
De Novo Assembly and Comparative Analysis of Mitochondrial Genomes of Two Pueraria montana Varieties

 , ,
, ,
Abstract
1. Introduction
2. Results
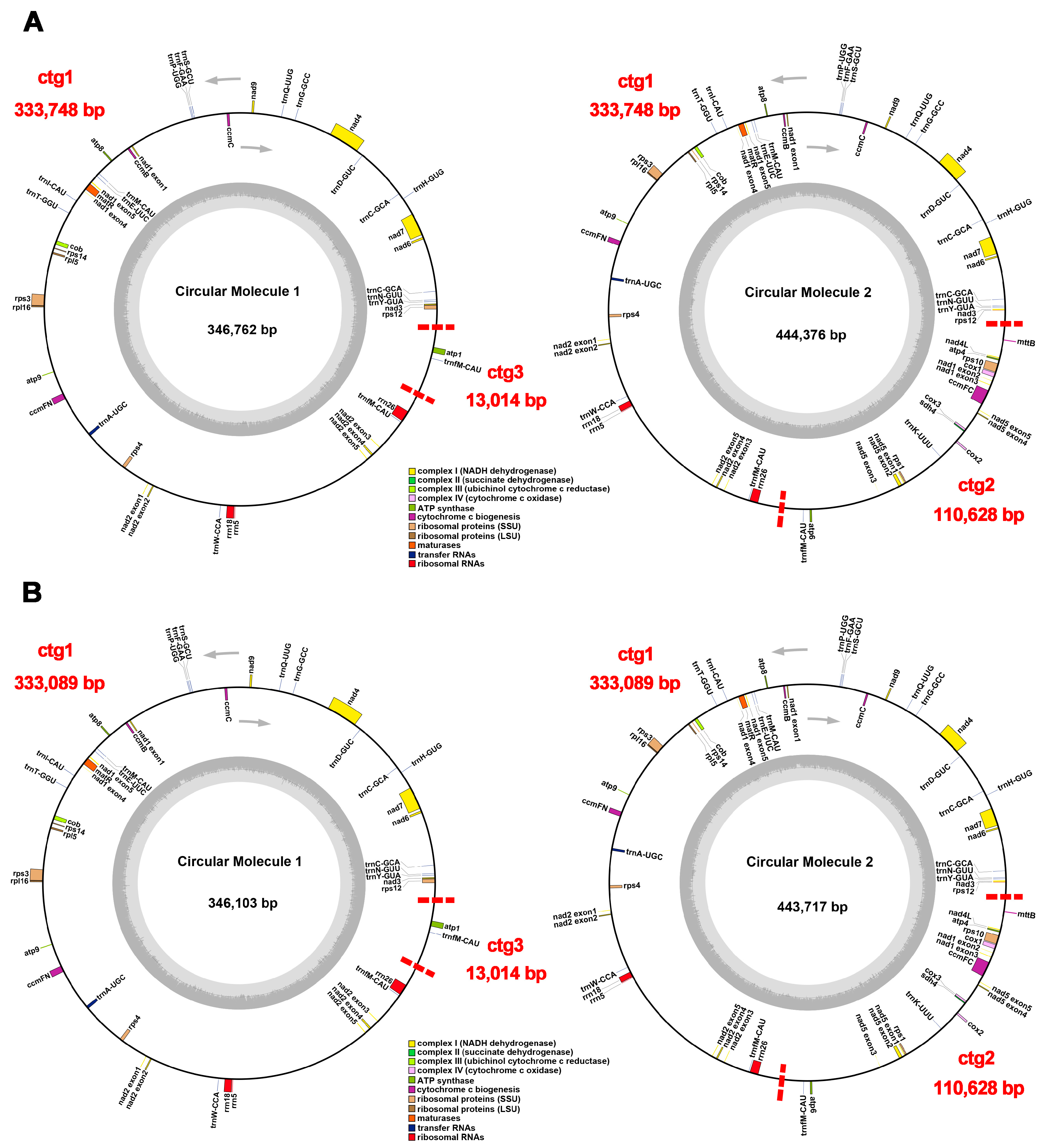
2.1. Mitochondrial Genome Assembly and Annotation
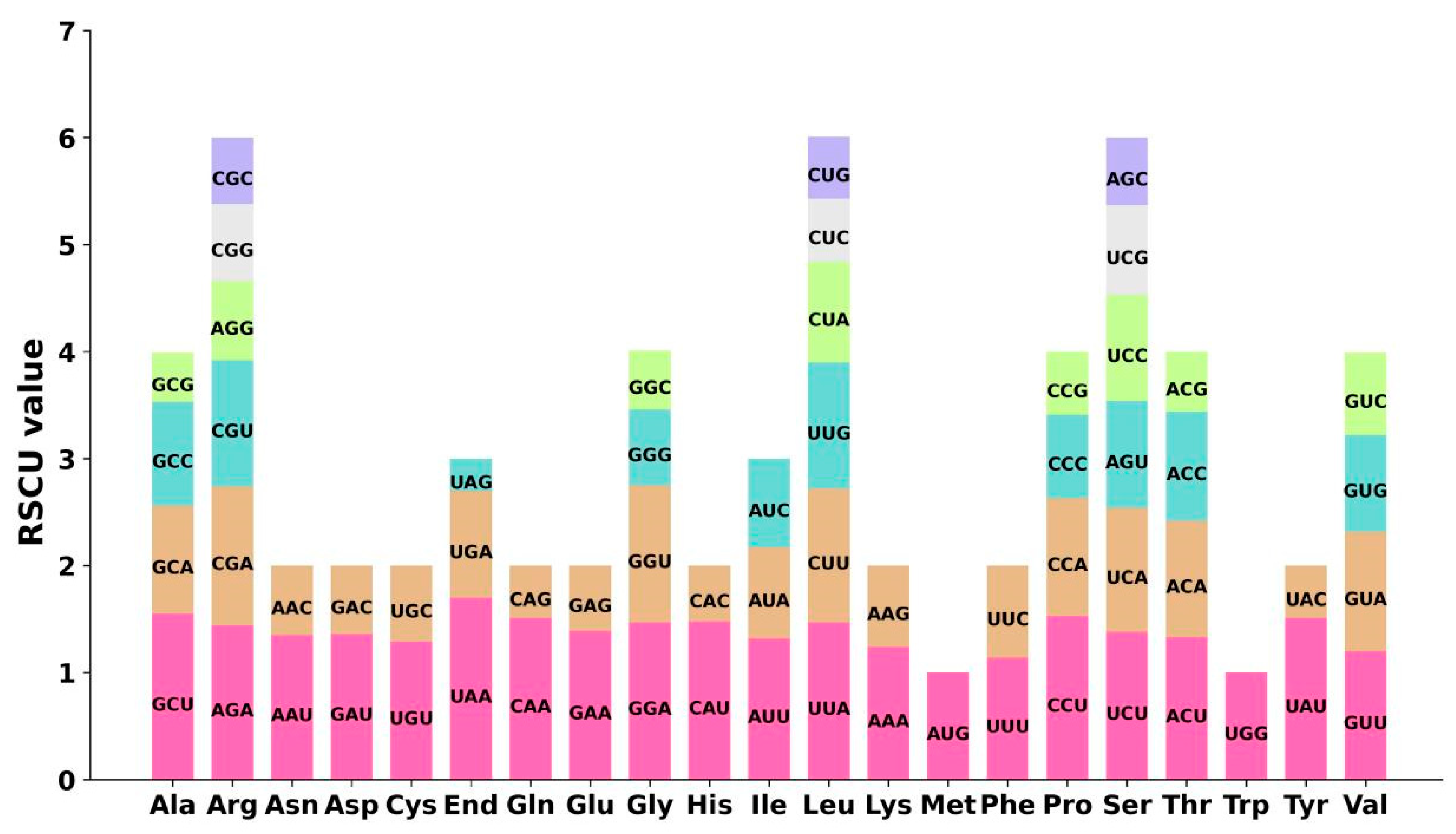
2.2. Codon Preference of the Mitogenome
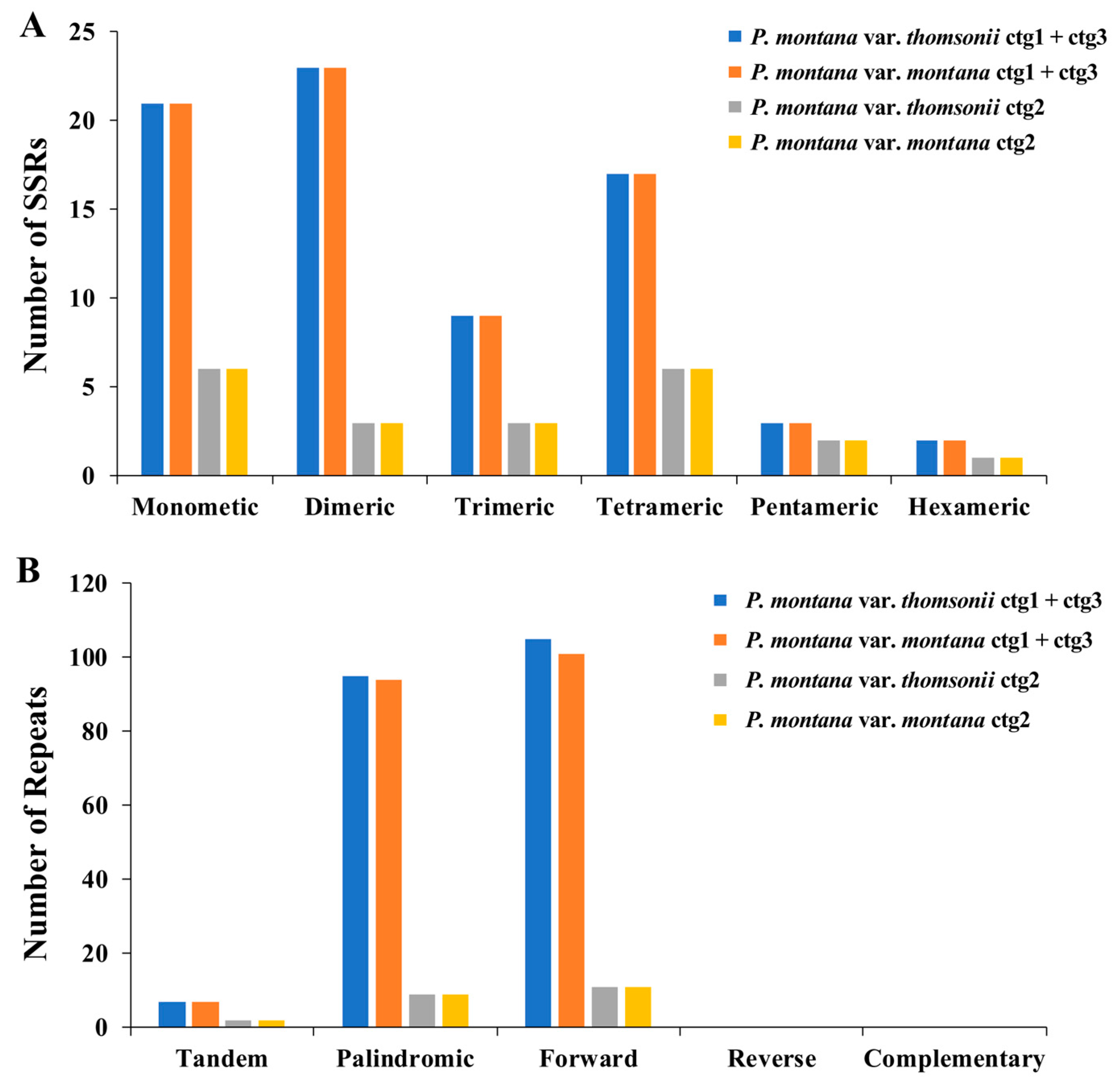
2.3. Repeat Elements
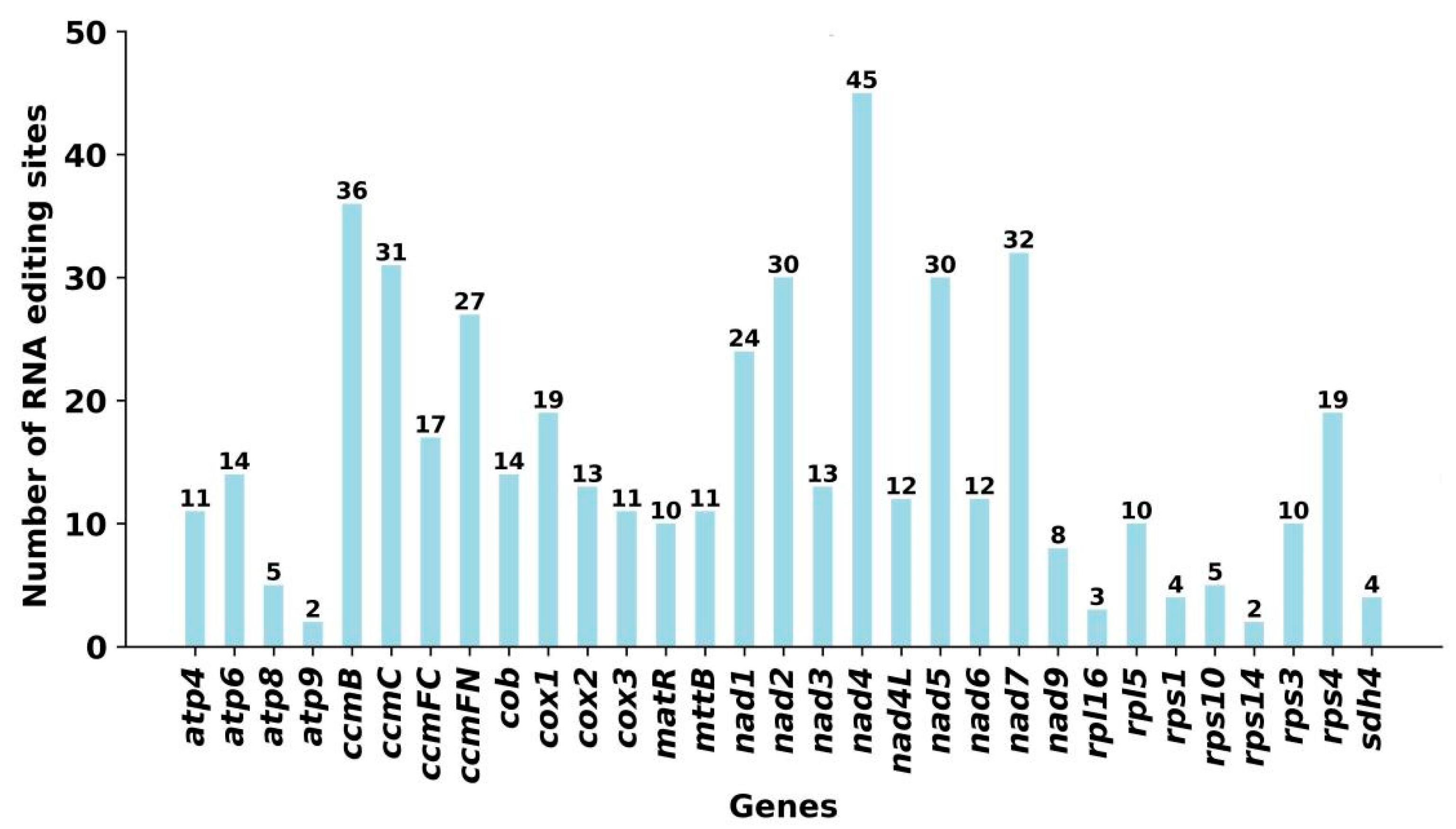
2.4. RNA-Editing Events
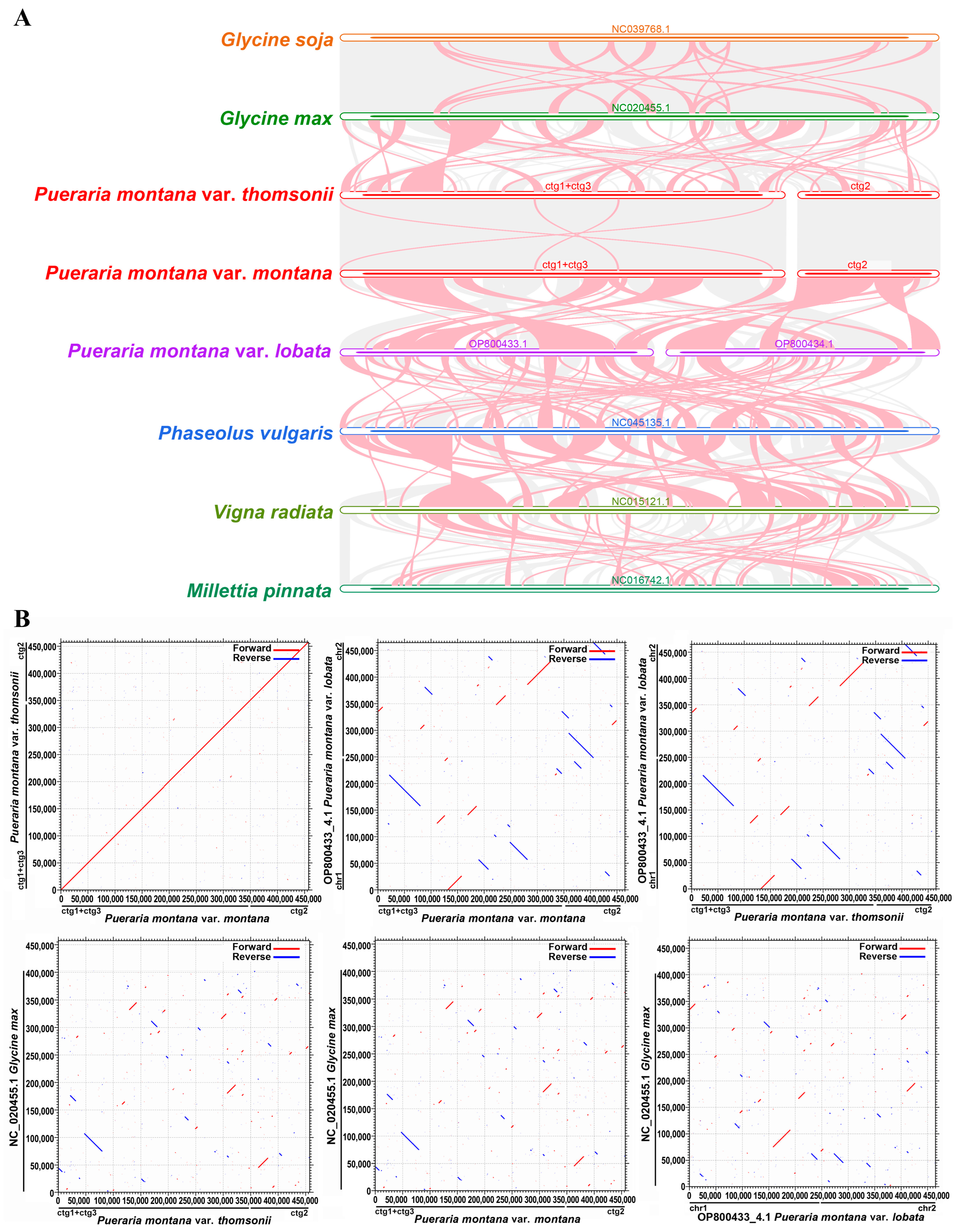
2.5. Synteny Analysis
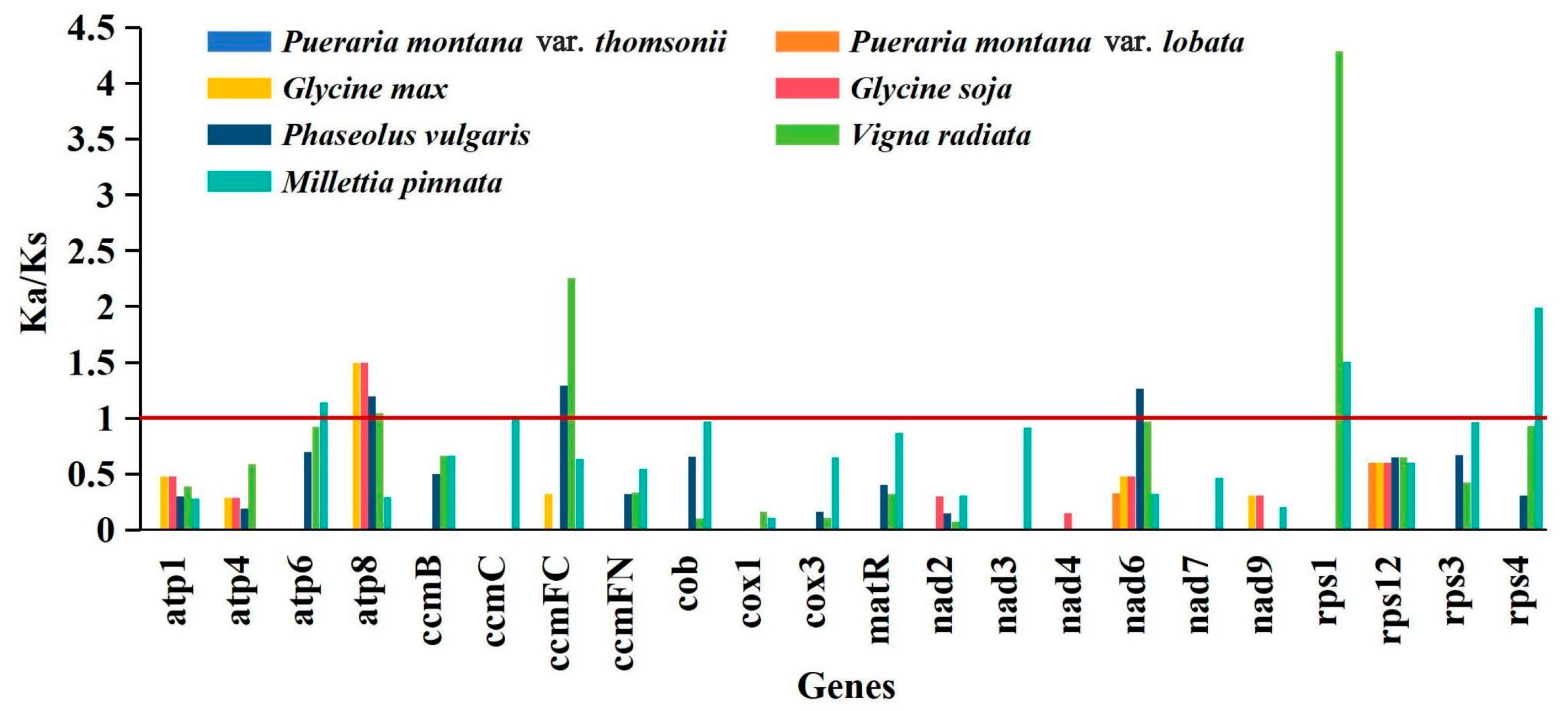
2.6. Substitution Rates of PCGs
3. Discussion
4. Materials and Methods
4.1. Plant Materials and Sequencing
4.2. Genome Assembly and Annotation
4.3. RNA-Editing Prediction
4.4. Codon Usage Bias and Repeat Analysis
4.5. Synteny Analysis
4.6. Non-Synonymous (Ka)/Synonymous (Ks) Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Van der Maesen, L. Pueraria: Botanical characteristics. In Pueraria: The Genus Pueraria; Taylor & Francis: New York, NY, USA, 2002; pp. 1–28. [Google Scholar]
- Haynsen, M.S.; Vatanparast, M.; Mahadwar, G.; Zhu, D.; Moger-Reischer, R.Z.; Doyle, J.J.; Crandall, K.A.; Egan, A.N. De novo transcriptome assembly of Pueraria montana var. lobata and Neustanthus phaseoloides for the development of eSSR and SNP markers: Narrowing the US origin (s) of the invasive kudzu. BMC Genom. 2018, 19, 439. [Google Scholar]
- Blaustein, R.J. Kudzu’s invasion into Southern United States life and culture. In The Great Reshuffling: Human Dimensions of Invasive Species; IUCN: Gland, Switzerland, 2001; pp. 55–62. [Google Scholar]
- Kato-Noguchi, H. The impact and invasive mechanisms of Pueraria montana var. lobata, one of the world’s worst alien species. Plants 2023, 12, 3066. [Google Scholar]
