
Comparative Evaluation of PCR-Based, LAMP and RPA-CRISPR/Cas12a Assays for the Rapid Detection of Diaporthe aspalathi


Abstract
:1. Introduction
2. Results
2.1. Optimization of LAMP and RPA-CRISPR/Cas12a Assays
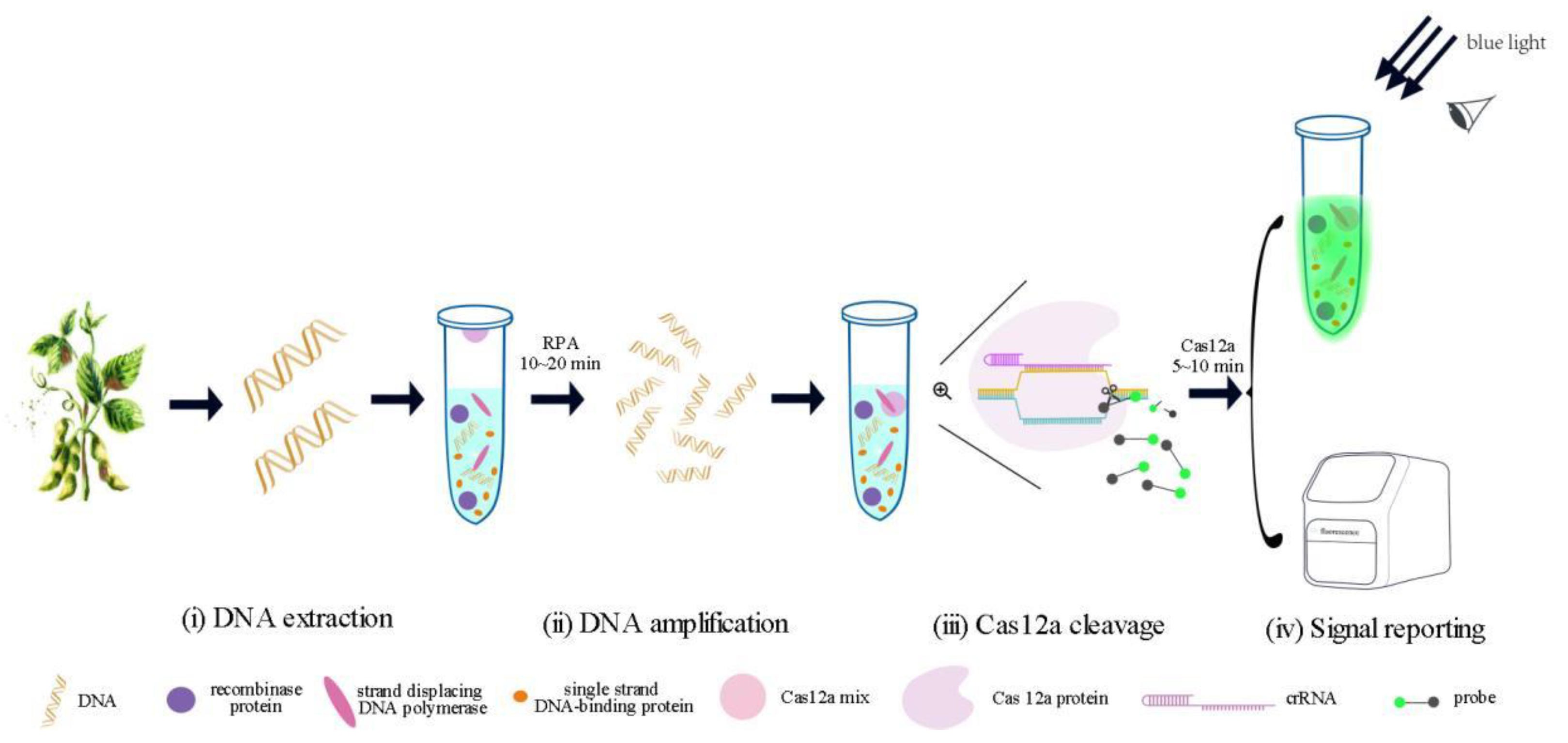
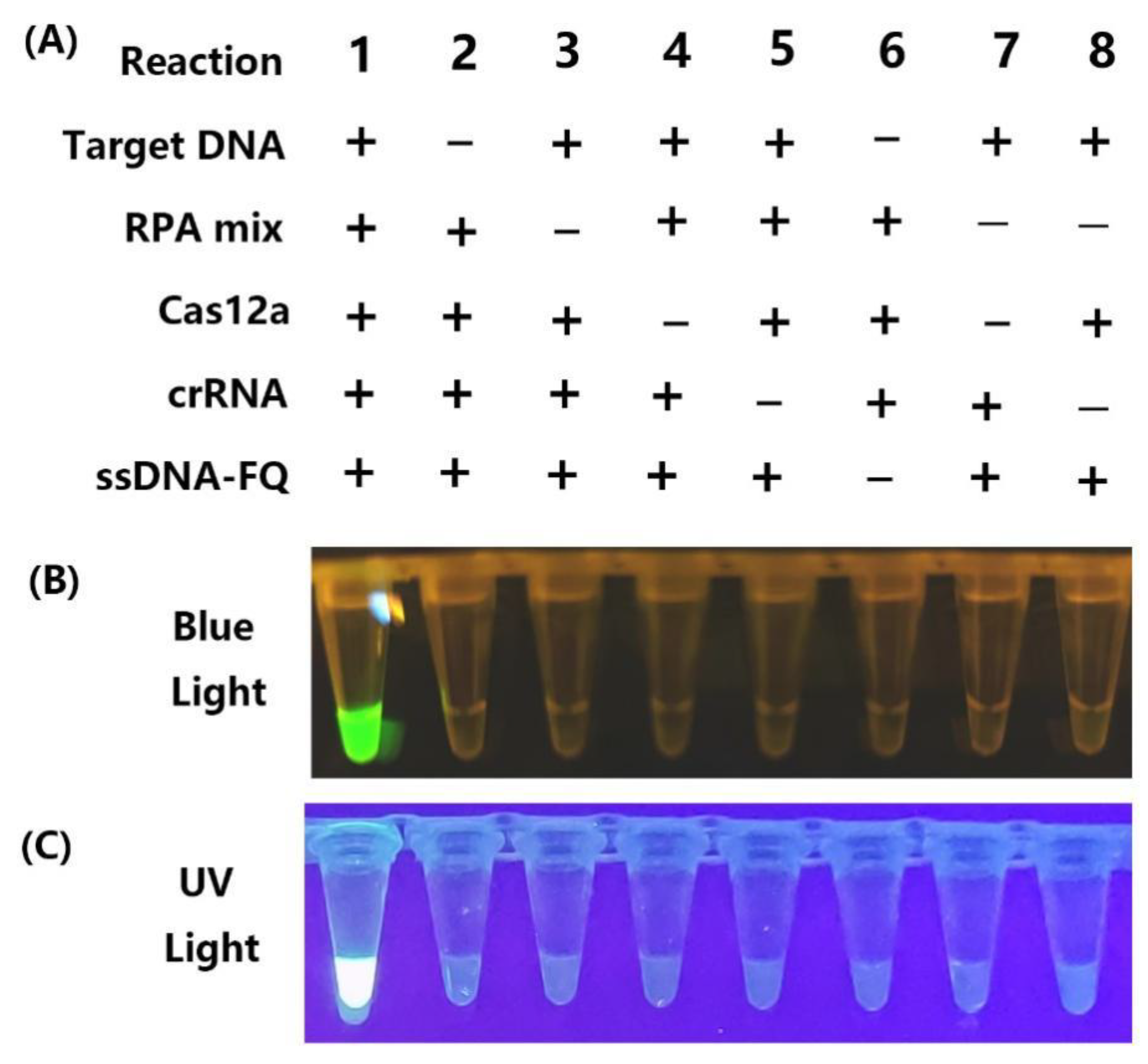
2.2. Establishment of One-Pot RPA-CRISPR/Cas12a Assay
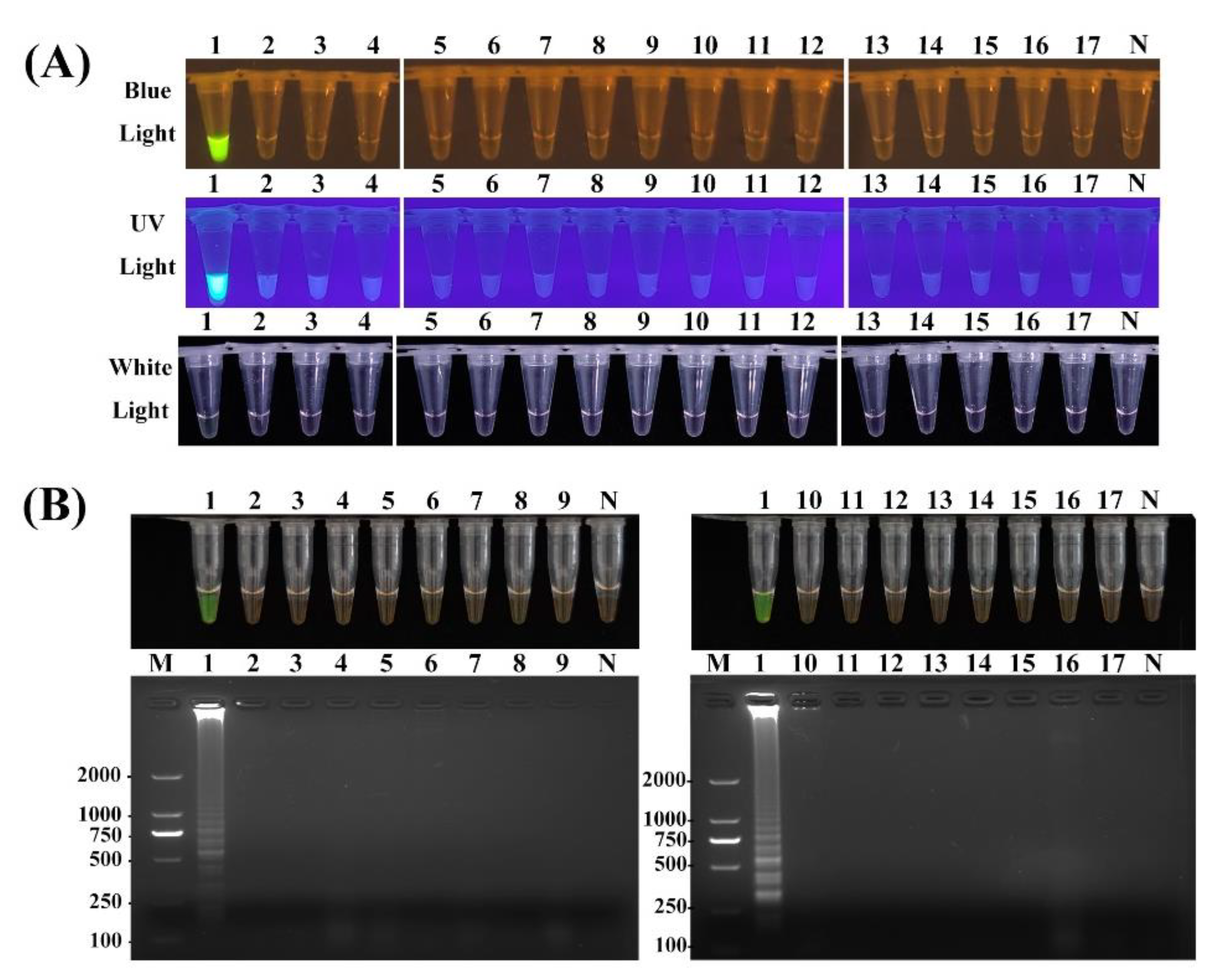
2.3. Specificity of PCR-Based, LAMP and RPA-CRISPR/Cas12a Assays
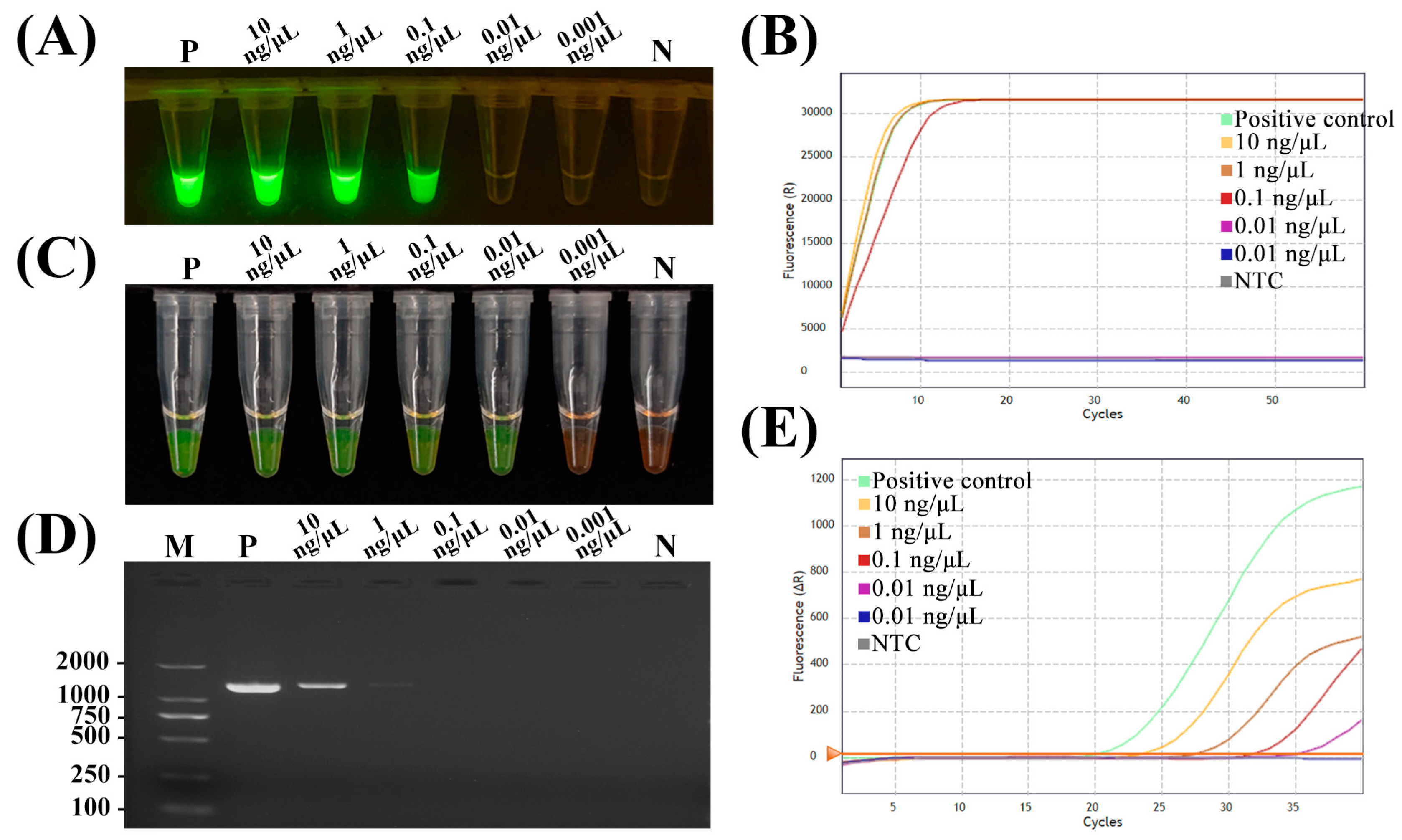
2.4. Comparison of Sensitivity
2.5. Validation of RPA-CRISPR/Cas12a and LAMP Detection Using Disease Samples
3. Discussion
4. Materials and Methods
4.1. Materials and Reagents
4.2. Isolates and DNA Extraction
4.3. Primer and crRNA Design
4.4. Conventional PCR Detection of D. aspalathi
4.5. Real-Time PCR Detection of D. aspalathi
4.6. Optimization of LAMP and RPA-CRISPR/Cas12a Assays
4.7. LAMP Assay
4.8. RPA-CRISPR/Cas12a Assay
4.9. Specificity Test
4.10. Comparison of Sensitivity
4.11. Feasibility of LAMP and RPA-CRISPR/Cas12a Detection Using Disease Samples
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hartman, G.L.; West, E.D.; Herman, T.K. Crops that feed the World 2. Soybean—Orldwide production, use, and constraints caused by pathogens and pests. Food Secur. 2011, 3, 5–17. [Google Scholar] [CrossRef]
- Wilson, R.F. Soybean: Market driven research needs. In Genetics and Genomics of Soybean; Stacey, G., Ed.; Springer: New York, NY, USA, 2008; Volume 2, pp. 3–15. [Google Scholar]
- Backman, P.A.; Weaver, D.B.; Morgan-Jones, G. Soybean stem canker: An emerging disease problem. Plant Dis. 1985, 69, 641–647. [Google Scholar] [CrossRef]
