, F.; Tilg, H.; Nguyen, M.H.; Sarin, S.K.; Fabrellas, N.; Zelber-Sagi, S.; Fan, J.G.; et al. Global multi-stakeholder endorsement of the MAFLD definition. Lancet Gastroenterol. Hepatol. 2022, 7, 388–390. [Google Scholar] [CrossRef]
- Rinella, M.E.; Lazarus, J.V.; Ratziu, V.; Francque, S.M.; Sanyal, A.J.; Kanwal, F.; Romero, D.; Abdelmalek, M.F.; Anstee, Q.M.; Arab, J.P.; et al. A multisociety Delphi consensus statement on new fatty liver disease nomenclature. Hepatology 2023, 78, 1966–1986. [Google Scholar] [CrossRef] [PubMed]
- Stepanova, M.; Rafiq, N.; Younossi, Z.M. Components of metabolic syndrome are independent predictors of mortality in patients with chronic liver disease: A population-based study. Gut 2010, 59, 1410–1415. [Google Scholar] [CrossRef] [PubMed]
- Ratziu, V.; Boursier, J.; AFEF Group for the Study of Liver Fibrosis. Confirmatory biomarker diagnostic studies are not needed when transitioning from NAFLD to MASLD. J. Hepatol. 2024, 80, e51–e52. [Google Scholar] [CrossRef]
- Song, S.J.; Lai, J.C.; Wong, G.L.; Wong, V.W.; Yip, T.C. Can we use old NAFLD data under the new MASLD definition? J. Hepatol. 2024, 80, e54–e56. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.M.; Yoon, E.L.; Kim, M.; Kang, B.K.; Cho, S.; Nah, E.H.; Jun, D.W. Prevalence, distribution, and hepatic fibrosis burden of the different subtypes of steatotic liver disease in primary care settings. Hepatology 2023, 79, 1393–1400. [Google Scholar] [CrossRef] [PubMed]
- Rinella, M.E.; Sookoian, S. From NAFLD to MASLD: Updated naming and diagnosis criteria for fatty liver disease. J. Lipid Res. 2024, 65, 100485. [Google Scholar] [CrossRef] [PubMed]
- Shao, Y.; Chen, S.; Han, L.; Liu, J. Pharmacotherapies of NAFLD: Updated opportunities based on metabolic intervention. Nutr. Metab. 2023, 20, 30. [Google Scholar] [CrossRef]
- Ramirez-Mejia, M.M.; Jimenez-Gutierrez, C.; Eslam, M.; George, J.; Mendez-Sanchez, N. Breaking new ground: MASLD vs. MAFLD-which holds the key for risk stratification? Hepatol. Int. 2024, 18, 168–178. [Google Scholar] [CrossRef]
- Spengler, E.K.; Loomba, R. Recommendations for Diagnosis, Referral for Liver Biopsy, and Treatment of Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis. Mayo Clin. Proc. 2015, 90, 1233–1246. [Google Scholar] [CrossRef]
- Palmentieri, B.; de Sio, I.; La Mura, V.; Masarone, M.; Vecchione, R.; Bruno, S.; Torella, R.; Persico, M. The role of bright liver echo pattern on ultrasound B-mode examination in the diagnosis of liver steatosis. Dig. Liver Dis. 2006, 38, 485–489. [Google Scholar] [CrossRef]
- Santoro, S.; Khalil, M.; Abdallah, H.; Farella, I.; Noto, A.; Dipalo, G.M.; Villani, P.; Bonfrate, L.; Di Ciaula, A.; Portincasa, P. Early and accurate diagnosis of steatotic liver by artificial intelligence (AI)-supported ultrasonography. Eur. J. Intern. Med. 2024, in press. [Google Scholar] [CrossRef] [PubMed]
- Torres, D.M.; Williams, C.D.; Harrison, S.A. Features, diagnosis, and treatment of nonalcoholic fatty liver disease. Clin. Gastroenterol. Hepatol. 2012, 10, 837–858. [Google Scholar] [CrossRef] [PubMed]
- Kleiner, D.E.; Brunt, E.M.; Van Natta, M.; Behling, C.; Contos, M.J.; Cummings, O.W.; Ferrell, L.D.; Liu, Y.C.; Torbenson, M.S.; Unalp-Arida, A.; et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005, 41, 1313–1321. [Google Scholar] [CrossRef] [PubMed]
- Vuppalanchi, R.; Noureddin, M.; Alkhouri, N.; Sanyal, A.J. Therapeutic pipeline in nonalcoholic steatohepatitis. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 373–392. [Google Scholar] [CrossRef] [PubMed]
- Caussy, C.; Soni, M.; Cui, J.; Bettencourt, R.; Schork, N.; Chen, C.H.; Ikhwan, M.A.; Bassirian, S.; Cepin, S.; Gonzalez, M.P.; et al. Nonalcoholic fatty liver disease with cirrhosis increases familial risk for advanced fibrosis. J. Clin. Investig. 2017, 127, 2697–2704. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Angulo, P.; Kleiner, D.E.; Dam-Larsen, S.; Adams, L.A.; Bjornsson, E.S.; Charatcharoenwitthaya, P.; Mills, P.R.; Keach, J.C.; Lafferty, H.D.; Stahler, A.; et al. Liver Fibrosis, but No Other Histologic Features, Is Associated with Long-term Outcomes of Patients with Nonalcoholic Fatty Liver Disease. Gastroenterology 2015, 149, 389–397.e10. [Google Scholar] [CrossRef] [PubMed]
- Targher, G.; Byrne, C.D.; Tilg, H. MASLD: A systemic metabolic disorder with cardiovascular and malignant complications. Gut 2024, 73, 691–702. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.A.; Kendall, B.J.; El-Serag, H.B.; Thrift, A.P.; Macdonald, G.A. Hepatocellular and extrahepatic cancer risk in people with non-alcoholic fatty liver disease. Lancet Gastroenterol. Hepatol. 2024, 9, 159–169. [Google Scholar] [CrossRef]
- Di Ciaula, A.; Passarella, S.; Shanmugam, H.; Noviello, M.; Bonfrate, L.; Wang, D.Q.-H.; Portincasa, P. Nonalcoholic Fatty Liver Disease (NAFLD). Mitochondria as Players and Targets of Therapies? Int. J. Mol. Sci. 2021, 22, 5375. [Google Scholar] [CrossRef]
- Grattagliano, I.; Di Ciaula, A.; Baj, J.; Molina-Molina, E.; Shanmugam, H.; Garruti, G.; Wang, D.Q.; Portincasa, P. Protocols for Mitochondria as the Target of Pharmacological Therapy in the Context of Nonalcoholic Fatty Liver Disease (NAFLD). Methods Mol. Biol. 2021, 2310, 201–246. [Google Scholar] [CrossRef] [PubMed]
- Molina-Molina, E.; Shanmugam, H.; Di Palo, D.; Grattagliano, I.; Portincasa, P. Exploring Liver Mitochondrial Function by (13)C-Stable Isotope Breath Tests: Implications in Clinical Biochemistry. Methods Mol. Biol. 2021, 2310, 179–199. [Google Scholar] [CrossRef] [PubMed]
- Di Ciaula, A.; Calamita, G.; Shanmugam, H.; Khalil, M.; Bonfrate, L.; Wang, D.Q.; Baffy, G.; Portincasa, P. Mitochondria Matter: Systemic Aspects of Nonalcoholic Fatty Liver Disease (NAFLD) and Diagnostic Assessment of Liver Function by Stable Isotope Dynamic Breath Tests. Int. J. Mol. Sci. 2021, 22, 7702. [Google Scholar] [CrossRef] [PubMed]
- Anstee, Q.M.; Seth, D.; Day, C.P. Genetic Factors That Affect Risk of Alcoholic and Nonalcoholic Fatty Liver Disease. Gastroenterology 2016, 150, 1728. [Google Scholar] [CrossRef] [PubMed]
- Krawczyk, M.; Portincasa, P.; Lammert, F. PNPLA3-associated steatohepatitis: Toward a gene-based classification of fatty liver disease. Semin. Liver Dis. 2013, 33, 369–379. [Google Scholar] [CrossRef]
- Krawczyk, M.; Bonfrate, L.; Portincasa, P. Nonalcoholic fatty liver disease. Best. Pract. Res. Clin. Gastroenterol. 2010, 24, 695–708. [Google Scholar] [CrossRef]
- Dongiovanni, P.; Donati, B.; Fares, R.; Lombardi, R.; Mancina, R.M.; Romeo, S.; Valenti, L. PNPLA3 I148M polymorphism and progressive liver disease. World J. Gastroenterol. 2013, 19, 6969–6978. [Google Scholar] [CrossRef]
- Dongiovanni, P.; Valenti, L.; Rametta, R.; Daly, A.K.; Nobili, V.; Mozzi, E.; Leathart, J.B.; Pietrobattista, A.; Burt, A.D.; Maggioni, M.; et al. Genetic variants regulating insulin receptor signalling are associated with the severity of liver damage in patients with non-alcoholic fatty liver disease. Gut 2010, 59, 267–273. [Google Scholar] [CrossRef]
- Kozlitina, J.; Smagris, E.; Stender, S.; Nordestgaard, B.G.; Zhou, H.H.; Tybjaerg-Hansen, A.; Vogt, T.F.; Hobbs, H.H.; Cohen, J.C. Exome-wide association study identifies a TM6SF2 variant that confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 2014, 46, 352–356. [Google Scholar] [CrossRef]
- Mancina, R.M.; Dongiovanni, P.; Petta, S.; Pingitore, P.; Meroni, M.; Rametta, R.; Boren, J.; Montalcini, T.; Pujia, A.; Wiklund, O.; et al. The MBOAT7-TMC4 Variant rs641738 Increases Risk of Nonalcoholic Fatty Liver Disease in Individuals of European Descent. Gastroenterology 2016, 150, 1219–1230.e6. [Google Scholar] [CrossRef]
- Beer, N.L.; Tribble, N.D.; McCulloch, L.J.; Roos, C.; Johnson, P.R.; Orho-Melander, M.; Gloyn, A.L. The P446L variant in GCKR associated with fasting plasma glucose and triglyceride levels exerts its effect through increased glucokinase activity in liver. Hum. Mol. Genet. 2009, 18, 4081–4088. [Google Scholar] [CrossRef] [PubMed]
- Vazquez-Chantada, M.; Gonzalez-Lahera, A.; Martinez-Arranz, I.; Garcia-Monzon, C.; Regueiro, M.M.; Garcia-Rodriguez, J.L.; Schlangen, K.A.; Mendibil, I.; Rodriguez-Ezpeleta, N.; Lozano, J.J.; et al. Solute carrier family 2 member 1 is involved in the development of nonalcoholic fatty liver disease. Hepatology 2013, 57, 505–514. [Google Scholar] [CrossRef] [PubMed]
- Abul-Husn, N.S.; Cheng, X.; Li, A.H.; Xin, Y.; Schurmann, C.; Stevis, P.; Liu, Y.; Kozlitina, J.; Stender, S.; Wood, G.C.; et al. A Protein-Truncating HSD17B13 Variant and Protection from Chronic Liver Disease. N. Engl. J. Med. 2018, 378, 1096–1106. [Google Scholar] [CrossRef]
- Portincasa, P.; Khalil, M.; Graziani, A.; Fruhbeck, G.; Baffy, G.; Garruti, G.; Di Ciaula, A.; Bonfrate, L. Gut microbes in metabolic disturbances. Promising role for therapeutic manipulations? Eur. J. Intern. Med. 2023, 119, 13–30. [Google Scholar] [CrossRef] [PubMed]
- Di Ciaula, A.; Baj, J.; Garruti, G.; Celano, G.; De Angelis, M.; Wang, H.H.; Di Palo, D.M.; Bonfrate, L.; Wang, D.Q.; Portincasa, P. Liver Steatosis, Gut-Liver Axis, Microbiome and Environmental Factors. A Never-Ending Bidirectional Cross-Talk. J. Clin. Med. 2020, 9, 2648. [Google Scholar] [CrossRef] [PubMed]
- Di Ciaula, A.; Bonfrate, L.; Portincasa, P. The role of microbiota in nonalcoholic fatty liver disease. Eur. J. Clin. Investig. 2022, 52, e13768. [Google Scholar] [CrossRef] [PubMed]
- Di Palo, D.M.; Garruti, G.; Di Ciaula, A.; Molina-Molina, E.; Shanmugam, H.; De Angelis, M.; Portincasa, P. Increased Colonic Permeability and Lifestyles as Contributing Factors to Obesity and Liver Steatosis. Nutrients 2020, 12, E564. [Google Scholar] [CrossRef]
- Molina-Molina, E.; Shanmugam, H.; Di Ciaula, A.; Grattagliano, I.; Di Palo, D.M.; Palmieri, V.O.; Portincasa, P. ((13)C)-Methacetin breath test provides evidence of subclinical liver dysfunction linked to fat storage but not lifestyle. JHEP Rep. 2021, 3, 100203. [Google Scholar] [CrossRef] [PubMed]
- Portincasa, P.; Bonfrate, L.; Khalil, M.; Angelis, M.; Calabrese, F.M.; D’Amato, M.; Wang, D.Q.; Di Ciaula, A. Intestinal Barrier and Permeability in Health, Obesity and NAFLD. Biomedicines 2021, 10, 83. [Google Scholar] [CrossRef]
- Bian, X.; Liu, R.; Meng, Y.; Xing, D.; Xu, D.; Lu, Z. Lipid metabolism and cancer. J. Exp. Med. 2021, 218, e20201606. [Google Scholar] [CrossRef]
- Lomonaco, R.; Ortiz-Lopez, C.; Orsak, B.; Webb, A.; Hardies, J.; Darland, C.; Finch, J.; Gastaldelli, A.; Harrison, S.; Tio, F.; et al. Effect of adipose tissue insulin resistance on metabolic parameters and liver histology in obese patients with nonalcoholic fatty liver disease. Hepatology 2012, 55, 1389–1397. [Google Scholar] [CrossRef]
- Ralston, J.C.; Lyons, C.L.; Kennedy, E.B.; Kirwan, A.M.; Roche, H.M. Fatty Acids and NLRP3 Inflammasome-Mediated Inflammation in Metabolic Tissues. Annu. Rev. Nutr. 2017, 37, 77–102. [Google Scholar] [CrossRef] [PubMed]
- Hirsova, P.; Ibrabim, S.H.; Gores, G.J.; Malhi, H. Lipotoxic lethal and sublethal stress signaling in hepatocytes: Relevance to NASH pathogenesis. J. Lipid Res. 2016, 57, 1758–1770. [Google Scholar] [CrossRef] [PubMed]
- Fu, S.; Watkins, S.M.; Hotamisligil, G.S. The role of endoplasmic reticulum in hepatic lipid homeostasis and stress signaling. Cell Metab. 2012, 15, 623–634. [Google Scholar] [CrossRef]
- Perry, R.J.; Samuel, V.T.; Petersen, K.F.; Shulman, G.I. The role of hepatic lipids in hepatic insulin resistance and type 2 diabetes. Nature 2014, 510, 84–91. [Google Scholar] [CrossRef]
- Shi, H.; Kokoeva, M.V.; Inouye, K.; Tzameli, I.; Yin, H.; Flier, J.S. TLR4 links innate immunity and fatty acid-induced insulin resistance. J. Clin. Investig. 2006, 116, 3015–3025. [Google Scholar] [CrossRef]
- Xia, J.Y.; Holland, W.L.; Kusminski, C.M.; Sun, K.; Sharma, A.X.; Pearson, M.J.; Sifuentes, A.J.; McDonald, J.G.; Gordillo, R.; Scherer, P.E. Targeted Induction of Ceramide Degradation Leads to Improved Systemic Metabolism and Reduced Hepatic Steatosis. Cell Metab. 2015, 22, 266–278. [Google Scholar] [CrossRef] [PubMed]
- Turpin, S.M.; Nicholls, H.T.; Willmes, D.M.; Mourier, A.; Brodesser, S.; Wunderlich, C.M.; Mauer, J.; Xu, E.; Hammerschmidt, P.; Bronneke, H.S.; et al. Obesity-induced CerS6-dependent C16:0 ceramide production promotes weight gain and glucose intolerance. Cell Metab. 2014, 20, 678–686. [Google Scholar] [CrossRef]
- Martinez, L.; Torres, S.; Baulies, A.; Alarcon-Vila, C.; Elena, M.; Fabrias, G.; Casas, J.; Caballeria, J.; Fernandez-Checa, J.C.; Garcia-Ruiz, C. Myristic acid potentiates palmitic acid-induced lipotoxicity and steatohepatitis associated with lipodystrophy by sustaning de novo ceramide synthesis. Oncotarget 2015, 6, 41479–41496. [Google Scholar] [CrossRef]
- Garcia-Ruiz, C.; Colell, A.; Mari, M.; Morales, A.; Fernandez-Checa, J.C. Direct effect of ceramide on the mitochondrial electron transport chain leads to generation of reactive oxygen species. Role of mitochondrial glutathione. J. Biol. Chem. 1997, 272, 11369–11377. [Google Scholar] [CrossRef]
- Sanyal, A.J.; Campbell-Sargent, C.; Mirshahi, F.; Rizzo, W.B.; Contos, M.J.; Sterling, R.K.; Luketic, V.A.; Shiffman, M.L.; Clore, J.N. Nonalcoholic steatohepatitis: Association of insulin resistance and mitochondrial abnormalities. Gastroenterology 2001, 120, 1183–1192. [Google Scholar] [CrossRef] [PubMed]
- Perez-Carreras, M.; Del Hoyo, P.; Martin, M.A.; Rubio, J.C.; Martin, A.; Castellano, G.; Colina, F.; Arenas, J.; Solis-Herruzo, J.A. Defective hepatic mitochondrial respiratory chain in patients with nonalcoholic steatohepatitis. Hepatology 2003, 38, 999–1007. [Google Scholar] [CrossRef] [PubMed]
- DiPilato, L.M.; Ahmad, F.; Harms, M.; Seale, P.; Manganiello, V.; Birnbaum, M.J. The Role of PDE3B Phosphorylation in the Inhibition of Lipolysis by Insulin. Mol. Cell Biol. 2015, 35, 2752–2760. [Google Scholar] [CrossRef] [PubMed]
- Ceddia, R.P.; Collins, S. A compendium of G-protein-coupled receptors and cyclic nucleotide regulation of adipose tissue metabolism and energy expenditure. Clin. Sci. 2020, 134, 473–512. [Google Scholar] [CrossRef] [PubMed]
- Gastaldelli, A.; Cusi, K.; Pettiti, M.; Hardies, J.; Miyazaki, Y.; Berria, R.; Buzzigoli, E.; Sironi, A.M.; Cersosimo, E.; Ferrannini, E. Relationship between hepatic/visceral fat and hepatic insulin resistance in nondiabetic and type 2 diabetic subjects. Gastroenterology 2007, 133, 496–506. [Google Scholar] [CrossRef] [PubMed]