- Tsugawa, H. Cultivation and Utilization of Kudzu-Vine (Pueraria lobata Ohwi): Taxonomy, geographical distribution, use, breeding and propagation. Jpn. J. Grassl. Sci. 1986, 31, 435–443. [Google Scholar]
- Haynsen, M.S. Population Genetics of Pueraria montana var. lobata. Master’s Thesis, The George Washington University, Washington, DC, USA, 2018. [Google Scholar]
- Cao, T.; Ma, X.; Zhang, Y.; Su, W.; Li, B.; Zhou, Q.; Zhu, Q. The complete chloroplast genome sequence of the Pueraria lobata (Willd.) Ohwi (Leguminosae). Mitochondrial DNA Part B 2020, 5, 3754–3756. [Google Scholar] [CrossRef]
- Miao, X.-R.; Niu, J.-Q.; Wang, A.-Q.; Wang, D.-B.; Fan, J. Complete chloroplast genome sequence of Pueraria thomsonii, an important traditional Chinese medicine plant. Mitochondrial DNA Part B 2019, 4, 4163–4165. [Google Scholar] [CrossRef]
- Zhou, Y.; Shang, X.-H.; Xiao, L.; Wu, Z.-D.; Cao, S.; Yan, H.-B. Comparative plastomes of Pueraria montana var. lobata (Leguminosae: Phaseoleae) and closely related taxa: Insights into phylogenomic implications and evolutionary divergence. BMC Genom. 2023, 24, 299. [Google Scholar]
- Egan, A.N.; Vatanparast, M.; Cagle, W. Parsing polyphyletic Pueraria: Delimiting distinct evolutionary lineages through phylogeny. Mol. Phylogenet. Evol. 2016, 104, 44–59. [Google Scholar] [CrossRef]
- Zeng, M.; Ma, Y.; Zheng, S.; Xu, J.; Di, X. Studies on ribosomal DNA sequence analyses of Radix puerariae and its sibling species. Chin. Pharm. J. Beijing 2003, 38, 173–175. [Google Scholar]
- Picard, M.; Shirihai, O.S. Mitochondrial signal transduction. Cell Metab. 2022, 34, 1620–1653. [Google Scholar] [CrossRef]