- Campbell, M.A.; Li, Z.; Buck, J.W. Development of southern stem canker disease on soybean seedlings in the greenhouse using a modified toothpick inoculation assay. Crop Prot. 2017, 100, 57–64. [Google Scholar] [CrossRef]
- Wrather, J.A.; Anderson, T.R.; Arsyad, D.M.; Gai, J.; Ploper, L.D.; Porta-Puglia, A.; Ram, H.H.; Yorinori, J.T. Soybean disease loss estimates for the top 10 soybean producing countries in 1994. Plant Dis. 1997, 81, 107–110. [Google Scholar] [CrossRef] [PubMed]
- Hosseini, B.; Voegele, R.T.; Link, T.I. Diagnosis of soybean diseases caused by fungal and oomycete pathogens: Existing methods and new developments. J. Fungi 2023, 9, 587. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, F.A.J.M. Morp11hological and RAPD analyses of Diaporthe phaseolorum from soybean. Mycologia 1996, 88, 425–440. [Google Scholar] [CrossRef]
- Hosseini, B.; Voegele, R.T.; Link, T.I. Establishment of a quadruplex real-time PCR assay to distinguish the fungal pathogens Diaporthe longicolla, D. caulivora, D. eres, and D. novem on soybean. PLoS ONE 2021, 16, e0257225. [Google Scholar] [CrossRef]
- Kontz, B.; Adhikari, S.; Subramanian, S.; Mathew, F.M. Optimization and application of a quantitative polymerase chain reaction assay to detect Diaporthe species in soybean plant tissue. Plant Dis. 2016, 100, 1669–1676. [Google Scholar] [CrossRef]
- Zhang, A.W.; Hartman, G.L.; Curio-Penny, B.; Pedersen, W.L.; Becker, K.B. Molecular detection of Diaporthe phaseolorum and Phomopsis longicolla from soybean seeds. Phytopathology 1999, 89, 796–804. [Google Scholar] [CrossRef]