- Fabbrini, E.; Magkos, F.; Mohammed, B.S.; Pietka, T.; Abumrad, N.A.; Patterson, B.W.; Okunade, A.; Klein, S. Intrahepatic fat, not visceral fat, is linked with metabolic complications of obesity. Proc. Natl. Acad. Sci. USA 2009, 106, 15430–15435. [Google Scholar] [CrossRef] [PubMed]
- Kotronen, A.; Juurinen, L.; Tiikkainen, M.; Vehkavaara, S.; Yki-Jarvinen, H. Increased liver fat, impaired insulin clearance, and hepatic and adipose tissue insulin resistance in type 2 diabetes. Gastroenterology 2008, 135, 122–130. [Google Scholar] [CrossRef]
- Samuel, V.T.; Shulman, G.I. The pathogenesis of insulin resistance: Integrating signaling pathways and substrate flux. J. Clin. Investig. 2016, 126, 12–22. [Google Scholar] [CrossRef]
- Shi, Y.; Cheng, D. Beyond triglyceride synthesis: The dynamic functional roles of MGAT and DGAT enzymes in energy metabolism. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E10–E18. [Google Scholar] [CrossRef]
- Wang, D.Q.H.; Neuschwander-Tetri, B.A.; Portincasa, P. The Biliary System, 2nd ed.; Morgan & Claypool Life Sciences: San Rafael, CA, USA, 2017; Volume 8, pp. 460–490. [Google Scholar]
- Samuel, V.T.; Shulman, G.I. Nonalcoholic Fatty Liver Disease as a Nexus of Metabolic and Hepatic Diseases. Cell Metab. 2018, 27, 22–41. [Google Scholar] [CrossRef]
- Lambert, J.E.; Ramos–Roman, M.A.; Browning, J.D.; Parks, E.J. Increased de novo lipogenesis is a distinct characteristic of individuals with nonalcoholic fatty liver disease. Gastroenterology 2014, 146, 726–735. [Google Scholar] [CrossRef] [PubMed]
- Coleman, R.A.; Lee, D.P. Enzymes of triacylglycerol synthesis and their regulation. Prog. Lipid Res. 2004, 43, 134–176. [Google Scholar] [CrossRef]
- Kawano, Y.; Cohen, D.E. Mechanisms of hepatic triglyceride accumulation in non-alcoholic fatty liver disease. J. Gastroenterol. 2013, 48, 434–441. [Google Scholar] [CrossRef] [PubMed]
- Oosterveer, M.H.; Schoonjans, K. Hepatic glucose sensing and integrative pathways in the liver. Cell Mol. Life Sci. 2014, 71, 1453–1467. [Google Scholar] [CrossRef]
- Bianchi, A.; Evans, J.L.; Iverson, A.J.; Nordlund, A.C.; Watts, T.D.; Witters, L.A. Identification of an isozymic form of acetyl-CoA carboxylase. J. Biol. Chem. 1990, 265, 1502–1509. [Google Scholar] [CrossRef] [PubMed]
- Smith, S. The animal fatty acid synthase: One gene, one polypeptide, seven enzymes. FASEB J. 1994, 8, 1248–1259. [Google Scholar] [CrossRef]
- Brindley, D.; Matsumura, S.; Bloch, K. Mycobacterium phlei fatty acid synthetase—A bacterial multienzyme complex. Nature 1969, 224, 666–669. [Google Scholar] [CrossRef]
- Majerus, P.W.; Alberts, A.W.; Vagelos, P.R. The Acyl Carrier Protein of Fatty Acid Synthesis: Purification, Physical Properties, and Substrate Binding Site. Proc. Natl. Acad. Sci. USA 1964, 51, 1231–1238. [Google Scholar] [CrossRef]
- Maier, T.; Leibundgut, M.; Ban, N. The crystal structure of a mammalian fatty acid synthase. Science 2008, 321, 1315–1322. [Google Scholar] [CrossRef]
- Smith, S.; Tsai, S.C. The type I fatty acid and polyketide synthases: A tale of two megasynthases. Nat. Prod. Rep. 2007, 24, 1041–1072. [Google Scholar] [CrossRef]
- Wakil, S.J. Fatty acid synthase, a proficient multifunctional enzyme. Biochemistry 1989, 28, 4523–4530. [Google Scholar] [CrossRef] [PubMed]
- Carey, E.M.; Dils, R.; Hansen, H.J. Short communications. Chain-length specificity for termination of atty acid biosynthesis by fatty acid synthetase complexes from lactating rabbit mamary gland and rat liver. Biochem. J. 1970, 117, 633–635. [Google Scholar] [CrossRef] [PubMed]
- Foster, D.W.; Bloom, B. The synthesis of fatty acids by rat liver slices in tritiated water. J. Biol. Chem. 1963, 238, 888–892. [Google Scholar] [CrossRef] [PubMed]
- Aguado, B.A.; Campbell, R.D. Characterization of a human lysophosphatidic acid acyltransferase that is encoded by a gene located in the class III region of the human major histocompatibility complex. J. Biol. Chem. 1998, 273, 4096–4105. [Google Scholar] [CrossRef] [PubMed]
- McGarry, J.D.; Mannaerts, G.P.; Foster, D.W. A possible role for malonyl-CoA in the regulation of hepatic fatty acid oxidation and ketogenesis. J. Clin. Investig. 1977, 60, 265–270. [Google Scholar] [CrossRef] [PubMed]
- Frayn, K.N.; Arner, P.; Yki-Jarvinen, H. Fatty acid metabolism in adipose tissue, muscle and liver in health and disease. Essays Biochem. 2006, 42, 89–103. [Google Scholar] [CrossRef] [PubMed]
- Yen, C.E.; Nelson, D.W.; Yen, M.I. Intestinal triacylglycerol synthesis in fat absorption and systemic energy metabolism. J. Lipid Res. 2015, 56, 489–501. [Google Scholar] [CrossRef] [PubMed]
- Laurencikiene, J.; Skurk, T.; Kulyte, A.; Heden, P.; Astrom, G.; Sjolin, E.; Ryden, M.; Hauner, H.; Arner, P. Regulation of lipolysis in small and large fat cells of the same subject. J. Clin. Endocrinol. Metab. 2011, 96, E2045–E2049. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, K.L.; Smith, C.I.; Schwarzenberg, S.J.; Jessurun, J.; Boldt, M.D.; Parks, E.J. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J. Clin. Investig. 2005, 115, 1343–1351. [Google Scholar] [CrossRef]
- Di Ciaula, A.; Garruti, G.; Lunardi Baccetto, R.; Molina-Molina, E.; Bonfrate, L.; Wang, D.Q.; Portincasa, P. Bile Acid Physiology. Ann. Hepatol. 2017, 16, s4–s14. [Google Scholar] [CrossRef]
- Chavez-Talavera, O.; Tailleux, A.; Lefebvre, P.; Staels, B. Bile Acid Control of Metabolism and Inflammation in Obesity, Type 2 Diabetes, Dyslipidemia, and Nonalcoholic Fatty Liver Disease. Gastroenterology 2017, 152, 1679–1694.e3. [Google Scholar] [CrossRef]
- Beaven, S.W.; Matveyenko, A.; Wroblewski, K.; Chao, L.; Wilpitz, D.; Hsu, T.W.; Lentz, J.; Drew, B.; Hevener, A.L.; Tontonoz, P. Reciprocal regulation of hepatic and adipose lipogenesis by liver X receptors in obesity and insulin resistance. Cell Metab. 2013, 18, 106–117. [Google Scholar] [CrossRef]
- Cave, M.C.; Clair, H.B.; Hardesty, J.E.; Falkner, K.C.; Feng, W.; Clark, B.J.; Sidey, J.; Shi, H.; Aqel, B.A.; McClain, C.J.; et al. Nuclear receptors and nonalcoholic fatty liver disease. Biochim. Biophys. Acta 2016, 1859, 1083–1099. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Shu, X.-B.; Yao, Z.; Ji, G.; Zhang, L. Is vitamin D receptor a druggable target for non-alcoholic steatohepatitis? World J. Gastroenterol. 2020, 26, 5812. [Google Scholar] [CrossRef]
- Watanabe, A.; Sohail, M.A.; Gomes, D.A.; Hashmi, A.; Nagata, J.; Sutterwala, F.S.; Mahmood, S.; Jhandier, M.N.; Shi, Y.; Flavell, R.A.; et al. Inflammasome-mediated regulation of hepatic stellate cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 296, G1248–G1257. [Google Scholar] [CrossRef] [PubMed]
- Flannery, C.; Dufour, S.; Rabol, R.; Shulman, G.I.; Petersen, K.F. Skeletal muscle insulin resistance promotes increased hepatic de novo lipogenesis, hyperlipidemia, and hepatic steatosis in the elderly. Diabetes 2012, 61, 2711–2717. [Google Scholar] [CrossRef]
- Brown, M.S.; Goldstein, J.L. Selective versus total insulin resistance: A pathogenic paradox. Cell Metab. 2008, 7, 95–96. [Google Scholar] [CrossRef]
- Uyeda, K.; Repa, J.J. Carbohydrate response element binding protein, ChREBP, a transcription factor coupling hepatic glucose utilization and lipid synthesis. Cell Metab. 2006, 4, 107–110. [Google Scholar] [CrossRef] [PubMed]
- Bindesboll, C.; Fan, Q.; Norgaard, R.C.; MacPherson, L.; Ruan, H.B.; Wu, J.; Pedersen, T.A.; Steffensen, K.R.; Yang, X.; Matthews, J.; et al. Liver X receptor regulates hepatic nuclear O-GlcNAc signaling and carbohydrate responsive element-binding protein activity. J. Lipid Res. 2015, 56, 771–785. [Google Scholar] [CrossRef]
- Nagai, Y.; Yonemitsu, S.; Erion, D.M.; Iwasaki, T.; Stark, R.; Weismann, D.; Dong, J.; Zhang, D.; Jurczak, M.J.; Loffler, M.G.; et al. The role of peroxisome proliferator-activated receptor gamma coactivator-1 beta in the pathogenesis of fructose-induced insulin resistance. Cell Metab. 2009, 9, 252–264. [Google Scholar] [CrossRef]
- Loomba, R.; Friedman, S.L.; Shulman, G.I. Mechanisms and disease consequences of nonalcoholic fatty liver disease. Cell 2021, 184, 2537–2564. [Google Scholar] [CrossRef] [PubMed]
- Richart, C.; Auguet, T.; Terra, X. Apolipoprotein C3 gene variants in nonalcoholic fatty liver disease. N. Engl. J. Med. 2010, 363, 193–194; author reply 195. [Google Scholar] [CrossRef] [PubMed]
- Petersen, M.C.; Shulman, G.I. Mechanisms of Insulin Action and Insulin Resistance. Physiol. Rev. 2018, 98, 2133–2223. [Google Scholar] [CrossRef] [PubMed]
- Bazick, J.; Donithan, M.; Neuschwander-Tetri, B.A.; Kleiner, D.; Brunt, E.M.; Wilson, L.; Doo, E.; Lavine, J.; Tonascia, J.; Loomba, R. Clinical Model for NASH and Advanced Fibrosis in Adult Patients with Diabetes and NAFLD: Guidelines for Referral in NAFLD. Diabetes Care 2015, 38, 1347–1355. [Google Scholar] [CrossRef] [PubMed]
- Portillo-Sanchez, P.; Bril, F.; Maximos, M.; Lomonaco, R.; Biernacki, D.; Orsak, B.; Subbarayan, S.; Webb, A.; Hecht, J.; Cusi, K. High Prevalence of Nonalcoholic Fatty Liver Disease in Patients with Type 2 Diabetes Mellitus and Normal Plasma Aminotransferase Levels. J. Clin. Endocrinol. Metab. 2015, 100, 2231–2238. [Google Scholar] [CrossRef]
- Liu, J.; Jiang, S.; Zhao, Y.; Sun, Q.; Zhang, J.; Shen, D.; Wu, J.; Shen, N.; Fu, X.; Sun, X.; et al. Geranylgeranyl diphosphate synthase (GGPPS) regulates non-alcoholic fatty liver disease (NAFLD)-fibrosis progression by determining hepatic glucose/fatty acid preference under high-fat diet conditions. J. Pathol. 2018, 246, 277–288. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.S.; Xiao, C.; Wang, R.H.; Lahusen, T.; Xu, X.; Vassilopoulos, A.; Vazquez-Ortiz, G.; Jeong, W.I.; Park, O.; Ki, S.H.; et al. Hepatic-specific disruption of SIRT6 in mice results in fatty liver formation due to enhanced glycolysis and triglyceride synthesis. Cell Metab. 2010, 12, 224–236. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Chen, K.; Yao, W.; Zheng, R.; He, Q.; Xia, J.; Li, J.; Shao, Y.; Zhang, L.; Huang, L.; et al. Acetylation of lactate dehydrogenase B drives NAFLD progression by impairing lactate clearance. J. Hepatol. 2021, 74, 1038–1052. [Google Scholar] [CrossRef] [PubMed]
- Satapati, S.; Sunny, N.E.; Kucejova, B.; Fu, X.; He, T.T.; Mendez-Lucas, A.; Shelton, J.M.; Perales, J.C.; Browning, J.D.; Burgess, S.C. Elevated TCA cycle function in the pathology of diet-induced hepatic insulin resistance and fatty liver. J. Lipid Res. 2012, 53, 1080–1092. [Google Scholar] [CrossRef]
- Passarella, S.; Schurr, A.; Portincasa, P. Mitochondrial Transport in Glycolysis and Gluconeogenesis: Achievements and Perspectives. Int. J. Mol. Sci. 2021, 22, 12620. [Google Scholar] [CrossRef]
- Samuel, V.T.; Liu, Z.X.; Qu, X.; Elder, B.D.; Bilz, S.; Befroy, D.; Romanelli, A.J.; Shulman, G.I. Mechanism of hepatic insulin resistance in non-alcoholic fatty liver disease. J. Biol. Chem. 2004, 279, 32345–32353. [Google Scholar] [CrossRef] [PubMed]
- Sunny, N.E.; Parks, E.J.; Browning, J.D.; Burgess, S.C. Excessive hepatic mitochondrial TCA cycle and gluconeogenesis in humans with nonalcoholic fatty liver disease. Cell Metab. 2011, 14, 804–810. [Google Scholar] [CrossRef] [PubMed]
- Kamagate, A.; Qu, S.; Perdomo, G.; Su, D.; Kim, D.H.; Slusher, S.; Meseck, M.; Dong, H.H. FoxO1 mediates insulin-dependent regulation of hepatic VLDL production in mice. J. Clin. Investig. 2008, 118, 2347–2364. [Google Scholar] [CrossRef] [PubMed]
- Avramoglu, R.K.; Basciano, H.; Adeli, K. Lipid and lipoprotein dysregulation in insulin resistant states. Clin. Chim. Acta 2006, 368, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.; Tian, X.; Wu, H.; Huang, J.; Li, M.; Mei, Z.; Zhou, L.; Xie, H.; Zheng, S. Metabolic Changes of Hepatocytes in NAFLD. Front. Physiol. 2021, 12, 710420. [Google Scholar] [CrossRef]
- Passarella, S.; Atlante, A.; Valenti, D.; de Bari, L. The role of mitochondrial transport in energy metabolism. Mitochondrion 2003, 2, 319–343. [Google Scholar] [CrossRef] [PubMed]
- Febbraio, M.A.; Karin, M. “Sweet death”: Fructose as a metabolic toxin that targets the gut-liver axis. Cell Metab. 2021, 33, 2316–2328. [Google Scholar] [CrossRef]
- Tappy, L.; Le, K.A. Metabolic effects of fructose and the worldwide increase in obesity. Physiol. Rev. 2010, 90, 23–46. [Google Scholar] [CrossRef]
- Kurtz, T.W.; Kabra, P.M.; Booth, B.E.; Al-Bander, H.A.; Portale, A.A.; Serena, B.G.; Tsai, H.C.; Morris, R.C., Jr. Liquid-chromatographic measurements of inosine, hypoxanthine, and xanthine in studies of fructose-induced degradation of adenine nucleotides in humans and rats. Clin. Chem. 1986, 32, 782–786. [Google Scholar] [CrossRef]
- Van den Berghe, G.; Bronfman, M.; Vanneste, R.; Hers, H.G. The mechanism of adenosine triphosphate depletion in the liver after a load of fructose. A kinetic study of liver adenylate deaminase. Biochem. J. 1977, 162, 601–609. [Google Scholar] [CrossRef]
- Smith, C.M.; Rovamo, L.M.; Raivio, K.O. Fructose-induced adenine nucleotide catabolism in isolated rat hepatocytes. Can. J. Biochem. 1977, 55, 1237–1240. [Google Scholar] [CrossRef] [PubMed]
- Maenpaa, P.H.; Raivio, K.O.; Kekomaki, M.P. Liver adenine nucleotides: Fructose-induced depletion and its effect on protein synthesis. Science 1968, 161, 1253–1254. [Google Scholar] [CrossRef] [PubMed]
- Bawden, S.J.; Stephenson, M.C.; Ciampi, E.; Hunter, K.; Marciani, L.; Macdonald, I.A.; Aithal, G.P.; Morris, P.G.; Gowland, P.A. Investigating the effects of an oral fructose challenge on hepatic ATP reserves in healthy volunteers: A (31)P MRS study. Clin. Nutr. 2016, 35, 645–649. [Google Scholar] [CrossRef] [PubMed]
- Lanaspa, M.A.; Sanchez-Lozada, L.G.; Choi, Y.J.; Cicerchi, C.; Kanbay, M.; Roncal-Jimenez, C.A.; Ishimoto, T.; Li, N.; Marek, G.; Duranay, M.; et al. Uric acid induces hepatic steatosis by generation of mitochondrial oxidative stress: Potential role in fructose-dependent and -independent fatty liver. J. Biol. Chem. 2012, 287, 40732–40744. [Google Scholar] [CrossRef]
- Softic, S.; Cohen, D.E.; Kahn, C.R. Role of Dietary Fructose and Hepatic De Novo Lipogenesis in Fatty Liver Disease. Dig. Dis. Sci. 2016, 61, 1282–1293. [Google Scholar] [CrossRef]
- Ritze, Y.; Bardos, G.; Claus, A.; Ehrmann, V.; Bergheim, I.; Schwiertz, A.; Bischoff, S.C. Lactobacillus rhamnosus GG protects against non-alcoholic fatty liver disease in mice. PLoS ONE 2014, 9, e80169. [Google Scholar] [CrossRef] [PubMed]