- Jiang, L. Male sterility in maize: A precise dialogue between the mitochondria and nucleus. Mol. Plant 2020, 13, 1237. [Google Scholar] [CrossRef]
- Petersen, G.; Seberg, O.; Davis, J.I.; Stevenson, D.W. RNA editing and phylogenetic reconstruction in two monocot mitochondrial genes. Taxon 2006, 55, 871–886. [Google Scholar] [CrossRef]
- Wallace, D.C.; Singh, G.; Lott, M.T.; Hodge, J.A.; Schurr, T.G.; Lezza, A.M.; Elsas, L.J.; Nikoskelainen, E.K. Mitochondrial DNA mutation associated with Leber’s hereditary optic neuropathy. Science 1988, 242, 1427–1430. [Google Scholar] [CrossRef]
- Drouin, G.; Daoud, H.; Xia, J. Relative rates of synonymous substitutions in the mitochondrial, chloroplast and nuclear genomes of seed plants. Mol. Phylogenet. Evol. 2008, 49, 827–831. [Google Scholar] [CrossRef]
- Ni, Y.; Li, J.; Chen, H.; Yue, J.; Chen, P.; Liu, C. Comparative analysis of the chloroplast and mitochondrial genomes of Saposhnikovia divaricata revealed the possible transfer of plastome repeat regions into the mitogenome. BMC Genom. 2022, 23, 570. [Google Scholar] [CrossRef]
- Bi, C.; Lu, N.; Xu, Y.; He, C.; Lu, Z. Characterization and analysis of the mitochondrial genome of common bean (Phaseolus vulgaris) by comparative genomic approaches. Int. J. Mol. Sci. 2020, 21, 3778. [Google Scholar] [CrossRef]
- Liu, D.; Qu, K.; Yuan, Y.; Zhao, Z.; Chen, Y.; Han, B.; Li, W.; El-Kassaby, Y.A.; Yin, Y.; Xie, X. Complete sequence and comparative analysis of the mitochondrial genome of the rare and endangered Clematis acerifolia, the first clematis mitogenome to provide new insights into the phylogenetic evolutionary status of the genus. Front. Genet. 2023, 13, 1050040. [Google Scholar] [CrossRef]