- Khan, M.; Li, B.; Jiang, Y.; Weng, Q.; Chen, Q. Evaluation of different PCR-based assays and lamp method for rapid detection of Phytophthora infestans by targeting the Ypt1 gene. Front. Microbiol. 2017, 8, 1920. [Google Scholar] [CrossRef]
- Soroka, M.; Wasowicz, B.; Rymaszewska, A. Loop-mediated isothermal amplification (LAMP): The better sibling of PCR? Cells 2021, 10, 1931. [Google Scholar] [CrossRef] [PubMed]
- Dai, T.; Yang, X.; Hu, T.; Jiao, B.; Xu, Y.; Zheng, X.; Shen, D. comparative evaluation of a novel recombinase polymerase amplification-lateral flow dipstick (RPA-LFD) assay, LAMP, conventional PCR, and leaf-disc baiting methods for detection of Phytophthora sojae. Front. Microbiol. 2019, 10, 1884. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Yin, K.; Li, Z.; Lalla, R.V.; Ballesteros, E.; Sfeir, M.M.; Liu, C. Ultrasensitive and visual detection of SARS-CoV-2 using all-in-one dual CRISPR-Cas12a assay. Nat. Commun. 2020, 11, 4711. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Macdonald, J.; von Stetten, F. Review: A comprehensive summary of a decade development of the recombinase polymerase amplification. Analyst 2018, 144, 31–67. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.S.; Ma, E.; Harrington, L.B.; Da Costa, M.; Tian, X.; Palefsky, J.M.; Doudna, J.A. CRISPR-Cas12a target binding unleashes indiscriminate single-stranded DNase activity. Science 2018, 360, 436–439. [Google Scholar] [CrossRef] [PubMed]
- Eisenstein, M. Seven technologies to watch in 2022. Nature 2022, 601, 658–661. [Google Scholar] [CrossRef] [PubMed]