- Jegatheesan, P.; Beutheu, S.; Ventura, G.; Sarfati, G.; Nubret, E.; Kapel, N.; Waligora-Dupriet, A.J.; Bergheim, I.; Cynober, L.; De-Bandt, J.P. Effect of specific amino acids on hepatic lipid metabolism in fructose-induced non-alcoholic fatty liver disease. Clin. Nutr. 2016, 35, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Jegatheesan, P.; Beutheu, S.; Freese, K.; Waligora-Dupriet, A.J.; Nubret, E.; Butel, M.J.; Bergheim, I.; De Bandt, J.P. Preventive effects of citrulline on Western diet-induced non-alcoholic fatty liver disease in rats. Brit. J. Nutr. 2016, 116, 191–203. [Google Scholar] [CrossRef]
- Massafra, V.; Pellicciari, R.; Gioiello, A.; van Mil, S.W.C. Progress and challenges of selective Farnesoid X receptor modulation. Pharmacol. Ther. 2018, 191, 162–177. [Google Scholar] [CrossRef]
- Jung, Y.; Koo, B.K.; Jang, S.Y.; Kim, D.; Lee, H.; Lee, D.H.; Joo, S.K.; Jung, Y.J.; Park, J.H.; Yoo, T.; et al. Association between circulating bile acid alterations and nonalcoholic steatohepatitis independent of obesity and diabetes mellitus. Liver Int. 2021, 41, 2892–2902. [Google Scholar] [CrossRef]
- Rao, A.; Kosters, A.; Mells, J.E.; Zhang, W.; Setchell, K.D.; Amanso, A.M.; Wynn, G.M.; Xu, T.; Keller, B.T.; Yin, H.; et al. Inhibition of ileal bile acid uptake protects against nonalcoholic fatty liver disease in high-fat diet-fed mice. Sci. Transl. Med. 2016, 8, 357ra122. [Google Scholar] [CrossRef] [PubMed]
- Pineda Torra, I.; Claudel, T.; Duval, C.; Kosykh, V.; Fruchart, J.C.; Staels, B. Bile acids induce the expression of the human peroxisome proliferator-activated receptor alpha gene via activation of the farnesoid X receptor. Mol. Endocrinol. 2003, 17, 259–272. [Google Scholar] [CrossRef]
- Watanabe, M.; Houten, S.M.; Wang, L.; Moschetta, A.; Mangelsdorf, D.J.; Heyman, R.A.; Moore, D.D.; Auwerx, J. Bile acids lower triglyceride levels via a pathway involving FXR, SHP, and SREBP-1c. J. Clin. Investig. 2004, 113, 1408–1418. [Google Scholar] [CrossRef] [PubMed]
- Savkur, R.S.; Bramlett, K.S.; Michael, L.F.; Burris, T.P. Regulation of pyruvate dehydrogenase kinase expression by the farnesoid X receptor. Biochem. Biophys. Res. Commun. 2005, 329, 391–396. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wong, K.; Walsh, K.; Gao, B.; Zang, M. Retinoic acid receptor beta stimulates hepatic induction of fibroblast growth factor 21 to promote fatty acid oxidation and control whole-body energy homeostasis in mice. J. Biol. Chem. 2013, 288, 10490–10504. [Google Scholar] [CrossRef]
- Badman, M.K.; Pissios, P.; Kennedy, A.R.; Koukos, G.; Flier, J.S.; Maratos-Flier, E. Hepatic fibroblast growth factor 21 is regulated by PPARalpha and is a key mediator of hepatic lipid metabolism in ketotic states. Cell Metab. 2007, 5, 426–437. [Google Scholar] [CrossRef] [PubMed]
- Kir, S.; Beddow, S.A.; Samuel, V.T.; Miller, P.; Previs, S.F.; Suino-Powell, K.; Xu, H.E.; Shulman, G.I.; Kliewer, S.A.; Mangelsdorf, D.J. FGF19 as a postprandial, insulin-independent activator of hepatic protein and glycogen synthesis. Science 2011, 331, 1621–1624. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; John, L.M.; Adams, S.H.; Yu, X.X.; Tomlinson, E.; Renz, M.; Williams, P.M.; Soriano, R.; Corpuz, R.; Moffat, B.; et al. Fibroblast growth factor 19 increases metabolic rate and reverses dietary and leptin-deficient diabetes. Endocrinology 2004, 145, 2594–2603. [Google Scholar] [CrossRef]
- Alvarez-Sola, G.; Uriarte, I.; Latasa, M.U.; Fernandez-Barrena, M.G.; Urtasun, R.; Elizalde, M.; Barcena-Varela, M.; Jimenez, M.; Chang, H.C.; Barbero, R.; et al. Fibroblast growth factor 15/19 (FGF15/19) protects from diet-induced hepatic steatosis: Development of an FGF19-based chimeric molecule to promote fatty liver regeneration. Gut 2017, 66, 1818–1828. [Google Scholar] [CrossRef]
- Tomlinson, E.; Fu, L.; John, L.; Hultgren, B.; Huang, X.; Renz, M.; Stephan, J.P.; Tsai, S.P.; Powell-Braxton, L.; French, D.; et al. Transgenic mice expressing human fibroblast growth factor-19 display increased metabolic rate and decreased adiposity. Endocrinology 2002, 143, 1741–1747. [Google Scholar] [CrossRef]
- Watanabe, M.; Horai, Y.; Houten, S.M.; Morimoto, K.; Sugizaki, T.; Arita, E.; Mataki, C.; Sato, H.; Tanigawara, Y.; Schoonjans, K.; et al. Lowering bile acid pool size with a synthetic farnesoid X receptor (FXR) agonist induces obesity and diabetes through reduced energy expenditure. J. Biol. Chem. 2011, 286, 26913–26920. [Google Scholar] [CrossRef]
- Zhang, Y.; Ge, X.; Heemstra, L.A.; Chen, W.D.; Xu, J.; Smith, J.L.; Ma, H.; Kasim, N.; Edwards, P.A.; Novak, C.M. Loss of FXR protects against diet-induced obesity and accelerates liver carcinogenesis in ob/ob mice. Mol. Endocrinol. 2012, 26, 272–280. [Google Scholar] [CrossRef]
- Prawitt, J.; Abdelkarim, M.; Stroeve, J.H.; Popescu, I.; Duez, H.; Velagapudi, V.R.; Dumont, J.; Bouchaert, E.; van Dijk, T.H.; Lucas, A.; et al. Farnesoid X receptor deficiency improves glucose homeostasis in mouse models of obesity. Diabetes 2011, 60, 1861–1871. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.J.; Kim, S.C.; Kim, J.; Anakk, S.; Lee, J.M.; Tseng, H.T.; Yechoor, V.; Park, J.; Choi, J.S.; Jang, H.C.; et al. Dissociation of diabetes and obesity in mice lacking orphan nuclear receptor small heterodimer partner. J. Lipid Res. 2011, 52, 2234–2244. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Jiang, C.; Shi, J.; Gao, X.; Sun, D.; Sun, L.; Wang, T.; Takahashi, S.; Anitha, M.; Krausz, K.W.; et al. An Intestinal Farnesoid X Receptor-Ceramide Signaling Axis Modulates Hepatic Gluconeogenesis in Mice. Diabetes 2017, 66, 613–626. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Xie, C.; Li, F.; Zhang, L.; Nichols, R.G.; Krausz, K.W.; Cai, J.; Qi, Y.; Fang, Z.Z.; Takahashi, S.; et al. Intestinal farnesoid X receptor signaling promotes nonalcoholic fatty liver disease. J. Clin. Investig. 2015, 125, 386–402. [Google Scholar] [CrossRef]
- Sayin, S.I.; Wahlstrom, A.; Felin, J.; Jantti, S.; Marschall, H.U.; Bamberg, K.; Angelin, B.; Hyotylainen, T.; Oresic, M.; Backhed, F. Gut microbiota regulates bile acid metabolism by reducing the levels of tauro-beta-muricholic acid, a naturally occurring FXR antagonist. Cell Metab. 2013, 17, 225–235. [Google Scholar] [CrossRef]
- Trabelsi, M.S.; Daoudi, M.; Prawitt, J.; Ducastel, S.; Touche, V.; Sayin, S.I.; Perino, A.; Brighton, C.A.; Sebti, Y.; Kluza, J.; et al. Farnesoid X receptor inhibits glucagon-like peptide-1 production by enteroendocrine L cells. Nat. Commun. 2015, 6, 7629. [Google Scholar] [CrossRef]
- Kawamata, Y.; Fujii, R.; Hosoya, M.; Harada, M.; Yoshida, H.; Miwa, M.; Fukusumi, S.; Habata, Y.; Itoh, T.; Shintani, Y.; et al. A G protein-coupled receptor responsive to bile acids. J. Biol. Chem. 2003, 278, 9435–9440. [Google Scholar] [CrossRef]
- Van Nierop, F.S.; Scheltema, M.J.; Eggink, H.M.; Pols, T.W.; Sonne, D.P.; Knop, F.K.; Soeters, M.R. Clinical relevance of the bile acid receptor TGR5 in metabolism. Lancet Diabetes Endocrinol. 2017, 5, 224–233. [Google Scholar] [CrossRef]
- Yuan, L.; Bambha, K. Bile acid receptors and nonalcoholic fatty liver disease. World J. Hepatol. 2015, 7, 2811–2818. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Li, M.; Han, B.; Qi, X. Association of non-alcoholic fatty liver disease with thyroid function: A systematic review and meta-analysis. Dig. Liver Dis. 2018, 50, 1153–1162. [Google Scholar] [CrossRef] [PubMed]
- Forrest, D.; Vennstrom, B. Functions of thyroid hormone receptors in mice. Thyroid. 2000, 10, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Lammel Lindemann, J.A.; Angajala, A.; Engler, D.A.; Webb, P.; Ayers, S.D. Thyroid hormone induction of human cholesterol 7 alpha-hydroxylase (Cyp7a1) in vitro. Mol. Cell Endocrinol. 2014, 388, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Ahn, H.Y.; Kim, H.H.; Kim, Y.A.; Kim, M.; Ohn, J.H.; Chung, S.S.; Lee, Y.K.; Park, D.J.; Park, K.S.; Moore, D.D.; et al. Thyroid Hormone Regulates the mRNA Expression of Small Heterodimer Partner through Liver Receptor Homolog-1. Endocrinol. Metab. 2015, 30, 584–592. [Google Scholar] [CrossRef]
- Sinha, R.A.; Bruinstroop, E.; Singh, B.K.; Yen, P.M. Nonalcoholic Fatty Liver Disease and Hypercholesterolemia: Roles of Thyroid Hormones, Metabolites, and Agonists. Thyroid 2019, 29, 1173–1191. [Google Scholar] [CrossRef]
- Shneider, B.L.; Dawson, P.A.; Christie, D.M.; Hardikar, W.; Wong, M.H.; Suchy, F.J. Cloning and molecular characterization of the ontogeny of a rat ileal sodium-dependent bile acid transporter. J. Clin. Investig. 1995, 95, 745–754. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Chiang, J.Y. Transcriptional regulation of the human sterol 12alpha-hydroxylase gene (CYP8B1): Roles of heaptocyte nuclear factor 4alpha in mediating bile acid repression. J. Biol. Chem. 2001, 276, 41690–41699. [Google Scholar] [CrossRef]
- Goodwin, B.; Jones, S.A.; Price, R.R.; Watson, M.A.; McKee, D.D.; Moore, L.B.; Galardi, C.; Wilson, J.G.; Lewis, M.C.; Roth, M.E.; et al. A regulatory cascade of the nuclear receptors FXR, SHP-1, and LRH-1 represses bile acid biosynthesis. Mol. Cell 2000, 6, 517–526. [Google Scholar] [CrossRef]
- Soroka, C.J.; Ballatori, N.; Boyer, J.L. Organic solute transporter, OSTα-OSTβ: Its role in bile acid transport and cholestasis. In Seminars in Liver Disease; Thieme Medical Publishers: New York, NY, USA, 2010; pp. 178–185. [Google Scholar]
- Song, K.H.; Li, T.; Owsley, E.; Strom, S.; Chiang, J.Y. Bile acids activate fibroblast growth factor 19 signaling in human hepatocytes to inhibit cholesterol 7alpha-hydroxylase gene expression. Hepatology 2009, 49, 297–305. [Google Scholar] [CrossRef]
- Zhou, J.; Martin, R.J.; Tulley, R.T.; Raggio, A.M.; McCutcheon, K.L.; Shen, L.; Danna, S.C.; Tripathy, S.; Hegsted, M.; Keenan, M.J. Dietary resistant starch upregulates total GLP-1 and PYY in a sustained day-long manner through fermentation in rodents. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E1160–E1166. [Google Scholar] [CrossRef] [PubMed]
- Leung, C.; Rivera, L.; Furness, J.B.; Angus, P.W. The role of the gut microbiota in NAFLD. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 412–425. [Google Scholar] [CrossRef] [PubMed]
- Di Ciaula, A.; Bonfrate, L.; Khalil, M.; Garruti, G.; Portincasa, P. Contribution of the microbiome for better phenotyping of people living with obesity. Rev. Endocr. Metab. Disord. 2023, 24, 839–870. [Google Scholar] [CrossRef] [PubMed]
- Portincasa, P.; Bonfrate, L.; Vacca, M.; De Angelis, M.; Farella, I.; Lanza, E.; Khalil, M.; Wang, D.Q.; Sperandio, M.; Di Ciaula, A. Gut Microbiota and Short Chain Fatty Acids: Implications in Glucose Homeostasis. Int. J. Mol. Sci. 2022, 23, 1105. [Google Scholar] [CrossRef] [PubMed]
- Sharpton, S.R.; Schnabl, B.; Knight, R.; Loomba, R. Current Concepts, Opportunities, and Challenges of Gut Microbiome-Based Personalized Medicine in Nonalcoholic Fatty Liver Disease. Cell Metab. 2021, 33, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S. Inflammation and metabolic disorders. Nature 2006, 444, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Cani, P.D.; Amar, J.; Iglesias, M.A.; Poggi, M.; Knauf, C.; Bastelica, D.; Neyrinck, A.M.; Fava, F.; Tuohy, K.M.; Chabo, C.; et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 2007, 56, 1761–1772. [Google Scholar] [CrossRef]
- Cani, P.D.; Bibiloni, R.; Knauf, C.; Waget, A.; Neyrinck, A.M.; Delzenne, N.M.; Burcelin, R. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes 2008, 57, 1470–1481. [Google Scholar] [CrossRef] [PubMed]
- Fei, N.; Bruneau, A.; Zhang, X.; Wang, R.; Wang, J.; Rabot, S.; Gerard, P.; Zhao, L. Endotoxin Producers Overgrowing in Human Gut Microbiota as the Causative Agents for Nonalcoholic Fatty Liver Disease. mBio 2020, 11, 10-1128. [Google Scholar] [CrossRef]
- Loomba, R.; Seguritan, V.; Li, W.; Long, T.; Klitgord, N.; Bhatt, A.; Dulai, P.S.; Caussy, C.; Bettencourt, R.; Highlander, S.K.; et al. Gut Microbiome-Based Metagenomic Signature for Non-invasive Detection of Advanced Fibrosis in Human Nonalcoholic Fatty Liver Disease. Cell Metab. 2017, 25, 1054–1062.e5. [Google Scholar] [CrossRef]
- Lee, G.; You, H.J.; Bajaj, J.S.; Joo, S.K.; Yu, J.; Park, S.; Kang, H.; Park, J.H.; Kim, J.H.; Lee, D.H.; et al. Distinct signatures of gut microbiome and metabolites associated with significant fibrosis in non-obese NAFLD. Nat. Commun. 2020, 11, 4982. [Google Scholar] [CrossRef] [PubMed]
- Frost, F.; Kacprowski, T.; Ruhlemann, M.; Pietzner, M.; Bang, C.; Franke, A.; Nauck, M.; Volker, U.; Volzke, H.; Dorr, M.; et al. Long-term instability of the intestinal microbiome is associated with metabolic liver disease, low microbiota diversity, diabetes mellitus and impaired exocrine pancreatic function. Gut 2021, 70, 522–530. [Google Scholar] [CrossRef]
- Cornejo-Pareja, I.; Amiar, M.R.; Ocana-Wilhelmi, L.; Soler-Humanes, R.; Arranz-Salas, I.; Garrido-Sanchez, L.; Gutierrez-Repiso, C.; Tinahones, F.J. Non-alcoholic fatty liver disease in patients with morbid obesity: The gut microbiota axis as a potential pathophysiology mechanism. J. Gastroenterol. 2024, 59, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Blaak, E.E.; Canfora, E.E.; Theis, S.; Frost, G.; Groen, A.K.; Mithieux, G.; Nauta, A.; Scott, K.; Stahl, B.; van Harsselaar, J.; et al. Short chain fatty acids in human gut and metabolic health. Benef. Microbes 2020, 11, 411–455. [Google Scholar] [CrossRef]
- Den Besten, G.; Bleeker, A.; Gerding, A.; van Eunen, K.; Havinga, R.; van Dijk, T.H.; Oosterveer, M.H.; Jonker, J.W.; Groen, A.K.; Reijngoud, D.J.; et al. Short-Chain Fatty Acids Protect against High-Fat Diet-Induced Obesity via a PPARgamma-Dependent Switch from Lipogenesis to Fat Oxidation. Diabetes 2015, 64, 2398–2408. [Google Scholar] [CrossRef]
- Litvak, Y.; Byndloss, M.X.; Tsolis, R.M.; Baumler, A.J. Dysbiotic Proteobacteria expansion: A microbial signature of epithelial dysfunction. Curr. Opin. Microbiol. 2017, 39, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Litvak, Y.; Mon, K.K.Z.; Nguyen, H.; Chanthavixay, G.; Liou, M.; Velazquez, E.M.; Kutter, L.; Alcantara, M.A.; Byndloss, M.X.; Tiffany, C.R.; et al. Commensal Enterobacteriaceae Protect against Salmonella Colonization through Oxygen Competition. Cell Host Microbe 2019, 25, 128–139.e5. [Google Scholar] [CrossRef]
- De Vos, W.M.; Tilg, H.; Van Hul, M.; Cani, P.D. Gut microbiome and health: Mechanistic insights. Gut 2022, 71, 1020–1032. [Google Scholar] [CrossRef]
- Keenan, M.J.; Zhou, J.; McCutcheon, K.L.; Raggio, A.M.; Bateman, H.G.; Todd, E.; Jones, C.K.; Tulley, R.T.; Melton, S.; Martin, R.J.; et al. Effects of resistant starch, a non-digestible fermentable fiber, on reducing body fat. Obesity 2006, 14, 1523–1534. [Google Scholar] [CrossRef]