- Gualberto, J.M.; Newton, K.J. Plant mitochondrial genomes: Dynamics and mechanisms of mutation. Annu. Rev. Plant Biol. 2017, 68, 225–252. [Google Scholar] [CrossRef]
- Fay, J.C.; Wu, C.-I. Sequence divergence, functional constraint, and selection in protein evolution. Annu. Rev. Genom. Hum. Genet. 2003, 4, 213–235. [Google Scholar] [CrossRef]
- Zhang, Z.; Li, J.; Zhao, X.-Q.; Wang, J.; Wong, G.K.-S.; Yu, J. KaKs_Calculator: Calculating Ka and Ks through model selection and model averaging. Genom. Proteom. Bioinform. 2006, 4, 259–263. [Google Scholar] [CrossRef]
- Kozik, A.; Rowan, B.A.; Lavelle, D.; Berke, L.; Schranz, M.E.; Michelmore, R.W.; Christensen, A.C. The alternative reality of plant mitochondrial DNA: One ring does not rule them all. PLoS Genet. 2019, 15, e1008373. [Google Scholar] [CrossRef]
- Shen, Z.; Sheng, Q.; Jin, Z.; Zhang, Y.; Lv, H. Mitogenome Characterization of a Vulnerable Gastromyzontid Fish, Beaufortia pingi (Teleostei: Gastromyzontidae): Genome Description and Phylogenetic Considerations. J. Ichthyol. 2023, 63, 735–746. [Google Scholar] [CrossRef]
- Kubo, T.; Newton, K.J. Angiosperm mitochondrial genomes and mutations. Mitochondrion 2008, 8, 5–14. [Google Scholar] [CrossRef]
- Kitazaki, K.; Kubo, T. Cost of having the largest mitochondrial genome: Evolutionary mechanism of plant mitochondrial genome. J. Bot. 2010, 2010, 620137. [Google Scholar] [CrossRef]
- Liu, H.; Yu, J.; Yu, X.; Zhang, D.; Chang, H.; Li, W.; Song, H.; Cui, Z.; Wang, P.; Luo, Y. Structural variation of mitochondrial genomes sheds light on evolutionary history of soybeans. Plant J. 2021, 108, 1456–1472. [Google Scholar] [CrossRef]
- Li, Y.; Zhao, H.; Tan, H.; Liu, X.; Zhang, C.; Dong, Y. Analysis and comparison on characteristic of mitochondrial genome of eight plants. Biotechnol. Bull. 2011, 10, 156–162. [Google Scholar]