- Gootenberg, J.S.; Abudayyeh, O.O.; Lee, J.W.; Essletzbichler, P.; Dy, A.J.; Joung, J.; Verdine, V.; Donghia, N.; Daringer, N.M.; Freije, C.A.; et al. Nucleic acid detection with CRISPR-Cas13a/C2c2. Science 2017, 356, 438–442. [Google Scholar] [CrossRef] [PubMed]
- Li, S.Y.; Cheng, Q.X.; Liu, J.K.; Nie, X.Q.; Zhao, G.P.; Wang, J. CRISPR-Cas12a has both cis- and trans-cleavage activities on single-stranded DNA. Cell Res. 2018, 28, 491–493. [Google Scholar] [CrossRef] [PubMed]
- Wheatley, M.S.; Yang, Y. Versatile Applications of the CRISPR/Cas toolkit in plant pathology and disease management. Phytopathology 2021, 111, 1080–1090. [Google Scholar] [CrossRef]
- Kellner, M.J.; Koob, J.G.; Gootenberg, J.S.; Abudayyeh, O.O.; Zhang, F. SHERLOCK: Nucleic acid detection with CRISPR nucleases. Nat. Protoc. 2019, 14, 2986–3012. [Google Scholar] [CrossRef]
- Pickar-Oliver, A.; Gersbach, C.A. The next generation of CRISPR-Cas technologies and applications. Nat. Rev. Mol. Cell Biol. 2019, 20, 490–507. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.Y.; Du, Y.C.; Wang, D.X.; Ma, J.Y.; Tang, A.N.; Kong, D.M. Signal amplification and output of CRISPR/Cas-based biosensing systems: A review. Anal. Chim. Acta 2021, 1185, 338882. [Google Scholar] [CrossRef] [PubMed]
- Aman, R.; Mahas, A.; Marsic, T.; Hassan, N.; Mahfouz, M.M. Efficient, rapid, and sensitive detection of plant RNA viruses with one-pot RT-RPA-CRISPR/Cas12a assay. Front. Microbiol. 2020, 11, 610872. [Google Scholar] [CrossRef] [PubMed]
- Hao, J.; Xie, L.; Yang, T.; Huo, Z.; Liu, G.; Liu, Y.; Xiong, W.; Zeng, Z. Naked-eye on-site detection platform for Pasteurella multocida based on the CRISPR-Cas12a system coupled with recombinase polymerase amplification. Talanta 2023, 255, 124220. [Google Scholar] [CrossRef]