- Aziz, A.A.; Kenney, L.S.; Goulet, B.; Abdel-Aal, E.S. Dietary starch type affects body weight and glycemic control in freely fed but not energy-restricted obese rats. J. Nutr. 2009, 139, 1881–1889. [Google Scholar] [CrossRef]
- Cho, I.; Yamanishi, S.; Cox, L.; Methe, B.A.; Zavadil, J.; Li, K.; Gao, Z.; Mahana, D.; Raju, K.; Teitler, I.; et al. Antibiotics in early life alter the murine colonic microbiome and adiposity. Nature 2012, 488, 621–626. [Google Scholar] [CrossRef]
- Gradisteanu Pircalabioru, G.; Ilie, I.; Oprea, L.; Picu, A.; Petcu, L.M.; Burlibasa, L.; Chifiriuc, M.C.; Musat, M. Microbiome, Mycobiome and Related Metabolites Alterations in Patients with Metabolic Syndrome-A Pilot Study. Metabolites 2022, 12, 218. [Google Scholar] [CrossRef] [PubMed]
- Le Roy, T.; Moens de Hase, E.; Van Hul, M.; Paquot, A.; Pelicaen, R.; Regnier, M.; Depommier, C.; Druart, C.; Everard, A.; Maiter, D.; et al. Dysosmobacter welbionis is a newly isolated human commensal bacterium preventing diet-induced obesity and metabolic disorders in mice. Gut 2022, 71, 534–543. [Google Scholar] [CrossRef]
- Jia, W.; Xie, G.; Jia, W. Bile acid-microbiota crosstalk in gastrointestinal inflammation and carcinogenesis. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 111–128. [Google Scholar] [CrossRef]
- Chitturi, S.; Wong, V.W.; Chan, W.K.; Wong, G.L.; Wong, S.K.; Sollano, J.; Ni, Y.H.; Liu, C.J.; Lin, Y.C.; Lesmana, L.A.; et al. The Asia-Pacific Working Party on Non-alcoholic Fatty Liver Disease guidelines 2017-Part 2: Management and special groups. J. Gastroenterol. Hepatol. 2018, 33, 86–98. [Google Scholar] [CrossRef] [PubMed]
- Linge, J.; Nasr, P.; Sanyal, A.J.; Dahlqvist Leinhard, O.; Ekstedt, M. Adverse muscle composition is a significant risk factor for all-cause mortality in NAFLD. JHEP Rep. 2023, 5, 100663. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.K.; Bae, S.J.; Lee, M.J.; Kim, E.H.; Park, H.; Kim, H.S.; Cho, Y.K.; Jung, C.H.; Lee, W.J.; Choe, J. Association of Visceral Fat Obesity, Sarcopenia, and Myosteatosis with Non-Alcoholic Fatty Liver Disease without Obesity. Clin. Mol. Hepatol. 2023, 29, 987–1001. [Google Scholar] [CrossRef]
- Molina-Molina, E.; Lunardi Baccetto, R.; Wang, D.Q.; de Bari, O.; Krawczyk, M.; Portincasa, P. Exercising the hepatobiliary-gut axis. The impact of physical activity performance. Eur. J. Clin. Investig. 2018, 48, e12958. [Google Scholar] [CrossRef]
- Van Gaal, L.F.; Mertens, J.; Francque, S.; De Block, C. Therapeutic approaches for non-alcoholic steatohepatitis. Ther. Adv. Endocrinol. Metab. 2021, 12, 20420188211034300. [Google Scholar] [CrossRef]
- Vilar-Gomez, E.; Martinez-Perez, Y.; Calzadilla-Bertot, L.; Torres-Gonzalez, A.; Gra-Oramas, B.; Gonzalez-Fabian, L.; Friedman, S.L.; Diago, M.; Romero-Gomez, M. Weight Loss through Lifestyle Modification Significantly Reduces Features of Nonalcoholic Steatohepatitis. Gastroenterology 2015, 149, 367–378.e5. [Google Scholar] [CrossRef]
- Markova, M.; Pivovarova, O.; Hornemann, S.; Sucher, S.; Frahnow, T.; Wegner, K.; Machann, J.; Petzke, K.J.; Hierholzer, J.; Lichtinghagen, R.; et al. Isocaloric Diets High in Animal or Plant Protein Reduce Liver Fat and Inflammation in Individuals with Type 2 Diabetes. Gastroenterology 2017, 152, 571–585.e8. [Google Scholar] [CrossRef]
- Haigh, L.; Kirk, C.; El Gendy, K.; Gallacher, J.; Errington, L.; Mathers, J.C.; Anstee, Q.M. The effectiveness and acceptability of Mediterranean diet and calorie restriction in non-alcoholic fatty liver disease (NAFLD): A systematic review and meta-analysis. Clin. Nutr. 2022, 41, 1913–1931. [Google Scholar] [CrossRef] [PubMed]
- Saleh, S.A.K.; Santos, H.O.; Gaman, M.A.; Cerqueira, H.S.; Zaher, E.A.; Alromaih, W.R.; Arafat, N.S.; Adi, A.R.; Adly, H.M.; Alyoubi, R.; et al. Effects of intermittent fasting regimens on glycemic, hepatic, anthropometric, and clinical markers in patients with non-alcoholic fatty liver disease: Systematic review and meta-analysis of randomized controlled trials. Clin. Nutr. ESPEN 2024, 59, 70–80. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.; So, R.; Shida, T.; Matsuo, T.; Kim, B.; Akiyama, K.; Isobe, T.; Okamoto, Y.; Tanaka, K.; Shoda, J. High-Intensity Aerobic Exercise Improves both Hepatic Fat Content and Stiffness in Sedentary Obese Men with Nonalcoholic Fatty Liver Disease. Sci. Rep. 2017, 7, 43029. [Google Scholar] [CrossRef]
- Orci, L.A.; Gariani, K.; Oldani, G.; Delaune, V.; Morel, P.; Toso, C. Exercise-based Interventions for Nonalcoholic Fatty Liver Disease: A Meta-analysis and Meta-regression. Clin. Gastroenterol. Hepatol. 2016, 14, 1398–1411. [Google Scholar] [CrossRef]
- Sung, K.C.; Ryu, S.; Lee, J.Y.; Kim, J.Y.; Wild, S.H.; Byrne, C.D. Effect of exercise on the development of new fatty liver and the resolution of existing fatty liver. J. Hepatol. 2016, 65, 791–797. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.; Shida, T.; Yamagishi, K.; Tanaka, K.; So, R.; Tsujimoto, T.; Shoda, J. Moderate to vigorous physical activity volume is an important factor for managing nonalcoholic fatty liver disease: A retrospective study. Hepatology 2015, 61, 1205–1215. [Google Scholar] [CrossRef]
- Hashida, R.; Kawaguchi, T.; Bekki, M.; Omoto, M.; Matsuse, H.; Nago, T.; Takano, Y.; Ueno, T.; Koga, H.; George, J.; et al. Aerobic vs. resistance exercise in non-alcoholic fatty liver disease: A systematic review. J. Hepatol. 2017, 66, 142–152. [Google Scholar] [CrossRef] [PubMed]
- Kistler, K.D.; Brunt, E.M.; Clark, J.M.; Diehl, A.M.; Sallis, J.F.; Schwimmer, J.B.; Group, N.C.R. Physical activity recommendations, exercise intensity, and histological severity of nonalcoholic fatty liver disease. Am. J. Gastroenterol. 2011, 106, 460–468; quiz 469. [Google Scholar] [CrossRef]
- Shanmugam, H.; Di Ciaula, A.; Di Palo, D.M.; Molina-Molina, E.; Garruti, G.; Faienza, M.F.; vanErpecum, K.; Portincasa, P. Multiplying effects of COVID-19 lockdown on metabolic risk and fatty liver. Eur. J. Clin. Investig. 2021, 51, e13597. [Google Scholar] [CrossRef]
- Chen, G.; Banini, B.A.; Do, A.; Gunderson, C.; Zaman, S.; Lim, J.K. Exercise Does Not Independently Improve Histological Outcomes in Biopsy-Proven Non-Alcoholic Fatty Liver Disease: A Systematic Review and Meta-Analysis. Genes 2023, 14, 1811. [Google Scholar] [CrossRef] [PubMed]
- Van Dijk, A.M.; de Vries, M.; El-Morabit, F.; Bac, S.T.; Mundt, M.W.; van der Schuit, L.E.; Hirdes, M.M.C.; Kara, M.; de Bruijne, J.; van Meer, S.; et al. Intra-gastric balloon with lifestyle modification: A promising therapeutic option for overweight and obese patients with metabolic dysfunction-associated steatotic liver disease. Intern. Emerg. Med. 2023, 18, 2271–2280. [Google Scholar] [CrossRef]
- Chavez-Tapia, N.C.; Tellez-Avila, F.I.; Barrientos-Gutierrez, T.; Mendez-Sanchez, N.; Lizardi-Cervera, J.; Uribe, M. Bariatric surgery for non-alcoholic steatohepatitis in obese patients. Cochrane Database Syst. Rev. 2010, 2010, CD007340. [Google Scholar] [CrossRef]
- Lee, Y.; Doumouras, A.G.; Yu, J.; Brar, K.; Banfield, L.; Gmora, S.; Anvari, M.; Hong, D. Complete Resolution of Nonalcoholic Fatty Liver Disease after Bariatric Surgery: A Systematic Review and Meta-analysis. Clin. Gastroenterol. Hepatol. 2019, 17, 1040–1060.e11. [Google Scholar] [CrossRef]
- Klein, S.; Mittendorfer, B.; Eagon, J.C.; Patterson, B.; Grant, L.; Feirt, N.; Seki, E.; Brenner, D.; Korenblat, K.; McCrea, J. Gastric bypass surgery improves metabolic and hepatic abnormalities associated with nonalcoholic fatty liver disease. Gastroenterology 2006, 130, 1564–1572. [Google Scholar] [CrossRef]
- Aguilar-Olivos, N.E.; Almeda-Valdes, P.; Aguilar-Salinas, C.A.; Uribe, M.; Mendez-Sanchez, N. The role of bariatric surgery in the management of nonalcoholic fatty liver disease and metabolic syndrome. Metabolism 2016, 65, 1196–1207. [Google Scholar] [CrossRef]
- Verrastro, O.; Panunzi, S.; Castagneto-Gissey, L.; De Gaetano, A.; Lembo, E.; Capristo, E.; Guidone, C.; Angelini, G.; Pennestri, F.; Sessa, L.; et al. Bariatric-metabolic surgery versus lifestyle intervention plus best medical care in non-alcoholic steatohepatitis (BRAVES): A multicentre, open-label, randomised trial. Lancet 2023, 401, 1786–1797. [Google Scholar] [CrossRef] [PubMed]
- Aoko, O.; Maharaj, T.; Boland, F.; Cheriyan, D.; Ryan, J. Meta-analysis: Impact of intragastric balloon therapy on NAFLD-related parameters in patients with obesity. Aliment. Pharmacol. Ther. 2024, 59, 8–22. [Google Scholar] [CrossRef] [PubMed]
- Di Ciaula, A.; Portincasa, P. Contrasting obesity: Is something missing here? Intern. Emerg. Med. 2024, 19, 265–269. [Google Scholar] [CrossRef]
- Sangro, P.; de la Torre Alaez, M.; Sangro, B.; D’Avola, D. Metabolic dysfunction-associated fatty liver disease (MAFLD): An update of the recent advances in pharmacological treatment. J. Physiol. Biochem. 2023, 79, 869–879. [Google Scholar] [CrossRef]
- Wong, R.J.; Aguilar, M.; Cheung, R.; Perumpail, R.B.; Harrison, S.A.; Younossi, Z.M.; Ahmed, A. Nonalcoholic Steatohepatitis Is the Second Leading Etiology of Liver Disease Among Adults Awaiting Liver Transplantation in the United States. Gastroenterology 2015, 148, 547–555. [Google Scholar] [CrossRef]
- Dufour, J.F.; Caussy, C.; Loomba, R. Combination therapy for non-alcoholic steatohepatitis: Rationale, opportunities and challenges. Gut 2020, 69, 1877–1884. [Google Scholar] [CrossRef] [PubMed]
- Newsome, P.N.; Buchholtz, K.; Cusi, K.; Linder, M.; Okanoue, T.; Ratziu, V.; Sanyal, A.J.; Sejling, A.S.; Harrison, S.A.; Investigators, N.N. A Placebo-Controlled Trial of Subcutaneous Semaglutide in Nonalcoholic Steatohepatitis. N. Engl. J. Med. 2021, 384, 1113–1124. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, M.J.; Gaunt, P.; Aithal, G.P.; Barton, D.; Hull, D.; Parker, R.; Hazlehurst, J.M.; Guo, K.; Abouda, G.; Aldersley, M.A.; et al. Liraglutide safety and efficacy in patients with non-alcoholic steatohepatitis (LEAN): A multicentre, double-blind, randomised, placebo-controlled phase 2 study. Lancet 2016, 387, 679–690. [Google Scholar] [CrossRef] [PubMed]
- Francque, S.M.; Bedossa, P.; Ratziu, V.; Anstee, Q.M.; Bugianesi, E.; Sanyal, A.J.; Loomba, R.; Harrison, S.A.; Balabanska, R.; Mateva, L.; et al. A Randomized, Controlled Trial of the Pan-PPAR Agonist Lanifibranor in NASH. N. Engl. J. Med. 2021, 385, 1547–1558. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, A.; Eguchi, Y.; Yoneda, M.; Imajo, K.; Tamaki, N.; Suganami, H.; Nojima, T.; Tanigawa, R.; Iizuka, M.; Iida, Y.; et al. Randomised clinical trial: Pemafibrate, a novel selective peroxisome proliferator-activated receptor alpha modulator (SPPARMalpha), versus placebo in patients with non-alcoholic fatty liver disease. Aliment. Pharmacol. Ther. 2021, 54, 1263–1277. [Google Scholar] [CrossRef] [PubMed]
- Ratziu, V.; de Guevara, L.; Safadi, R.; Poordad, F.; Fuster, F.; Flores-Figueroa, J.; Arrese, M.; Fracanzani, A.L.; Ben Bashat, D.; Lackner, K.; et al. Aramchol in patients with nonalcoholic steatohepatitis: A randomized, double-blind, placebo-controlled phase 2b trial. Nat. Med. 2021, 27, 1825–1835. [Google Scholar] [CrossRef]
- Loomba, R.; Sanyal, A.J.; Kowdley, K.V.; Bhatt, D.L.; Alkhouri, N.; Frias, J.P.; Bedossa, P.; Harrison, S.A.; Lazas, D.; Barish, R.; et al. Randomized, Controlled Trial of the FGF21 Analogue Pegozafermin in NASH. N. Engl. J. Med. 2023, 389, 998–1008. [Google Scholar] [CrossRef]
- Harrison, S.A.; Ruane, P.J.; Freilich, B.L.; Neff, G.; Patil, R.; Behling, C.A.; Hu, C.; Fong, E.; de Temple, B.; Tillman, E.J.; et al. Efruxifermin in non-alcoholic steatohepatitis: A randomized, double-blind, placebo-controlled, phase 2a trial. Nat. Med. 2021, 27, 1262–1271. [Google Scholar] [CrossRef]
- Patel, K.; Harrison, S.A.; Elkhashab, M.; Trotter, J.F.; Herring, R.; Rojter, S.E.; Kayali, Z.; Wong, V.W.; Greenbloom, S.; Jayakumar, S.; et al. Cilofexor, a Nonsteroidal FXR Agonist, in Patients With Noncirrhotic NASH: A Phase 2 Randomized Controlled Trial. Hepatology 2020, 72, 58–71. [Google Scholar] [CrossRef]
- Chalasani, N.P.; Sanyal, A.J.; Kowdley, K.V.; Robuck, P.R.; Hoofnagle, J.; Kleiner, D.E.; Unalp, A.; Tonascia, J.; Group, N.C.R. Pioglitazone versus vitamin E versus placebo for the treatment of non-diabetic patients with non-alcoholic steatohepatitis: PIVENS trial design. Contemp. Clin. Trials 2009, 30, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Gawrieh, S.; Noureddin, M.; Loo, N.; Mohseni, R.; Awasty, V.; Cusi, K.; Kowdley, K.V.; Lai, M.; Schiff, E.; Parmar, D.; et al. Saroglitazar, a PPAR-alpha/gamma Agonist, for Treatment of NAFLD: A Randomized Controlled Double-Blind Phase 2 Trial. Hepatology 2021, 74, 1809–1824. [Google Scholar] [CrossRef] [PubMed]
- Harrison, S.A.; Wong, V.W.; Okanoue, T.; Bzowej, N.; Vuppalanchi, R.; Younes, Z.; Kohli, A.; Sarin, S.; Caldwell, S.H.; Alkhouri, N.; et al. Selonsertib for patients with bridging fibrosis or compensated cirrhosis due to NASH: Results from randomized phase III STELLAR trials. J. Hepatol. 2020, 73, 26–39. [Google Scholar] [CrossRef] [PubMed]
- VanWagner, L.B.; Ning, H.; Allen, N.B.; Ajmera, V.; Lewis, C.E.; Carr, J.J.; Lloyd-Jones, D.M.; Terrault, N.A.; Siddique, J. Alcohol Use and Cardiovascular Disease Risk in Patients With Nonalcoholic Fatty Liver Disease. Gastroenterology 2017, 153, 1260–1272.e3. [Google Scholar] [CrossRef] [PubMed]
- Anstee, Q.M.; Neuschwander-Tetri, B.A.; Wong, V.W.; Abdelmalek, M.F.; Younossi, Z.M.; Yuan, J.; Pecoraro, M.L.; Seyedkazemi, S.; Fischer, L.; Bedossa, P.; et al. Cenicriviroc for the treatment of liver fibrosis in adults with nonalcoholic steatohepatitis: AURORA Phase 3 study design. Contemp. Clin. Trials 2020, 89, 105922. [Google Scholar] [CrossRef] [PubMed]
- Belfort, R.; Harrison, S.A.; Brown, K.; Darland, C.; Finch, J.; Hardies, J.; Balas, B.; Gastaldelli, A.; Tio, F.; Pulcini, J.; et al. A placebo-controlled trial of pioglitazone in subjects with nonalcoholic steatohepatitis. N. Engl. J. Med. 2006, 355, 2297–2307. [Google Scholar] [CrossRef] [PubMed]
- Cusi, K.; Orsak, B.; Bril, F.; Lomonaco, R.; Hecht, J.; Ortiz-Lopez, C.; Tio, F.; Hardies, J.; Darland, C.; Musi, N.; et al. Long-Term Pioglitazone Treatment for Patients With Nonalcoholic Steatohepatitis and Prediabetes or Type 2 Diabetes Mellitus: A Randomized Trial. Ann. Intern. Med. 2016, 165, 305–315. [Google Scholar] [CrossRef]
- Sanyal, A.J.; Chalasani, N.; Kowdley, K.V.; McCullough, A.; Diehl, A.M.; Bass, N.M.; Neuschwander-Tetri, B.A.; Lavine, J.E.; Tonascia, J.; Unalp, A.; et al. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N. Engl. J. Med. 2010, 362, 1675–1685. [Google Scholar] [CrossRef]