- Mazumder, G.A.; Uddin, A.; Chakraborty, S. Prediction of gene expression and codon usage in human parasitic helminths. Genes Genom. 2017, 39, 27–36. [Google Scholar] [CrossRef]
- Chakraborty, S.; Yengkhom, S.; Uddin, A. Analysis of codon usage bias of chloroplast genes in Oryza species: Codon usage of chloroplast genes in Oryza species. Planta 2020, 252, 67. [Google Scholar] [CrossRef]
- Zhao, S.; Deng, L.; Chen, F. Codon usage bias of chloroplast genome in Kandelia obovate. J. For. Environ. 2020, 40, 534–541. [Google Scholar]
- Ma, Q.; Wang, Y.; Li, S.; Wen, J.; Zhu, L.; Yan, K.; Du, Y.; Ren, J.; Li, S.; Chen, Z. Assembly and comparative analysis of the first complete mitochondrial genome of Acer truncatum Bunge: A woody oil-tree species producing nervonic acid. BMC Plant Biol. 2022, 22, 29. [Google Scholar] [CrossRef]
- Ke, S.-J.; Liu, D.-K.; Tu, X.-D.; He, X.; Zhang, M.-M.; Zhu, M.-J.; Zhang, D.-Y.; Zhang, C.-L.; Lan, S.-R.; Liu, Z.-J. Apostasia Mitochondrial Genome Analysis and Monocot Mitochondria Phylogenomics. Int. J. Mol. Sci. 2023, 24, 7837. [Google Scholar] [CrossRef]
- Liu, H.; Liu, W.; Xiao, Q.; Zhou, Q.; Li, X.; Yao, Z.; Wang, A.; Gao, Q.; Chen, S. Comparative Analysis of the Complete Mitochondrial Genome Sequence of an Alpine Plant Triosteum pinnatifidum. Cytol. Genet. 2023, 57, 335–346. [Google Scholar] [CrossRef]
- Stern, D.B.; Palmer, J.D. Recombination sequences in plant mitochondrial genomes: Diversity and homologies to known mitochondrial genes. Nucleic Acids Res. 1984, 12, 6141–6157. [Google Scholar] [CrossRef]
- Chen, H.; Deng, L.; Jiang, Y.; Lu, P.; Yu, J. RNA editing sites exist in protein-coding genes in the chloroplast genome of Cycas taitungensis. J. Integr. Plant Biol. 2011, 53, 961–970. [Google Scholar] [CrossRef]