- Liu, S.; Huang, S.; Li, F.; Sun, Y.; Fu, J.; Xiao, F.; Jia, N.; Huang, X.; Sun, C.; Zhou, J.; et al. Rapid detection of Pseudomonas aeruginosa by recombinase polymerase amplification combined with CRISPR-Cas12a biosensing system. Front. Cell Infect. Microbiol. 2023, 13, 1239269. [Google Scholar] [CrossRef] [PubMed]
- Peng, S.; Tan, Z.; Chen, S.; Lei, C.; Nie, Z. Integrating CRISPR-Cas12a with a DNA circuit as a generic sensing platform for amplified detection of microRNA. Chem. Sci. 2020, 11, 7362–7368. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.X.; Peng, Y.; Hua, K.Y.; Deng, Y.F.; Bellizzi, M.; Gupta, D.R.; Mahmud, N.U.; Urashima, A.S.; Paul, S.K.; Peterson, G.; et al. Rapid detection of wheat blast pathogen Magnaporthe oryzae triticum pathotype using genome-specific primers and Cas12a-mediated technology. Engineering 2021, 7, 10. [Google Scholar] [CrossRef]
- Bhat, A.I.; Aman, R.; Mahfouz, M. Onsite detection of plant viruses using isothermal amplification assays. Plant Biotechnol. J. 2022, 20, 1859–1873. [Google Scholar] [CrossRef]
- Wheatley, M.S.; Duan, Y.P.; Yang, Y. Highly sensitive and rapid detection of citrus huanglongbing pathogen (Candidatus liberibacter asiaticus) using Cas12a-Based methods. Phytopathology 2021, 111, 2375–2382. [Google Scholar] [CrossRef]
- Sun, X.; Lei, R.; Zhang, H.; Chen, W.; Jia, Q.; Guo, X.; Zhang, Y.; Wu, P.; Wang, X. Rapid and sensitive detection of two fungal pathogens in soybeans using the recombinase polymerase amplification/CRISPR-Cas12a method for potential on-site disease diagnosis. Pest. Manag. Sci. 2024, 80, 1168–1181. [Google Scholar] [CrossRef]