- LaMoia, T.E.; Butrico, G.M.; Kalpage, H.A.; Goedeke, L.; Hubbard, B.T.; Vatner, D.F.; Gaspar, R.C.; Zhang, X.M.; Cline, G.W.; Nakahara, K.; et al. Metformin, phenformin, and galegine inhibit complex IV activity and reduce glycerol-derived gluconeogenesis. Proc. Natl. Acad. Sci. USA 2022, 119, e2122287119. [Google Scholar] [CrossRef]
- European Association for the Study of The Liver; European Association for the Study of Diabetes; European Association for the Study of Obesity. EASL-EASD-EASO Clinical Practice Guidelines for the management of non-alcoholic fatty liver disease. Diabetologia 2016, 59, 1121–1140. [Google Scholar] [CrossRef]
- Cusi, K.; Isaacs, S.; Barb, D.; Basu, R.; Caprio, S.; Garvey, W.T.; Kashyap, S.; Mechanick, J.I.; Mouzaki, M.; Nadolsky, K.; et al. American Association of Clinical Endocrinology Clinical Practice Guideline for the Diagnosis and Management of Nonalcoholic Fatty Liver Disease in Primary Care and Endocrinology Clinical Settings: Co-Sponsored by the American Association for the Study of Liver Diseases (AASLD). Endocr. Pract. 2022, 28, 528–562. [Google Scholar] [CrossRef] [PubMed]
- Rena, G.; Hardie, D.G.; Pearson, E.R. The mechanisms of action of metformin. Diabetologia 2017, 60, 1577–1585. [Google Scholar] [CrossRef] [PubMed]
- DeFronzo, R.A.; Barzilai, N.; Simonson, D.C. Mechanism of metformin action in obese and lean noninsulin-dependent diabetic subjects. J. Clin. Endocrinol. Metab. 1991, 73, 1294–1301. [Google Scholar] [CrossRef] [PubMed]
- Jackson, R.A.; Hawa, M.I.; Jaspan, J.B.; Sim, B.M.; Disilvio, L.; Featherbe, D.; Kurtz, A.B. Mechanism of metformin action in non-insulin-dependent diabetes. Diabetes 1987, 36, 632–640. [Google Scholar] [CrossRef] [PubMed]
- Ferrannini, E. The target of metformin in type 2 diabetes. N. Engl. J. Med. 2014, 371, 1547–1548. [Google Scholar] [CrossRef]
- Madiraju, A.K.; Erion, D.M.; Rahimi, Y.; Zhang, X.M.; Braddock, D.T.; Albright, R.A.; Prigaro, B.J.; Wood, J.L.; Bhanot, S.; MacDonald, M.J.; et al. Metformin suppresses gluconeogenesis by inhibiting mitochondrial glycerophosphate dehydrogenase. Nature 2014, 510, 542–546. [Google Scholar] [CrossRef]
- McIntyre, H.D.; Ma, A.; Bird, D.M.; Paterson, C.A.; Ravenscroft, P.J.; Cameron, D.P. Metformin increases insulin sensitivity and basal glucose clearance in type 2 (non-insulin dependent) diabetes mellitus. Aust. N. Z. J. Med. 1991, 21, 714–719. [Google Scholar] [CrossRef]
- Stumvoll, M.; Nurjhan, N.; Perriello, G.; Dailey, G.; Gerich, J.E. Metabolic effects of metformin in non-insulin-dependent diabetes mellitus. N. Engl. J. Med. 1995, 333, 550–554. [Google Scholar] [CrossRef]
- Zhou, G.; Myers, R.; Li, Y.; Chen, Y.; Shen, X.; Fenyk-Melody, J.; Wu, M.; Ventre, J.; Doebber, T.; Fujii, N.; et al. Role of AMP-activated protein kinase in mechanism of metformin action. J. Clin. Investig. 2001, 108, 1167–1174. [Google Scholar] [CrossRef]
- Hawley, S.A.; Gadalla, A.E.; Olsen, G.S.; Hardie, D.G. The antidiabetic drug metformin activates the AMP-activated protein kinase cascade via an adenine nucleotide-independent mechanism. Diabetes 2002, 51, 2420–2425. [Google Scholar] [CrossRef]
- Shaw, R.J. Metformin trims fats to restore insulin sensitivity. Nat. Med. 2013, 19, 1570–1572. [Google Scholar] [CrossRef] [PubMed]
- Fullerton, M.D.; Galic, S.; Marcinko, K.; Sikkema, S.; Pulinilkunnil, T.; Chen, Z.P.; O’Neill, H.M.; Ford, R.J.; Palanivel, R.; O’Brien, M.; et al. Single phosphorylation sites in Acc1 and Acc2 regulate lipid homeostasis and the insulin-sensitizing effects of metformin. Nat. Med. 2013, 19, 1649–1654. [Google Scholar] [CrossRef] [PubMed]
- Glueck, C.J.; Fontaine, R.N.; Wang, P.; Subbiah, M.T.; Weber, K.; Illig, E.; Streicher, P.; Sieve-Smith, L.; Tracy, T.M.; Lang, J.E.; et al. Metformin reduces weight, centripetal obesity, insulin, leptin, and low-density lipoprotein cholesterol in nondiabetic, morbidly obese subjects with body mass index greater than 30. Metab. Clin. Exp. 2001, 50, 856–861. [Google Scholar] [CrossRef] [PubMed]
- Salpeter, S.R.; Buckley, N.S.; Kahn, J.A.; Salpeter, E.E. Meta-analysis: Metformin treatment in persons at risk for diabetes mellitus. Am. J. Med. 2008, 121, 149–157.e2. [Google Scholar] [CrossRef] [PubMed]
- Turner, R.C. The U.K. Prospective Diabetes Study. A review. Diabetes Care 1998, 21 (Suppl. S3), C35–C38. [Google Scholar] [CrossRef] [PubMed]
- Petersen, M.C.; Vatner, D.F.; Shulman, G.I. Regulation of hepatic glucose metabolism in health and disease. Nat. Rev. Endocrinol. 2017, 13, 572–587. [Google Scholar] [CrossRef] [PubMed]
- Bao, J.; Zhao, Y.; Xu, X.; Ling, S. Advance of Metformin in Liver Disease. Curr. Med. Chem. 2024, in press. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Liu, L.; Wang, B.; Wang, J.; Chen, D. Metformin in non-alcoholic fatty liver disease: A systematic review and meta-analysis. Biomed. Rep. 2013, 1, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Hegazi, O.E.; Alalalmeh, S.O.; Shahwan, M.; Jairoun, A.A.; Alourfi, M.M.; Bokhari, G.A.; Alkhattabi, A.; Alsharif, S.; Aljehani, M.A.; Alsabban, A.M.; et al. Exploring Promising Therapies for Non-Alcoholic Fatty Liver Disease: A ClinicalTrials.gov Analysis. Diabetes Metab. Syndr. Obes. 2024, 17, 545–561. [Google Scholar] [CrossRef]
- Jang, H.; Kim, Y.; Lee, D.H.; Joo, S.K.; Koo, B.K.; Lim, S.; Lee, W.; Kim, W. Outcomes of Various Classes of Oral Antidiabetic Drugs on Nonalcoholic Fatty Liver Disease. JAMA Intern. Med. 2024, 184, 375–383. [Google Scholar] [CrossRef]
- Galli, A.; Crabb, D.W.; Ceni, E.; Salzano, R.; Mello, T.; Svegliati-Baroni, G.; Ridolfi, F.; Trozzi, L.; Surrenti, C.; Casini, A. Antidiabetic thiazolidinediones inhibit collagen synthesis and hepatic stellate cell activation in vivo and in vitro. Gastroenterology 2002, 122, 1924–1940. [Google Scholar] [CrossRef] [PubMed]
- Daniele, G.; Xiong, J.; Solis-Herrera, C.; Merovci, A.; Eldor, R.; Tripathy, D.; DeFronzo, R.A.; Norton, L.; Abdul-Ghani, M. Dapagliflozin Enhances Fat Oxidation and Ketone Production in Patients with Type 2 Diabetes. Diabetes Care 2016, 39, 2036–2041. [Google Scholar] [CrossRef] [PubMed]
- Zachou, M.; Flevari, P.; Nasiri-Ansari, N.; Varytimiadis, C.; Kalaitzakis, E.; Kassi, E.; Androutsakos, T. The role of anti-diabetic drugs in NAFLD. Have we found the Holy Grail? A narrative review. Eur. J. Clin. Pharmacol. 2024, 80, 127–150. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, J.M.; Moore, L.B.; Smith-Oliver, T.A.; Wilkison, W.O.; Willson, T.M.; Kliewer, S.A. An Antidiabetic Thiazolidinedione Is a High Affinity Ligand for Peroxisome Proliferator-activated Receptor γ (PPARγ)∗. J. Biol. Chem. 1995, 270, 12953–12956. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Deng, C.; Gu, Y.; He, Y.; Yang, L.; Shi, J. Effects of dapagliflozin in patients with nonalcoholic fatty liver disease: A systematic review and meta-analysis of randomized controlled trials. Clin. Res. Hepatol. Gastroenterol. 2022, 46, 101876. [Google Scholar] [CrossRef]
- Juurlink, D.N.; Gomes, T.; Lipscombe, L.L.; Austin, P.C.; Hux, J.E.; Mamdani, M.M. Adverse cardiovascular events during treatment with pioglitazone and rosiglitazone: Population based cohort study. BMJ 2009, 339, b2942. [Google Scholar] [CrossRef] [PubMed]
- Staels, B.; Rubenstrunk, A.; Noel, B.; Rigou, G.; Delataille, P.; Millatt, L.J.; Baron, M.; Lucas, A.; Tailleux, A.; Hum, D.W.; et al. Hepatoprotective effects of the dual peroxisome proliferator-activated receptor alpha/delta agonist, GFT505, in rodent models of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. Hepatology 2013, 58, 1941–1952. [Google Scholar] [CrossRef] [PubMed]
- Cariou, B.; Hanf, R.; Lambert-Porcheron, S.; Zair, Y.; Sauvinet, V.; Noel, B.; Flet, L.; Vidal, H.; Staels, B.; Laville, M. Dual peroxisome proliferator-activated receptor alpha/delta agonist GFT505 improves hepatic and peripheral insulin sensitivity in abdominally obese subjects. Diabetes Care 2013, 36, 2923–2930. [Google Scholar] [CrossRef] [PubMed]
- Muller, T.D.; Finan, B.; Bloom, S.R.; D’Alessio, D.; Drucker, D.J.; Flatt, P.R.; Fritsche, A.; Gribble, F.; Grill, H.J.; Habener, J.F.; et al. Glucagon-like peptide 1 (GLP-1). Mol. Metab. 2019, 30, 72–130. [Google Scholar] [CrossRef]
- Ueda, P.; Svanstrom, H.; Melbye, M.; Eliasson, B.; Svensson, A.M.; Franzen, S.; Gudbjornsdottir, S.; Hveem, K.; Jonasson, C.; Pasternak, B. Sodium glucose cotransporter 2 inhibitors and risk of serious adverse events: Nationwide register based cohort study. BMJ 2018, 363, k4365. [Google Scholar] [CrossRef]
- Kahl, S.; Gancheva, S.; Strassburger, K.; Herder, C.; Machann, J.; Katsuyama, H.; Kabisch, S.; Henkel, E.; Kopf, S.; Lagerpusch, M.; et al. Empagliflozin Effectively Lowers Liver Fat Content in Well-Controlled Type 2 Diabetes: A Randomized, Double-Blind, Phase 4, Placebo-Controlled Trial. Diabetes Care 2020, 43, 298–305. [Google Scholar] [CrossRef] [PubMed]
- Latva-Rasku, A.; Honka, M.-J.; Kullberg, J.; Mononen, N.; Lehtimäki, T.; Saltevo, J.; Kirjavainen, A.K.; Saunavaara, V.; Iozzo, P.; Johansson, L. The SGLT2 inhibitor dapagliflozin reduces liver fat but does not affect tissue insulin sensitivity: A randomized, double-blind, placebo-controlled study with 8-week treatment in type 2 diabetes patients. Diabetes Care 2019, 42, 931–937. [Google Scholar] [CrossRef] [PubMed]
- Han, K.H.; Rha, S.W.; Kang, H.J.; Bae, J.W.; Choi, B.J.; Choi, S.Y.; Gwon, H.C.; Bae, J.H.; Hong, B.K.; Choi, D.H.; et al. Evaluation of short-term safety and efficacy of HMG-CoA reductase inhibitors in hypercholesterolemic patients with elevated serum alanine transaminase concentrations: PITCH study (PITavastatin versus atorvastatin to evaluate the effect on patients with hypercholesterolemia and mild to moderate hepatic damage). J. Clin. Lipidol. 2012, 6, 340–351. [Google Scholar] [CrossRef] [PubMed]
- Nakahara, T.; Hyogo, H.; Kimura, Y.; Ishitobi, T.; Arihiro, K.; Aikata, H.; Takahashi, S.; Chayama, K. Efficacy of rosuvastatin for the treatment of non-alcoholic steatohepatitis with dyslipidemia: An open-label, pilot study. Hepatol. Res. 2012, 42, 1065–1072. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.I.; Lee, H.W.; Lee, K.S.; Lee, H.S.; Park, J.Y. Effects of Statin Use on the Development and Progression of Nonalcoholic Fatty Liver Disease: A Nationwide Nested Case-Control Study. Am. J. Gastroenterol. 2021, 116, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Sfikas, G.; Psallas, M.; Koumaras, C.; Imprialos, K.; Perdikakis, E.; Doumas, M.; Giouleme, O.; Karagiannis, A.; Athyros, V.G. Prevalence, diagnosis, and treatment with 3 different statins of non-alcoholic fatty liver disease/non-alcoholic steatohepatitis in military personnel. Do genetics play a role? Curr. Vasc. Pharmacol. 2021, 19, 572–581. [Google Scholar] [CrossRef]
- Pineda, C.; Rios, R.; Raya, A.I.; Rodriguez, M.; Aguilera-Tejero, E.; Lopez, I. Hypocaloric Diet Prevents the Decrease in FGF21 Elicited by High Phosphorus Intake. Nutrients 2018, 10, 1496. [Google Scholar] [CrossRef]
- Kliewer, S.A.; Mangelsdorf, D.J. A Dozen Years of Discovery: Insights into the Physiology and Pharmacology of FGF21. Cell Metab. 2019, 29, 246–253. [Google Scholar] [CrossRef]
- Xu, J.; Lloyd, D.J.; Hale, C.; Stanislaus, S.; Chen, M.; Sivits, G.; Vonderfecht, S.; Hecht, R.; Li, Y.S.; Lindberg, R.A.; et al. Fibroblast growth factor 21 reverses hepatic steatosis, increases energy expenditure, and improves insulin sensitivity in diet-induced obese mice. Diabetes 2009, 58, 250–259. [Google Scholar] [CrossRef]
- Gong, Q.; Hu, Z.; Zhang, F.; Cui, A.; Chen, X.; Jiang, H.; Gao, J.; Chen, X.; Han, Y.; Liang, Q.; et al. Fibroblast growth factor 21 improves hepatic insulin sensitivity by inhibiting mammalian target of rapamycin complex 1 in mice. Hepatology 2016, 64, 425–438. [Google Scholar] [CrossRef]
- Sanyal, A.; Charles, E.D.; Neuschwander-Tetri, B.A.; Loomba, R.; Harrison, S.A.; Abdelmalek, M.F.; Lawitz, E.J.; Halegoua-DeMarzio, D.; Kundu, S.; Noviello, S.; et al. Pegbelfermin (BMS-986036), a PEGylated fibroblast growth factor 21 analogue, in patients with non-alcoholic steatohepatitis: A randomised, double-blind, placebo-controlled, phase 2a trial. Lancet 2019, 392, 2705–2717. [Google Scholar] [CrossRef] [PubMed]
- Cui, A.; Li, J.; Ji, S.; Ma, F.; Wang, G.; Xue, Y.; Liu, Z.; Gao, J.; Han, J.; Tai, P.; et al. The Effects of B1344, a Novel Fibroblast Growth Factor 21 Analog, on Nonalcoholic Steatohepatitis in Nonhuman Primates. Diabetes 2020, 69, 1611–1623. [Google Scholar] [CrossRef]
- Abdelmalek, M.F.; Charles, E.D.; Sanyal, A.J.; Harrison, S.A.; Neuschwander-Tetri, B.A.; Goodman, Z.; Ehman, R.A.; Karsdal, M.; Nakajima, A.; Du, S.; et al. The FALCON program: Two phase 2b randomized, double-blind, placebo-controlled studies to assess the efficacy and safety of pegbelfermin in the treatment of patients with nonalcoholic steatohepatitis and bridging fibrosis or compensated cirrhosis. Contemp. Clin. Trials 2021, 104, 106335. [Google Scholar] [CrossRef]
- Jani, R.H.; Pai, V.; Jha, P.; Jariwala, G.; Mukhopadhyay, S.; Bhansali, A.; Joshi, S. A multicenter, prospective, randomized, double-blind study to evaluate the safety and efficacy of Saroglitazar 2 and 4 mg compared with placebo in type 2 diabetes mellitus patients having hypertriglyceridemia not controlled with atorvastatin therapy (PRESS VI). Diabetes Technol. Ther. 2014, 16, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, M.S.; Idowu, M.O.; Parmar, D.; Borg, B.B.; Denham, D.; Loo, N.M.; Lazas, D.; Younes, Z.; Sanyal, A.J. A Phase 2 Double Blinded, Randomized Controlled Trial of Saroglitazar in Patients With Nonalcoholic Steatohepatitis. Clin. Gastroenterol. Hepatol. 2021, 19, 2670–2672. [Google Scholar] [CrossRef]
- Boeckmans, J.; Natale, A.; Rombaut, M.; Buyl, K.; Rogiers, V.; De Kock, J.; Vanhaecke, T.; Rodrigues, M.R. Anti-NASH Drug Development Hitches a Lift on PPAR Agonism. Cells 2019, 9, 37. [Google Scholar] [CrossRef]
- Gross, B.; Pawlak, M.; Lefebvre, P.; Staels, B. PPARs in obesity-induced T2DM, dyslipidaemia and NAFLD. Nat. Rev. Endocrinol. 2017, 13, 36–49. [Google Scholar] [CrossRef] [PubMed]
- Harrison, S.; Ratziu, V.; Bedossa, P.; Dufour, J.-F.; Kruger, F.; Schattenberg, J.; Francque, S.; Arrese, M.; George, J.; Bugianesi, E. RESOLVE-IT Phase 3 Trial of Elafibranor in NASH: Final Results of the Week 72 Interim Surrogate Efficacy Analysis. In Proceedings of the Liver Meeting Digital Experience 2020 of American Association for the Study of Liver Diseases, Online, 11–16 November 2020; Available online: https://www.natap.org/2020/AASLD/AASLD_162.htm (accessed on 1 April 2024).