- Raman, G.; Park, S. Analysis of the complete chloroplast genome of a medicinal plant, Dianthus superbus var. longicalyncinus, from a comparative genomics perspective. PLoS ONE 2015, 10, e0141329. [Google Scholar]
- Mower, J.P. PREP-Mt: Predictive RNA editor for plant mitochondrial genes. BMC Bioinform. 2005, 6, 96. [Google Scholar] [CrossRef]
- Xia, C.; Li, J.; Zuo, Y.; He, P.; Zhang, H.; Zhang, X.; Wang, B.; Zhang, J.; Yu, J.; Deng, H. Complete mitochondrial genome of Thuja sutchuenensis and its implications on evolutionary analysis of complex mitogenome architecture in Cupressaceae. BMC Plant Biol. 2023, 23, 84. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Kim, J.S.; Senol, D.; Xin, H.; Lee, D.; Ghose, S.; Alser, M.; Hassan, H.; Ergin, O.; Alkan, C.; Mutlu, O. GRIM-filter: Fast seed filtering in read mapping using emerging memory technologies. arXiv 2017, arXiv:1708.04329. [Google Scholar]
- Kolmogorov, M.; Yuan, J.; Lin, Y.; Pevzner, P.A. Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 2019, 37, 540–546. [Google Scholar] [CrossRef]
- Wick, R.R.; Schultz, M.B.; Zobel, J.; Holt, K.E. Bandage: Interactive visualization of de novo genome assemblies. Bioinformatics 2015, 31, 3350–3352. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Wick, R.R.; Judd, L.M.; Gorrie, C.L.; Holt, K.E. Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 2017, 13, e1005595. [Google Scholar] [CrossRef]
- Tillich, M.; Lehwark, P.; Pellizzer, T.; Ulbricht-Jones, E.S.; Fischer, A.; Bock, R.; Greiner, S. GeSeq–versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 2017, 45, W6–W11. [Google Scholar] [CrossRef]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef]