- Lobato, I.M.; O’Sullivan, C.K. Recombinase polymerase amplification: Basics, applications and recent advances. Trends Analyt. Chem. 2018, 98, 19–35. [Google Scholar] [CrossRef] [PubMed]
- Silva, G.; Oyekanmi, J.; Nkere, C.K.; Bömer, M.; Kumar, P.L.; Seal, S.E. Rapid detection of potyviruses from crude plant extracts. Anal. Biochem. 2018, 546, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Xiong, D.; Dai, W.; Gong, J.; Li, G.; Liu, N.; Wu, W.; Pan, J.; Chen, C.; Jiao, Y.; Deng, H.; et al. Rapid detection of SARS-CoV-2 with CRISPR-Cas12a. PLoS Biol. 2020, 18, e3000978. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Pandian, V.; Mauk, M.G.; Bau, H.H.; Cherry, S.; Tisi, L.C.; Liu, C. Smartphone-based mobile detection platform for molecular diagnostics and spatiotemporal disease mapping. Analyt Chem. 2018, 90, 4823–4831. [Google Scholar] [CrossRef]
- Yu, T.; Zhang, S.; Matei, R.; Marx, W.; Beisel, C.L.; Wei, Q. Coupling smartphone and CRISPR–Cas12a for digital and multiplexed nucleic acid detection. AIChE J. 2021, 67, e17365. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1.00 × 102 ng μL−1 | 1.00 × 101 ng μL−1 | 1.00 × 100 ng μL−1 | 1.00 × 10−1 ng μL−1 | 1.00 × 10−2 ng μL−1 | 1.00 × 10−3 ng μL−1 | |
|---|---|---|---|---|---|---|
| Conventional PCR | + | + | + | − | − | − |
| qPCR | + | + | + | + | − | − |
| LAMP | + | + | + | + | + | − |
| RPA-Cas12a | + | + | + | + | − | − |
| Primer | Sequence | Reference |
|---|---|---|
| Da-1F | GAATCCTTGTGGGTATTTG | For PCR |
| Da-1R | GTCAATATGCTATGGTCAC | |
| Da-Q3-F1 | GCGATTTGTGGGATTGAC | For qPCR |
| Da-Q3-R1 | CATGCTGAATAGGAGAGG | |