- Mayerson, A.B.; Hundal, R.S.; Dufour, S.; Lebon, V.; Befroy, D.; Cline, G.W.; Enocksson, S.; Inzucchi, S.E.; Shulman, G.I.; Petersen, K.F. The effects of rosiglitazone on insulin sensitivity, lipolysis, and hepatic and skeletal muscle triglyceride content in patients with type 2 diabetes. Diabetes 2002, 51, 797–802. [Google Scholar] [CrossRef]
- Stayrook, K.R.; Bramlett, K.S.; Savkur, R.S.; Ficorilli, J.; Cook, T.; Christe, M.E.; Michael, L.F.; Burris, T.P. Regulation of carbohydrate metabolism by the farnesoid X receptor. Endocrinology 2005, 146, 984–991. [Google Scholar] [CrossRef]
- Di Ciaula, A.; Bonfrate, L.; Baj, J.; Khalil, M.; Garruti, G.; Stellaard, F.; Wang, H.H.; Wang, D.Q.; Portincasa, P. Recent Advances in the Digestive, Metabolic and Therapeutic Effects of Farnesoid X Receptor and Fibroblast Growth Factor 19: From Cholesterol to Bile Acid Signaling. Nutrients 2022, 14, 4950. [Google Scholar] [CrossRef]
- Di Ciaula, A.; Bonfrate, L.; Khalil, M.; Portincasa, P. The interaction of bile acids and gut inflammation influences the pathogenesis of inflammatory bowel disease. Intern. Emerg. Med. 2023, 18, 2181–2197. [Google Scholar] [CrossRef] [PubMed]
- Di Ciaula, A.; Wang, D.Q.; Molina-Molina, E.; Lunardi Baccetto, R.; Calamita, G.; Palmieri, V.O.; Portincasa, P. Bile Acids and Cancer: Direct and Environmental-Dependent Effects. Ann. Hepatol. 2017, 16, s87–s105. [Google Scholar] [CrossRef] [PubMed]
- Garruti, G.; Di Ciaula, A.; Wang, H.H.; Wang, D.Q.; Portincasa, P. Cross-Talk Between Bile Acids and Gastro-Intestinal and Thermogenic Hormones: Clues from Bariatric Surgery. Ann. Hepatol. 2017, 16, s68–s82. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Lu, Y.; Li, X.Y. Farnesoid X receptor: A master regulator of hepatic triglyceride and glucose homeostasis. Acta Pharmacol. Sin. 2015, 36, 44–50. [Google Scholar] [CrossRef]
- Panzitt, K.; Wagner, M. FXR in liver physiology: Multiple faces to regulate liver metabolism. Biochim. Biophys. Acta Mol. Basis Dis. 2021, 1867, 166133. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Ratziu, V.; Loomba, R.; Rinella, M.; Anstee, Q.M.; Goodman, Z.; Bedossa, P.; Geier, A.; Beckebaum, S.; Newsome, P.N.; et al. Obeticholic acid for the treatment of non-alcoholic steatohepatitis: Interim analysis from a multicentre, randomised, placebo-controlled phase 3 trial. Lancet 2019, 394, 2184–2196. [Google Scholar] [CrossRef] [PubMed]
- Anstee, Q.M.; Lucas, K.J.; Francque, S.M.; Abdelmalek, M.F.; Sanyal, A.J.; Ratziu, V.; Gadano, A.C.; Rinella, M.E.; Charlton, M.R.; Loomba, R. Safety and efficacy of Tropifexor plus Cenicriviroc combination therapy in adult patients with fibrotic NASH: 48 week results from the phase 2b tandem study. Hepatology 2021, 74, 96A–97A. [Google Scholar]
- Alkhouri, N.; Herring, R.; Kabler, H.; Kayali, Z.; Hassanein, T.; Kohli, A.; Huss, R.S.; Zhu, Y.; Billin, A.N.; Damgaard, L.H.; et al. Safety and efficacy of combination therapy with semaglutide, cilofexor and firsocostat in patients with non-alcoholic steatohepatitis: A randomised, open-label phase II trial. J. Hepatol. 2022, 77, 607–618. [Google Scholar] [CrossRef]
- Lawitz, E.J.; Poordad, F.; Coste, A.; Loo, N.; Djedjos, C.S.; McColgan, B.; Jia, C.; Xu, R.; Myers, R.P.; Subramanian, G.M.; et al. Acetyl-CoA carboxylase (ACC) inhibitor GS-0976 leads to suppression of hepatic de novo lipogenesis and significant improvements in MRI-PDFF, MRE, and markers of fibrosis after 12 weeks of therapy in patients with NASH. J. Hepatol. 2017, 66, S34. [Google Scholar] [CrossRef]
- Calle, R.A.; Amin, N.B.; Carvajal-Gonzalez, S.; Ross, T.T.; Bergman, A.; Aggarwal, S.; Crowley, C.; Rinaldi, A.; Mancuso, J.; Aggarwal, N.; et al. ACC inhibitor alone or co-administered with a DGAT2 inhibitor in patients with non-alcoholic fatty liver disease: Two parallel, placebo-controlled, randomized phase 2a trials. Nat. Med. 2021, 27, 1836–1848. [Google Scholar] [CrossRef]
- Ratziu, V.; Rinella, M.E.; Neuschwander-Tetri, B.A.; Lawitz, E.; Denham, D.; Kayali, Z.; Sheikh, A.; Kowdley, K.V.; Desta, T.; Elkhashab, M.; et al. EDP-305 in patients with NASH: A phase II double-blind placebo-controlled dose-ranging study. J. Hepatol. 2022, 76, 506–517. [Google Scholar] [CrossRef]
- Loomba, R.; Kowdley, K.V.; Vuppalanchi, R.; Hassanein, T.; Rojter, S.E.; Sheikh, M.Y.; Moussa, S.; Chung, D.; Eng, C.; Marmon, T.; et al. Liver-distributed FXR agonist TERN-101 demonstrates favorable safety and efficacy profile in NASH phase 2a lift study. Hepatology 2021, 74, 97a–98a. [Google Scholar]
- Talukdar, S.; Kharitonenkov, A. FGF19 and FGF21: In NASH we trust. Mol. Metab. 2021, 46, 101152. [Google Scholar] [CrossRef] [PubMed]
- Harrison, S.A.; Neff, G.; Guy, C.D.; Bashir, M.R.; Paredes, A.H.; Frias, J.P.; Younes, Z.; Trotter, J.F.; Gunn, N.T.; Moussa, S.E.; et al. Efficacy and Safety of Aldafermin, an Engineered FGF19 Analog, in a Randomized, Double-Blind, Placebo-Controlled Trial of Patients With Nonalcoholic Steatohepatitis. Gastroenterology 2021, 160, 219–231.e1. [Google Scholar] [CrossRef] [PubMed]
- Harrison, S.A.; Abdelmalek, M.F.; Neff, G.; Gunn, N.; Guy, C.D.; Alkhouri, N.; Bashir, M.R.; Freilich, B.; Kohli, A.; Khazanchi, A.; et al. Aldafermin in patients with non-alcoholic steatohepatitis (ALPINE 2/3): A randomised, double-blind, placebo-controlled, phase 2b trial. Lancet Gastroenterol. Hepatol. 2022, 7, 603–616. [Google Scholar] [CrossRef] [PubMed]
- Cusi, K. Selective Agonists of Thyroid Hormone Receptor Beta for the Treatment of NASH. N. Engl. J. Med. 2024, 390, 559–561. [Google Scholar] [CrossRef]
- Lonardo, A.; Ballestri, S.; Mantovani, A.; Nascimbeni, F.; Lugari, S.; Targher, G. Pathogenesis of hypothyroidism-induced NAFLD: Evidence for a distinct disease entity? Dig. Liver Dis. 2019, 51, 462–470. [Google Scholar] [CrossRef]
- Wirth, E.K.; Puengel, T.; Spranger, J.; Tacke, F. Thyroid hormones as a disease modifier and therapeutic target in nonalcoholic steatohepatitis. Expert Rev. Endocrinol. Metab. 2022, 17, 425–434. [Google Scholar] [CrossRef]
- Harrison, S.A.; Bashir, M.R.; Guy, C.D.; Zhou, R.; Moylan, C.A.; Frias, J.P.; Alkhouri, N.; Bansal, M.B.; Baum, S.; Neuschwander-Tetri, B.A.; et al. Resmetirom (MGL-3196) for the treatment of non-alcoholic steatohepatitis: A multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet 2019, 394, 2012–2024. [Google Scholar] [CrossRef]
- Harrison, S.; Taub, R.; Neff, G.; Moussa, S.; Alkhouri, N.; Bashir, M. Primary data analyses of MAESTRO-NAFLD-1 a 52 week double-blind placebo-controlled phase 3 clinical trial of resmetirom in patients with NAFLD. J. Hepatol. 2022, 77, S14. [Google Scholar] [CrossRef]
- Harrison, S.A.; Bedossa, P.; Guy, C.D.; Schattenberg, J.M.; Loomba, R.; Taub, R.; Labriola, D.; Moussa, S.E.; Neff, G.W.; Rinella, M.E.; et al. A Phase 3, Randomized, Controlled Trial of Resmetirom in NASH with Liver Fibrosis. N. Engl. J. Med. 2024, 390, 497–509. [Google Scholar] [CrossRef] [PubMed]
- Lian, B.; Loomba, R.; Neutel, J.; Margaritescu, C.; Homer, K.; Luk, A.; Mancini, M.; Ji, S.; Barker, G.; Severance, R.; et al. VK2809, a novel liver-directed thyroid receptor agonist, produces durable reductions in liver fat in patients with non-alcoholic fatty liver disease: Results of 4-week follow-up assessment from a 12-week phase 2 randomized, placebo-controlled trial. J. Hepatol. 2020, 73, S53. [Google Scholar] [CrossRef]
- Pedrosa, M.; Seyedkazemi, S.; Francque, S.; Sanyal, A.; Rinella, M.; Charlton, M.; Loomba, R.; Ratziu, V.; Kochuparampil, J.; Fischer, L.; et al. A randomized, double-blind, multicenter, phase 2b study to evaluate the safety and efficacy of a combination of tropifexor and cenicriviroc in patients with nonalcoholic steatohepatitis and liver fibrosis: Study design of the TANDEM trial. Contemp. Clin. Trials 2020, 88, 105889. [Google Scholar] [CrossRef]