- Chen, Y.; Ye, W.; Zhang, Y.; Xu, Y. High speed BLASTN: An accelerated MegaBLAST search tool. Nucleic Acids Res. 2015, 43, 7762–7768. [Google Scholar] [CrossRef]
- Lewis, S.E.; Searle, S.; Harris, N.; Gibson, M.; Iyer, V.; Richter, J.; Wiel, C.; Bayraktaroglu, L.; Birney, E.; Crosby, M. Apollo: A sequence annotation editor. Genome Biol. 2002, 3, RESEARCH0082. [Google Scholar] [CrossRef]
- Edera, A.A.; Small, I.; Milone, D.H.; Sanchez-Puerta, M.V. Deepred-Mt: Deep representation learning for predicting C-to-U RNA editing in plant mitochondria. Comput. Biol. Med. 2021, 136, 104682. [Google Scholar] [CrossRef]
- Beier, S.; Thiel, T.; Münch, T.; Scholz, U.; Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef]
- Kurtz, S.; Choudhuri, J.V.; Ohlebusch, E.; Schleiermacher, C.; Stoye, J.; Giegerich, R. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef]
- Zhang, H.; Meltzer, P.; Davis, S. RCircos: An R package for Circos 2D track plots. BMC Bioinform. 2013, 14, 244. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Gao, F.; Jakovlić, I.; Zou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Wang, Y.; Tang, H.; DeBarry, J.D.; Tan, X.; Li, J.; Wang, X.; Lee, T.-h.; Jin, H.; Marler, B.; Guo, H. MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012, 40, e49. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genome Feature | P. montana var. thomsonii | P. montana var. montana |
|---|---|---|
| Genome size (bp) | 457,390 | 456,731 |
| Numbers of contigs | 2 | 2 |
| Contig length(bp) | Molecule 1: 346,762 Molecule 2: 444,376 | Molecule 1: 346,103 Molecule 2: 443,717 |
| GenBank Nos | ctg1 + ctg3: PP275071 ctg2: PP275072 | ctg1 + ctg3: PP275073 ctg2: PP275074 |