| Da-Q3 | ROX-CCGTCAAGGCTACACTCGTCG-BHQ2 | |
| D1-F3 | GTGCCTCGAACGTTGTCTC | For LAMP |
| D1-B3 | GGCGACACAAACACGGAC | |
| D1-FIP | ACCCGAGGGGGAAGTTCAAACTTCGTAGTTCTGCCAAGAAGC | |
| D1-BIP | CTGCGGTCCCTGGAGAGGATACTTCATGTAGCGCCCGA | |
| D-RPA-1F | CTTTGTCTGAGGCTGAACCCCAGGCGTATT | For RPA |
| D-PRA-1R | CGCATCAACAACCAAGATCACGGGCAAGAC | |
| D-Cas-C | UAAUUUCUACUAAGUGUAGAUCCGGAUGCGUACAACAUGACA | For Cas12a cleavage |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dong, J.; Feng, W.; Lin, M.; Chen, S.; Liu, X.; Wang, X.; Chen, Q. Comparative Evaluation of PCR-Based, LAMP and RPA-CRISPR/Cas12a Assays for the Rapid Detection of Diaporthe aspalathi. Int. J. Mol. Sci. 2024, 25, 5773. https://doi.org/10.3390/ijms25115773
Dong J, Feng W, Lin M, Chen S, Liu X, Wang X, Chen Q. Comparative Evaluation of PCR-Based, LAMP and RPA-CRISPR/Cas12a Assays for the Rapid Detection of Diaporthe aspalathi. International Journal of Molecular Sciences. 2024; 25(11):5773. https://doi.org/10.3390/ijms25115773
Chicago/Turabian StyleDong, Jiali, Wanzhen Feng, Mingze Lin, Shuzhe Chen, Xiaozhen Liu, Xiaodan Wang, and Qinghe Chen. 2024. "Comparative Evaluation of PCR-Based, LAMP and RPA-CRISPR/Cas12a Assays for the Rapid Detection of Diaporthe aspalathi" International Journal of Molecular Sciences 25, no. 11: 5773. https://doi.org/10.3390/ijms25115773
APA StyleDong, J., Feng, W., Lin, M., Chen, S., Liu, X., Wang, X., & Chen, Q. (2024). Comparative Evaluation of PCR-Based, LAMP and RPA-CRISPR/Cas12a Assays for the Rapid Detection of Diaporthe aspalathi. International Journal of Molecular Sciences, 25(11), 5773. https://doi.org/10.3390/ijms25115773