- Di Lella, S.; Sundblad, V.; Cerliani, J.P.; Guardia, C.M.; Estrin, D.A.; Vasta, G.R.; Rabinovich, G.A. When galectins recognize glycans: From biochemistry to physiology and back again. Biochemistry 2011, 50, 7842–7857. [Google Scholar] [CrossRef]
- Chalasani, N.; Abdelmalek, M.F.; Garcia-Tsao, G.; Vuppalanchi, R.; Alkhouri, N.; Rinella, M.; Noureddin, M.; Pyko, M.; Shiffman, M.; Sanyal, A.; et al. Effects of Belapectin, an Inhibitor of Galectin-3, in Patients With Nonalcoholic Steatohepatitis With Cirrhosis and Portal Hypertension. Gastroenterology 2020, 158, 1334–1345.e5. [Google Scholar] [CrossRef]
- Iruarrizaga-Lejarreta, M.; Varela-Rey, M.; Fernandez-Ramos, D.; Martinez-Arranz, I.; Delgado, T.C.; Simon, J.; Juan, V.G.; delaCruz-Villar, L.; Azkargorta, M.; Lavin, J.L.; et al. Role of Aramchol in steatohepatitis and fibrosis in mice. Hepatol. Commun. 2017, 1, 911–927. [Google Scholar] [CrossRef]
- Miyazaki, M.; Flowers, M.T.; Sampath, H.; Chu, K.; Otzelberger, C.; Liu, X.; Ntambi, J.M. Hepatic stearoyl-CoA desaturase-1 deficiency protects mice from carbohydrate-induced adiposity and hepatic steatosis. Cell Metab. 2007, 6, 484–496. [Google Scholar] [CrossRef] [PubMed]
- Safadi, R.; Konikoff, F.M.; Mahamid, M.; Zelber-Sagi, S.; Halpern, M.; Gilat, T.; Oren, R.; Group, F. The fatty acid-bile acid conjugate Aramchol reduces liver fat content in patients with nonalcoholic fatty liver disease. Clin. Gastroenterol. Hepatol. 2014, 12, 2085–2091.e1. [Google Scholar] [CrossRef]
- Malik, A.; Nadeem, M.; Amjad, W.; Malik, M.I.; Javaid, S.; Farooq, U.; Naseem, K.; Khan, A. Effects of Aramchol in patients with nonalcoholic fatty liver disease (NAFLD). A systematic review and meta-analysis. Prz. Gastroenterol. 2023, 18, 67–75. [Google Scholar] [CrossRef]
- Yen, C.L.; Stone, S.J.; Koliwad, S.; Harris, C.; Farese, R.V., Jr. Thematic review series: Glycerolipids. DGAT enzymes and triacylglycerol biosynthesis. J. Lipid Res. 2008, 49, 2283–2301. [Google Scholar] [CrossRef]
- Gluchowski, N.L.; Gabriel, K.R.; Chitraju, C.; Bronson, R.T.; Mejhert, N.; Boland, S.; Wang, K.; Lai, Z.W.; Farese, R.V., Jr.; Walther, T.C. Hepatocyte Deletion of Triglyceride-Synthesis Enzyme Acyl CoA: Diacylglycerol Acyltransferase 2 Reduces Steatosis Without Increasing Inflammation or Fibrosis in Mice. Hepatology 2019, 70, 1972–1985. [Google Scholar] [CrossRef]
- Saxena, A.; Chidsey, K.; Somayaji, V.; Ogden, A.; Duvvuri, S. Diacylglycerol acyltransferase 2 (DGAT2) inhibitor PF-06865571 reduces liver fat by MRI-PDFF after 2 weeks in adults with NAFLD. Hepatology 2019, 70, 1260A. [Google Scholar]
- Amin, N.B.; Darekar, A.; Anstee, Q.M.; Wong, V.W.; Tacke, F.; Vourvahis, M.; Lee, D.S.; Charlton, M.; Alkhouri, N.; Nakajima, A.; et al. Efficacy and safety of an orally administered DGAT2 inhibitor alone or coadministered with a liver-targeted ACC inhibitor in adults with non-alcoholic steatohepatitis (NASH): Rationale and design of the phase II, dose-ranging, dose-finding, randomised, placebo-controlled MIRNA (Metabolic Interventions to Resolve NASH with fibrosis) study. BMJ Open 2022, 12, e056159. [Google Scholar] [CrossRef]
- Loomba, R.; Mohseni, R.; Lucas, K.J.; Gutierrez, J.A.; Perry, R.G.; Trotter, J.F.; Rahimi, R.S.; Harrison, S.A.; Ajmera, V.; Wayne, J.D.; et al. TVB-2640 (FASN Inhibitor) for the Treatment of Nonalcoholic Steatohepatitis: FASCINATE-1, a Randomized, Placebo-Controlled Phase 2a Trial. Gastroenterology 2021, 161, 1475–1486. [Google Scholar] [CrossRef]
- Cao, J.; Lockwood, J.; Burn, P.; Shi, Y. Cloning and functional characterization of a mouse intestinal acyl-CoA:monoacylglycerol acyltransferase, MGAT2. J. Biol. Chem. 2003, 278, 13860–13866. [Google Scholar] [CrossRef]
- Yen, C.L.; Farese, R.V., Jr. MGAT2, a monoacylglycerol acyltransferase expressed in the small intestine. J. Biol. Chem. 2003, 278, 18532–18537. [Google Scholar] [CrossRef]
- Cheng, D.; Zinker, B.A.; Luo, Y.; Shipkova, P.; De Oliveira, C.H.; Krishna, G.; Brown, E.A.; Boehm, S.L.; Tirucherai, G.S.; Gu, H.; et al. MGAT2 inhibitor decreases liver fibrosis and inflammation in murine NASH models and reduces body weight in human adults with obesity. Cell Metab. 2022, 34, 1732–1748.e5. [Google Scholar] [CrossRef]
- Gastaldelli, A.; Gaggini, M.; Daniele, G.; Ciociaro, D.; Cersosimo, E.; Tripathy, D.; Triplitt, C.; Fox, P.; Musi, N.; DeFronzo, R.; et al. Exenatide Improves both Hepatic and Adipose Tissue Insulin Resistance: A Dynamic Positron Emission Tomography Study. Hepatology 2016, 64, 2028–2037. [Google Scholar] [CrossRef]
- Gimeno, R.E.; Briere, D.A.; Seeley, R.J. Leveraging the Gut to Treat Metabolic Disease. Cell Metab. 2020, 31, 679–698. [Google Scholar] [CrossRef]
- Mells, J.E.; Fu, P.P.; Sharma, S.; Olson, D.; Cheng, L.; Handy, J.A.; Saxena, N.K.; Sorescu, D.; Anania, F.A. Glp-1 analog, liraglutide, ameliorates hepatic steatosis and cardiac hypertrophy in C57BL/6J mice fed a Western diet. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 302, G225–G235. [Google Scholar] [CrossRef]
- Armstrong, M.J.; Hull, D.; Guo, K.; Barton, D.; Hazlehurst, J.M.; Gathercole, L.L.; Nasiri, M.; Yu, J.; Gough, S.C.; Newsome, P.N.; et al. Glucagon-like peptide 1 decreases lipotoxicity in non-alcoholic steatohepatitis. J. Hepatol. 2016, 64, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Tetri, L.H.; Basaranoglu, M.; Brunt, E.M.; Yerian, L.M.; Neuschwander-Tetri, B.A. Severe NAFLD with hepatic necroinflammatory changes in mice fed trans fats and a high-fructose corn syrup equivalent. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 295, G987–G995. [Google Scholar] [CrossRef] [PubMed]
- Abdelmalek, M.F.; Suzuki, A.; Guy, C.; Unalp-Arida, A.; Colvin, R.; Johnson, R.J.; Diehl, A.M.; Clini, N.S. Increased Fructose Consumption Is Associated with Fibrosis Severity in Patients with Nonalcoholic Fatty Liver Disease. Hepatology 2010, 51, 1961–1971. [Google Scholar] [CrossRef] [PubMed]
- Basaranoglu, M.; Basaranoglu, G.; Sabuncu, T.; Senturk, H. Fructose as a key player in the development of fatty liver disease. World J. Gastroenterol. 2013, 19, 1166–1172. [Google Scholar] [CrossRef]
- Ding, X.; Saxena, N.K.; Lin, S.; Gupta, N.A.; Anania, F.A. Exendin-4, a glucagon-like protein-1 (GLP-1) receptor agonist, reverses hepatic steatosis in ob/ob mice. Hepatology 2006, 43, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Svegliati-Baroni, G.; Saccomanno, S.; Rychlicki, C.; Agostinelli, L.; De Minicis, S.; Candelaresi, C.; Faraci, G.; Pacetti, D.; Vivarelli, M.; Nicolini, D.; et al. Glucagon-like peptide-1 receptor activation stimulates hepatic lipid oxidation and restores hepatic signalling alteration induced by a high-fat diet in nonalcoholic steatohepatitis. Liver Int. 2011, 31, 1285–1297. [Google Scholar] [CrossRef] [PubMed]
- Harrison, S.A.; Alkhouri, N.; Davison, B.A.; Sanyal, A.; Edwards, C.; Colca, J.R.; Lee, B.H.; Loomba, R.; Cusi, K.; Kolterman, O.; et al. Insulin sensitizer MSDC-0602K in non-alcoholic steatohepatitis: A randomized, double-blind, placebo-controlled phase IIb study. J. Hepatol. 2020, 72, 613–626. [Google Scholar] [CrossRef]
- Harrison, S. PXL065 (deuterium-stabilized R-enantiomer of pioglitazone) reduces liver fat content and improves liver histology without PPARG-mediated side effects in patients with NASH: Analysis of a 36 week placebo-controlled Phase 2 trial (DESTINY1). In Proceedings of the 2022 AASLD the Liver Meeting, Washington, DC, USA, 4–8 November 2022; pp. 4–8. [Google Scholar]
- Stagi, S.; Ricci, F.; Bianconi, M.; Sammarco, M.A.; Municchi, G.; Toni, S.; Lenzi, L.; Verrotti, A.; de Martino, M. Retrospective Evaluation of Metformin and/or Metformin Plus a New Polysaccharide Complex in Treating Severe Hyperinsulinism and Insulin Resistance in Obese Children and Adolescents with Metabolic Syndrome. Nutrients 2017, 9, 524. [Google Scholar] [CrossRef]