| GC content (%) | 45.05 | 45.05 |
| Length of the protein-coding region (bp) | 30,234 | 30,234 |
| GC content of the protein-coding region (%) | 42.91 | 42.91 |
| Length of rRNA genes (bp) | 5272 | 5272 |
| GC content of rRNA genes (%) | 51.75 | 51.75 |
| Length of tRNA genes (bp) | 1487 | 1487 |
| GC content of tRNA genes (%) | 51.78 | 51.78 |
| Number of protein-coding genes (native) | 33 | 33 |
| Number of protein-coding genes (plastid-derived) | 2 | 2 |
| Number of rRNA genes | 3 | 3 |
| Number of tRNA genes (native) | 13 | 13 |
| Number of tRNA genes (plastid-derived) | 5 | 5 |
| Total genes | 56 | 56 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, L.; Lao, G.; He, L.; Xiao, D.; Zhan, J.; Wang, A. De Novo Assembly and Comparative Analysis of Mitochondrial Genomes of Two Pueraria montana Varieties. Int. J. Mol. Sci. 2024, 25, 5656. https://doi.org/10.3390/ijms25115656
Guo L, Lao G, He L, Xiao D, Zhan J, Wang A. De Novo Assembly and Comparative Analysis of Mitochondrial Genomes of Two Pueraria montana Varieties. International Journal of Molecular Sciences. 2024; 25(11):5656. https://doi.org/10.3390/ijms25115656
Chicago/Turabian StyleGuo, Lijun, Guoren Lao, Longfei He, Dong Xiao, Jie Zhan, and Aiqin Wang. 2024. "De Novo Assembly and Comparative Analysis of Mitochondrial Genomes of Two Pueraria montana Varieties" International Journal of Molecular Sciences 25, no. 11: 5656. https://doi.org/10.3390/ijms25115656
APA StyleGuo, L., Lao, G., He, L., Xiao, D., Zhan, J., & Wang, A. (2024). De Novo Assembly and Comparative Analysis of Mitochondrial Genomes of Two Pueraria montana Varieties. International Journal of Molecular Sciences, 25(11), 5656. https://doi.org/10.3390/ijms25115656