- Stagi, S.; Lapi, E.; Seminara, S.; Pelosi, P.; Del Greco, P.; Capirchio, L.; Strano, M.; Giglio, S.; Chiarelli, F.; de Martino, M. Policaptil Gel Retard significantly reduces body mass index and hyperinsulinism and may decrease the risk of type 2 diabetes mellitus (T2DM) in obese children and adolescents with family history of obesity and T2DM. Ital. J. Pediatr. 2015, 41, 10. [Google Scholar] [CrossRef]
- Guarino, G.; Della Corte, T.; Strollo, F.; Gentile, S.; Nefrocenter Research Study, G. Policaptil Gel Retard in adult subjects with the metabolic syndrome: Efficacy, safety, and tolerability compared to metformin. Diabetes Metab. Syndr. 2021, 15, 901–907. [Google Scholar] [CrossRef]
- Guarino, G.; Strollo, F.; Della Corte, T.; Satta, E.; Gentile, S. Effect of Policaptil Gel Retard on Liver Fat Content and Fibrosis in Adults with Metabolic Syndrome and Type 2 Diabetes: A Non-invasive Approach to MAFLD. Diabetes Ther. 2023, 14, 2089–2108. [Google Scholar] [CrossRef] [PubMed]
- Horvath, A.; Leber, B.; Feldbacher, N.; Tripolt, N.; Rainer, F.; Blesl, A.; Trieb, M.; Marsche, G.; Sourij, H.; Stadlbauer, V. Effects of a multispecies synbiotic on glucose metabolism, lipid marker, gut microbiome composition, gut permeability, and quality of life in diabesity: A randomized, double-blind, placebo-controlled pilot study. Eur. J. Nutr. 2020, 59, 2969–2983. [Google Scholar] [CrossRef] [PubMed]
- Craven, L.; Rahman, A.; Nair Parvathy, S.; Beaton, M.; Silverman, J.; Qumosani, K.; Hramiak, I.; Hegele, R.; Joy, T.; Meddings, J.; et al. Allogenic Fecal Microbiota Transplantation in Patients with Nonalcoholic Fatty Liver Disease Improves Abnormal Small Intestinal Permeability: A Randomized Control Trial. Am. J. Gastroenterol. 2020, 115, 1055–1065. [Google Scholar] [CrossRef] [PubMed]
- Koopen, A.; Witjes, J.; Wortelboer, K.; Majait, S.; Prodan, A.; Levin, E.; Herrema, H.; Winkelmeijer, M.; Aalvink, S.; Bergman, J.; et al. Duodenal Anaerobutyricum soehngenii infusion stimulates GLP-1 production, ameliorates glycaemic control and beneficially shapes the duodenal transcriptome in metabolic syndrome subjects: A randomised double-blind placebo-controlled cross-over study. Gut 2022, 71, 1577–1587. [Google Scholar] [CrossRef]
- Witjes, J.; Koopen, A.; Wortelboer, K.; Majait, S.; Prodan, A.; Levin, E.; Herrema, H.; Winkelmeijer, M.; Aalvink, S.; Bergman, J. Duodenal Infusion of Anaerobutyricum Soehngenii Ameliorates Glycemic Control and Postprandial GLP-1 Responses and Alters the Transcriptional Profile of Small Intestine in Subjects with Metabolic Syndrome. In Proceedings of the 1st International Electronic Conference on Nutrients—Nutritional and Microbiota Effects on Chronic Disease, Online, 2–15 November 2020. [Google Scholar]
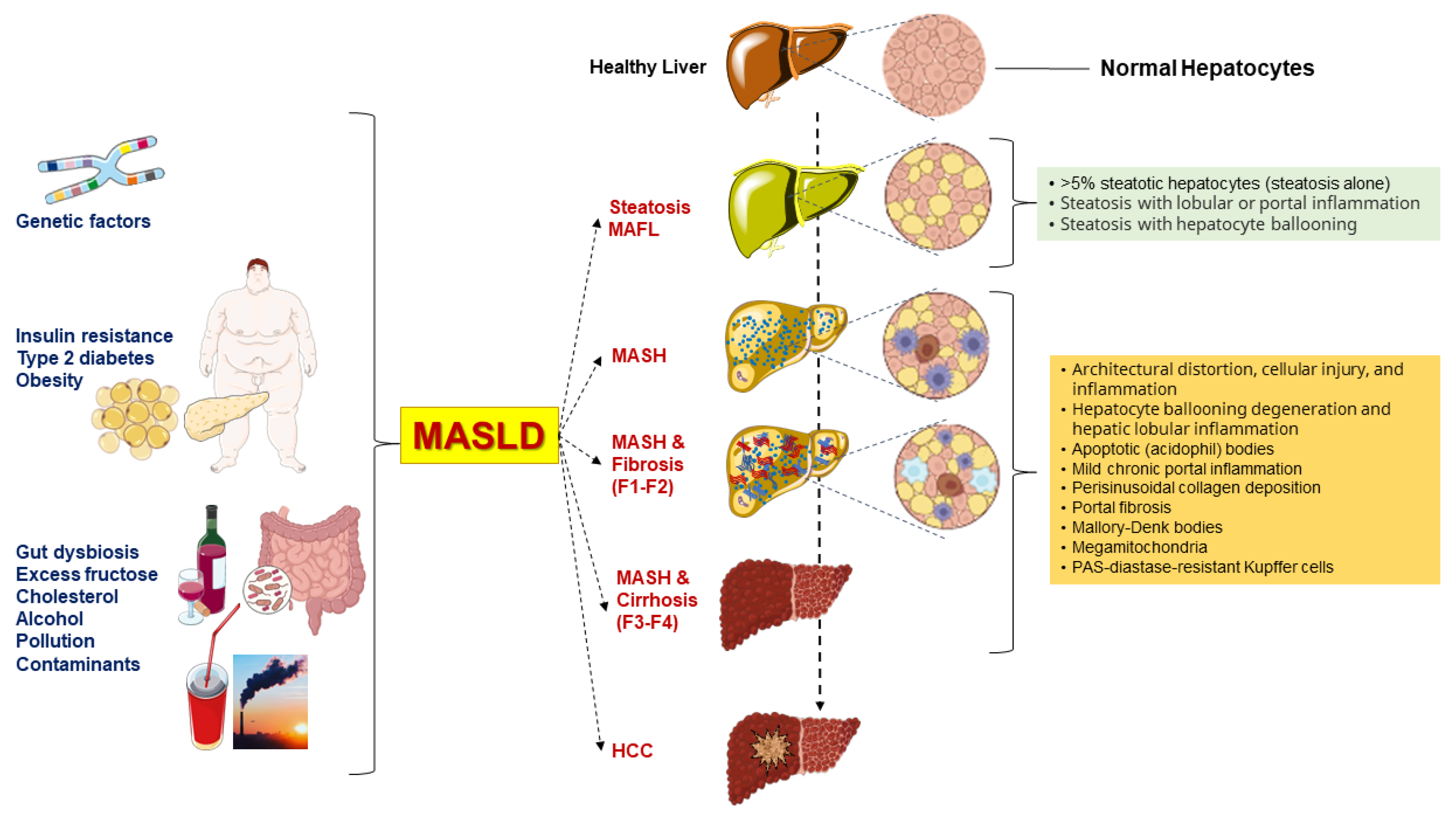
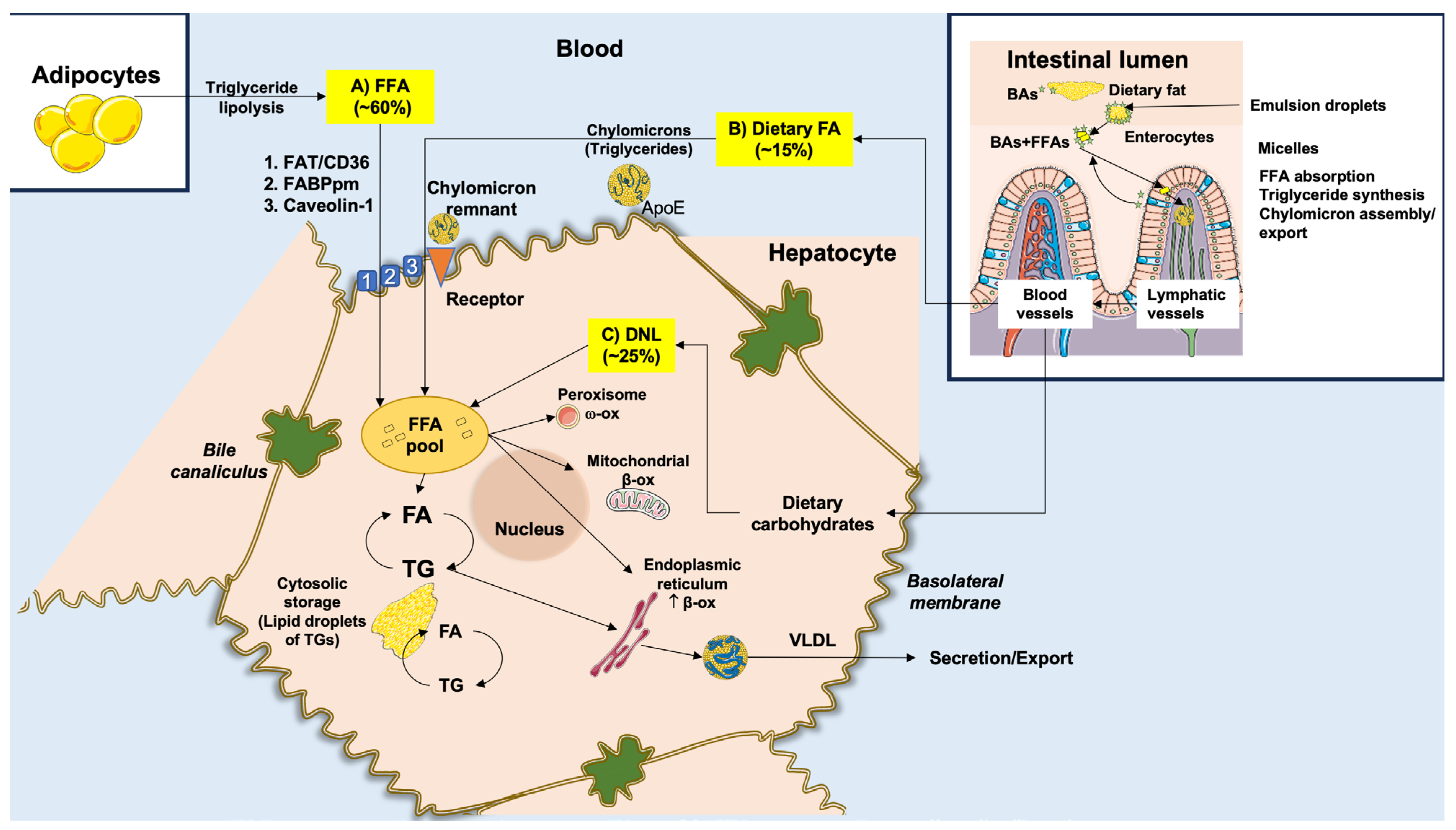
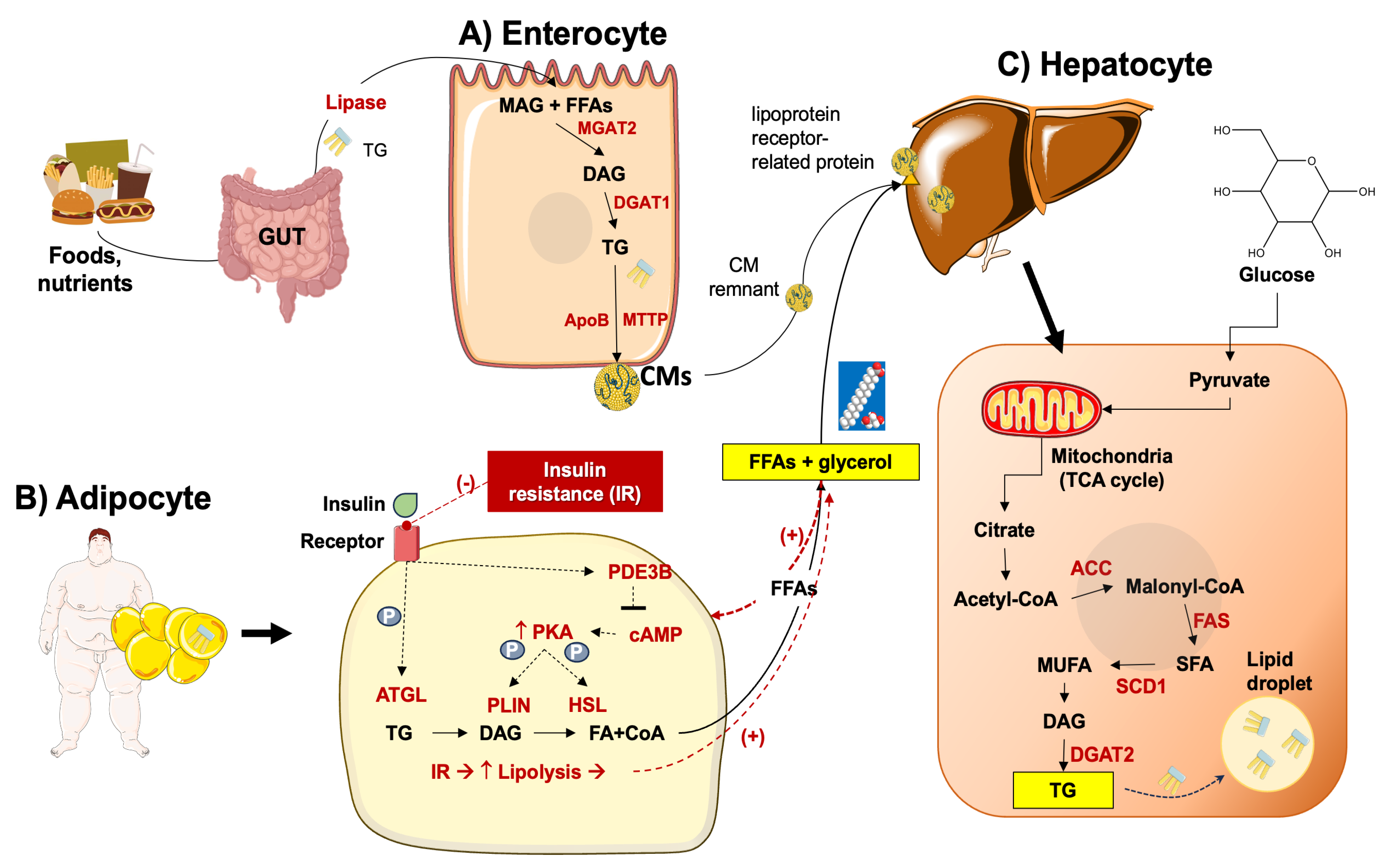
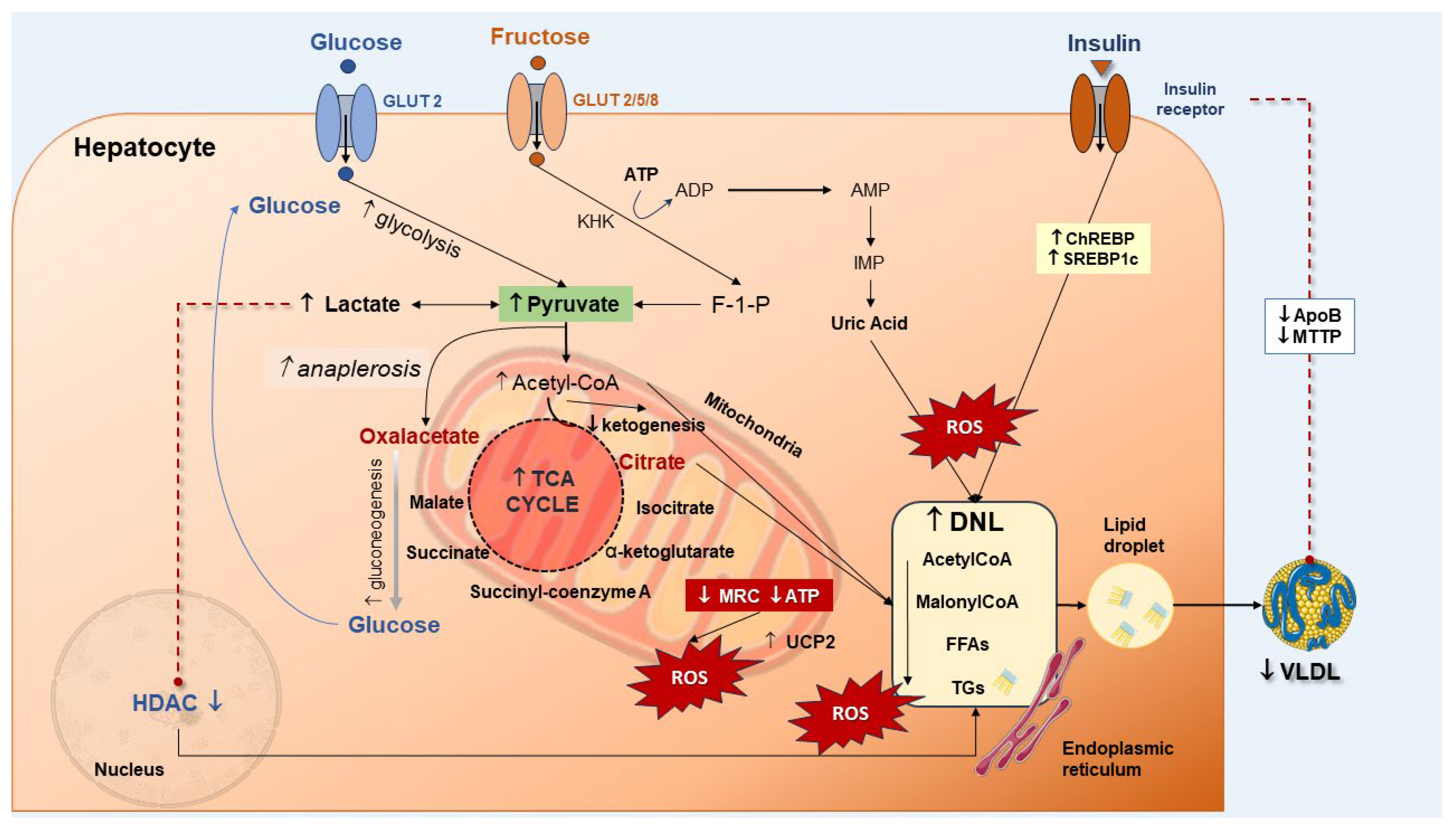
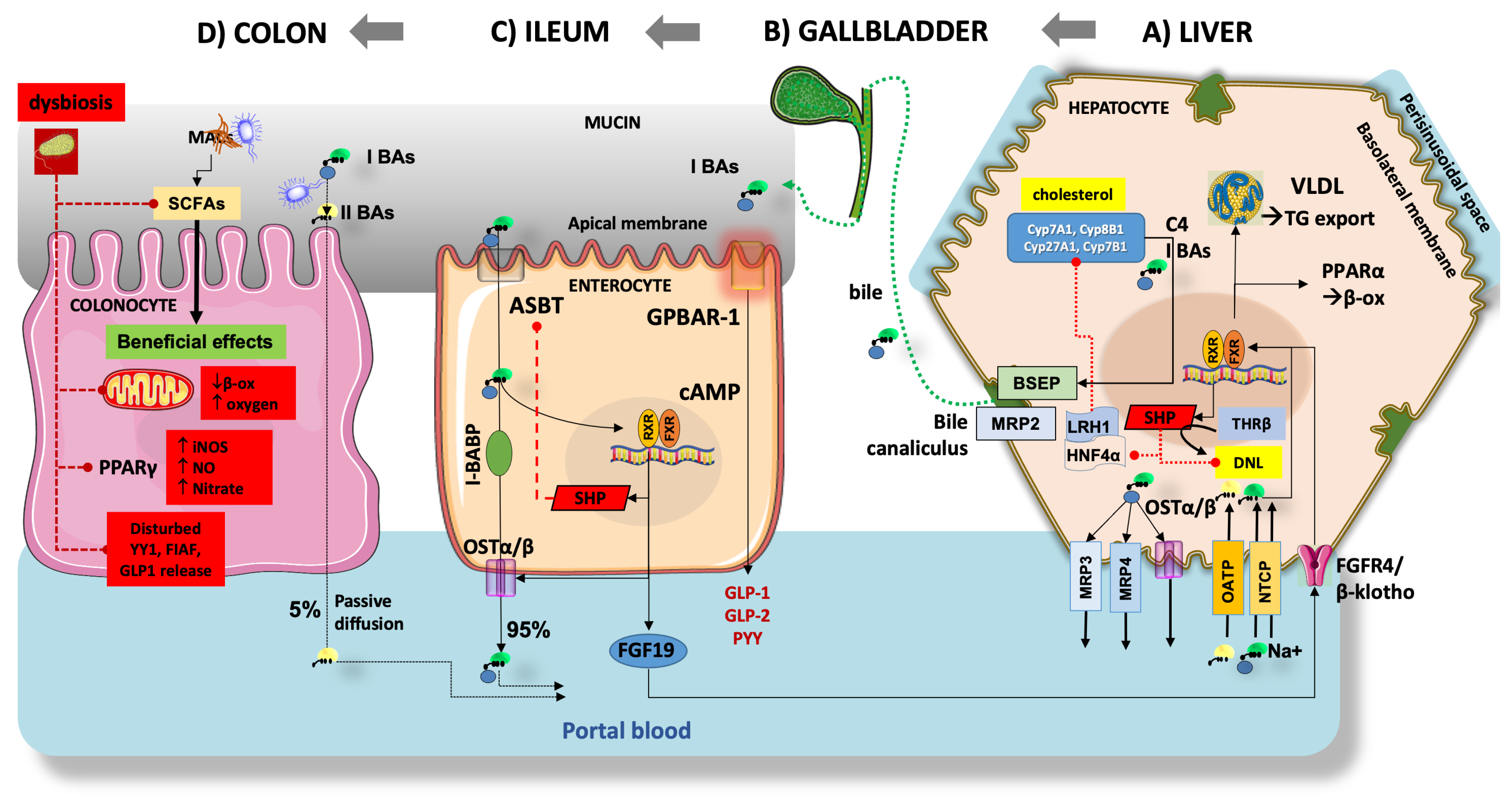

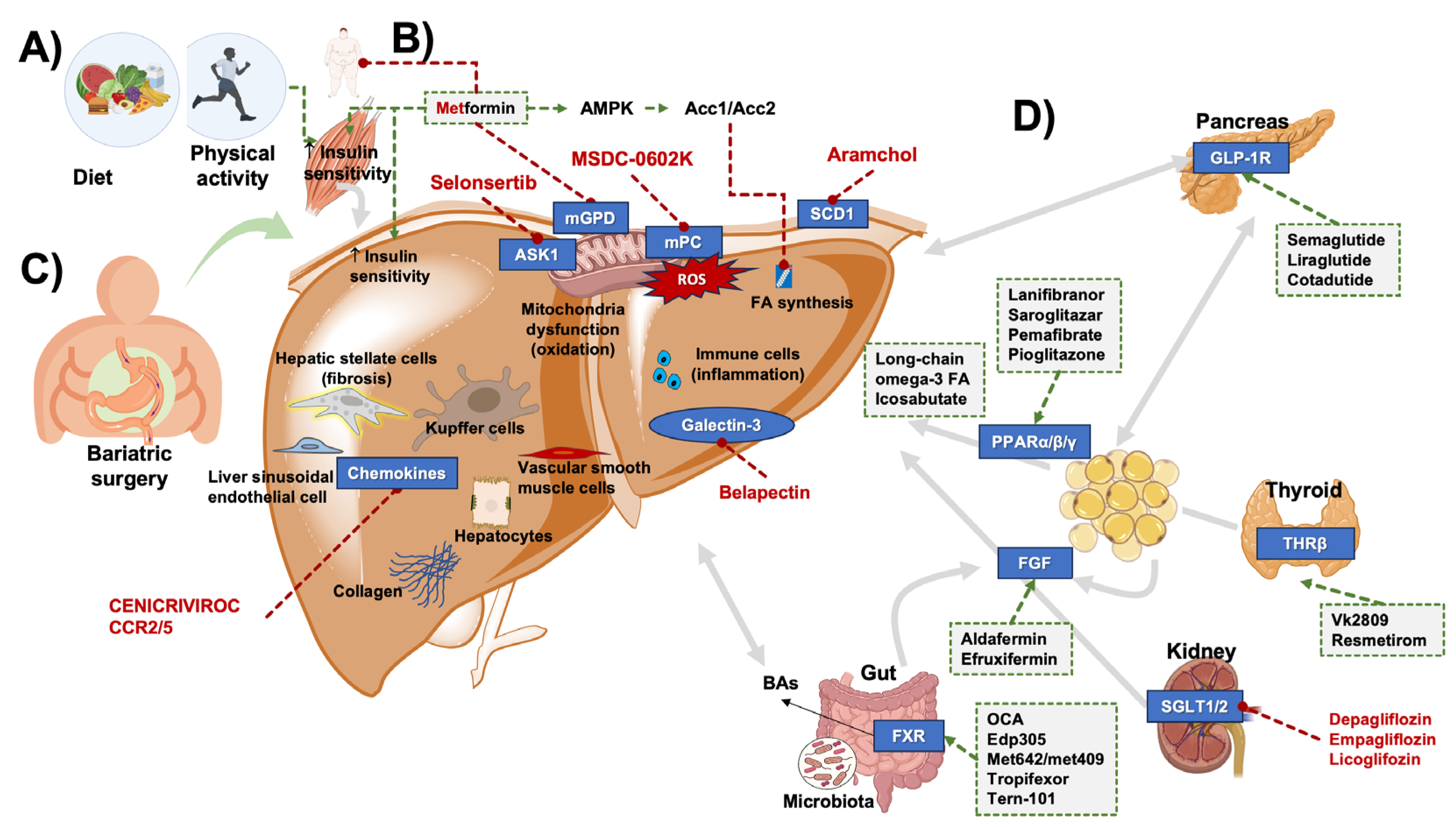
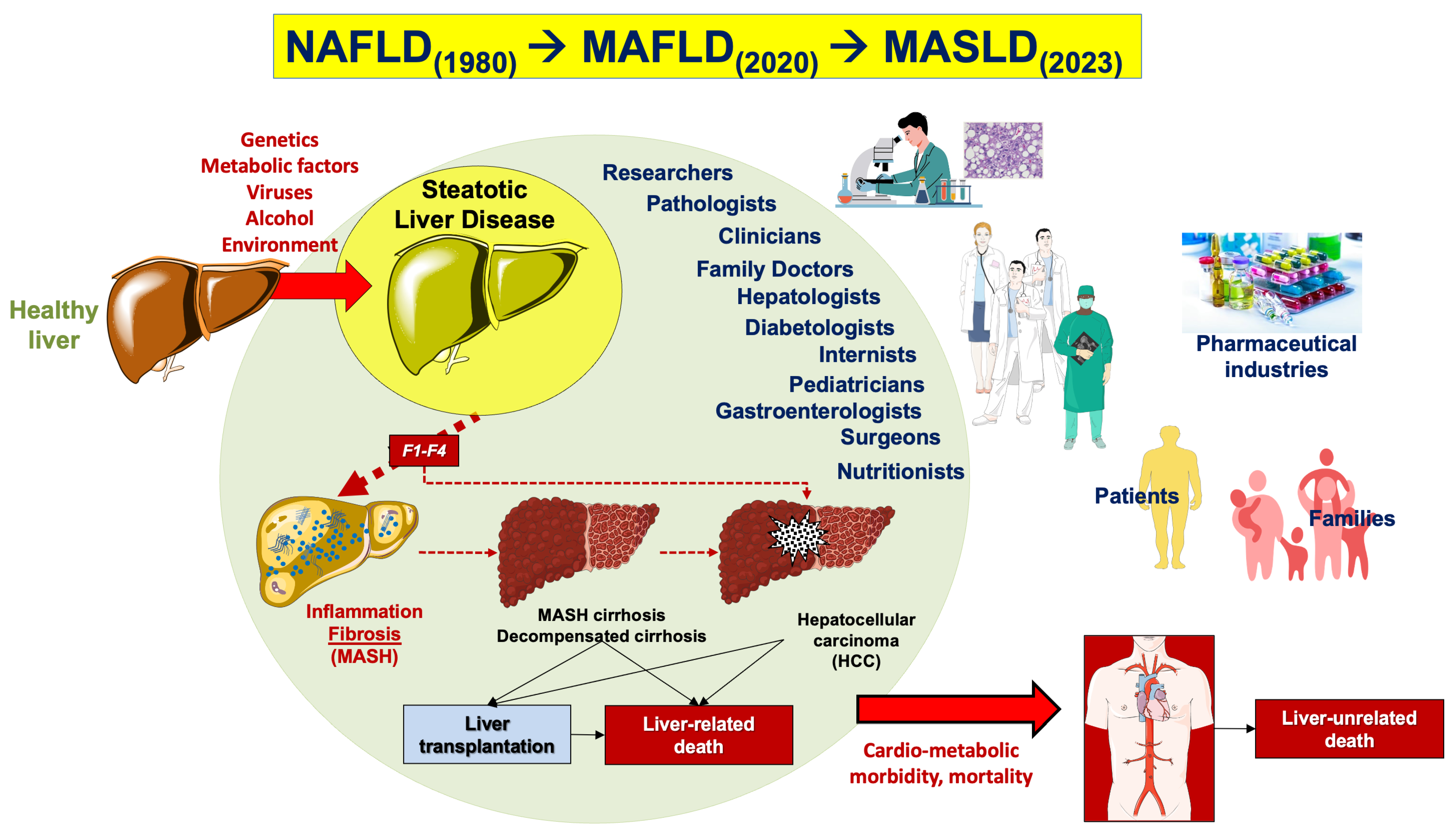

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trial Phases | ClinicalTrials.gov | Start Date of Trial (Year-Month-Day) | Drug | Molecular Mechanism (Target) | Patients | Main Findings | Adverse Effect | References |
|---|---|---|---|---|---|---|---|---|
| II | NCT02970942 | 2016-11-30 | Semaglutide | Agonist GLP1 | 320 NASH patients | NASH resolution without fibrosis improvement | Nausea, diarrhea, abdominal discomfort, reaction site injections | [226] |
| II | NCT01237119 | 2010-08 | Liraglutide | Agonist GLP1 | 52 NASH, NAFLD, Obese, and T2DM patients | NASH resolution but with worsening of fibrosis | Nausea, diarrhea, abdominal discomfort, reaction site injections | [227] |
| II | NCT03008070 | 2017-02-07 | Lanifbranor | Pan-PPAR agonist | 247 NASH patients having stable T2DM | Improvement of NASH and fibrosis per liver biopsy | Nausea, diarrhea, peripheral edema, anemia, and weight gain | [228] |
| II | NCT03350165 | 2017-12-27 | Pemafbrate | PPARα agonist | 118 NASH and NAFLD patients | No differences were observed in liver fat content but a reduction in stiffness was achieved | Mild-or-moderate adverse events | [229] |
| II | NCT02279524 | 2015-04-29 | Aramchol | Partial inhibitor of hepatic stearoyl-CoA desaturase | 247 Obese, T2DM diagnosed with NASH | No changes in liver fat but there was an improvement in liver fibrosis by ≥1 without worsening of NASH on liver histology | Well tolerated | [230] |
| II | NCT04929483 | 2021-06-04 | Pegozafermin | FGFR1,2,3,Stimulants | 222 diagnosed with NASH | improvements in fibrosis | nausea and diarrhea. | [231] |
| II | NCT03976401 | 2019-05-28 | Efruxifermin | FGF21R Agonist | 110 NASH, NAFLD Patients | Generally safe; significantly reduced liver fat | Gastrointestinal abnormalities, pulmonary embolism, acute pancreatitis with subsequent diabetic ketoacidosis | [232] |
| II | NCT02854605 | 2016-10-26 | Cilofexor tromethamine, GS-9674 | FXR Agonists, insulin sensitizers | 140 NAFLD-diagnosed patients | significant reductions in hepatic steatosis, liver biochemistry, and serum bile acids in NASH patients | Vertigo, abdominal pain, diarrhea, fatigue, pruritus, headache | [233] |
| III | NCT00063622 | 2005-01 | Pioglitazone/vitamin E | PPARγ agonist | 247 NASH patients | Pioglitazone was effective | Weight gain | [234] |
| III | NCT03061721 | 2017-04-06 | Saroglitazar | Dual PPARα and PPARγ agonist | 106 NAFLD patients | significantly improved all of ALT, LFC, insulin resistance, and atherogenic dyslipidemia | Diarrhea and cough | [235] |
| III | NCT03053050 | 2017-02-13 | Selonsertib | ASK1 Inhibitors | 808 NASH patients | no anti-fibrotic effect | Back pain, pruritus, cough, nasopharyngitis, cirrhosis, Diarrhoea. | [236] |
| III | NCT00267670 | 2005-03 | Pentoxifylline | PDE inhibitors, TNF-α inhibitor | 26 NASH Patients | improvement in serum aminotransferases as well as some histological features of NASH when compared to baseline measurements, no effect on fibrosis | Headache and abdominal cramps | [237] |
| III | NCT03028740 | 2017-04-05 | Cenicriviroc | CCR2 antagonists, CCR5 antagonists | 1778 NASH patients | Safe, and there was an improvement in fibrosis, no worsening of steatohepatitis | Nausea, Diarrhoea, abdominal pain, fatigue, arthralgia, headache | [238] |
| III | NCT04104321 | 2019-09-23 | Aramchol | SCD1 inhibitors | 150 diagnosed with NASH | Suspended | Suspended | |
| IV | NCT00227110 | 2002-10 | Pioglitazone | PPARγ Agonist | 55 NASH patients | metabolic and histologic improvement in subjects with non-alcoholic steatohepatitis | Fatigue and mild lower-extremity edema | [239] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Portincasa, P.; Khalil, M.; Mahdi, L.; Perniola, V.; Idone, V.; Graziani, A.; Baffy, G.; Di Ciaula, A. Metabolic Dysfunction–Associated Steatotic Liver Disease: From Pathogenesis to Current Therapeutic Options. Int. J. Mol. Sci. 2024, 25, 5640. https://doi.org/10.3390/ijms25115640
Portincasa P, Khalil M, Mahdi L, Perniola V, Idone V, Graziani A, Baffy G, Di Ciaula A. Metabolic Dysfunction–Associated Steatotic Liver Disease: From Pathogenesis to Current Therapeutic Options. International Journal of Molecular Sciences. 2024; 25(11):5640. https://doi.org/10.3390/ijms25115640
Chicago/Turabian StylePortincasa, Piero, Mohamad Khalil, Laura Mahdi, Valeria Perniola, Valeria Idone, Annarita Graziani, Gyorgy Baffy, and Agostino Di Ciaula. 2024. "Metabolic Dysfunction–Associated Steatotic Liver Disease: From Pathogenesis to Current Therapeutic Options" International Journal of Molecular Sciences 25, no. 11: 5640. https://doi.org/10.3390/ijms25115640
APA StylePortincasa, P., Khalil, M., Mahdi, L., Perniola, V., Idone, V., Graziani, A., Baffy, G., & Di Ciaula, A. (2024). Metabolic Dysfunction–Associated Steatotic Liver Disease: From Pathogenesis to Current Therapeutic Options. International Journal of Molecular Sciences, 25(11), 5640. https://doi.org/10.3390/ijms25115640