
Prediction of Protein–Protein Interactions Based on Integrating Deep Learning and Feature Fusion †


Abstract
1. Introduction
2. Results and Discussion
2.1. Datasets
2.2. Evaluation Metrics
2.3. Effect of Amino Acid Embedding Vector Dimensions
2.4. Comparison between APAAC and APAACplus Descriptors
2.5. Effect of Feature Fusion Models
2.6. Effect of the Channel Weight
2.7. Comparison with Other Protein Sequence Embedding Approaches
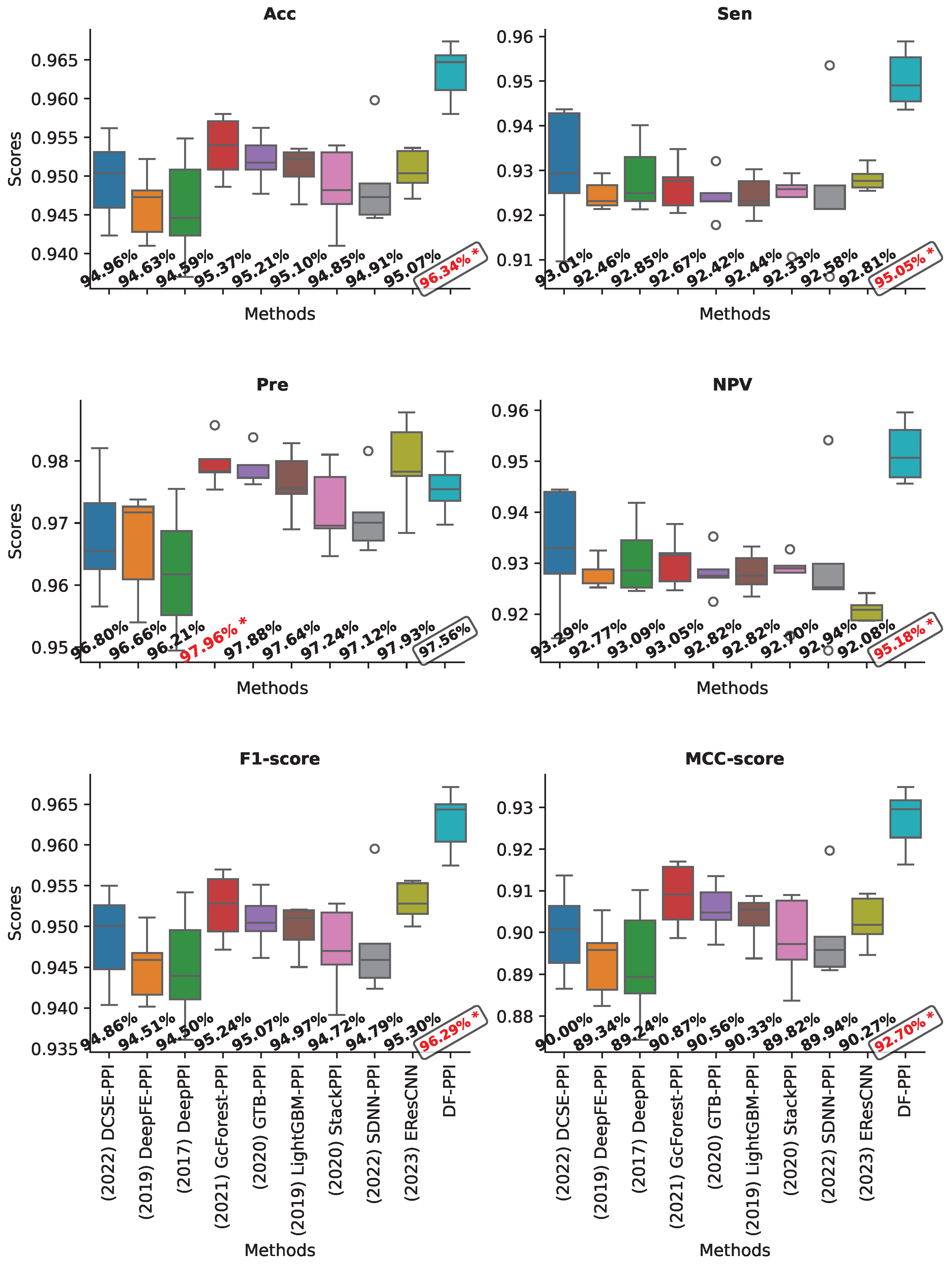
2.8. Validation on the Yeast Core Dataset
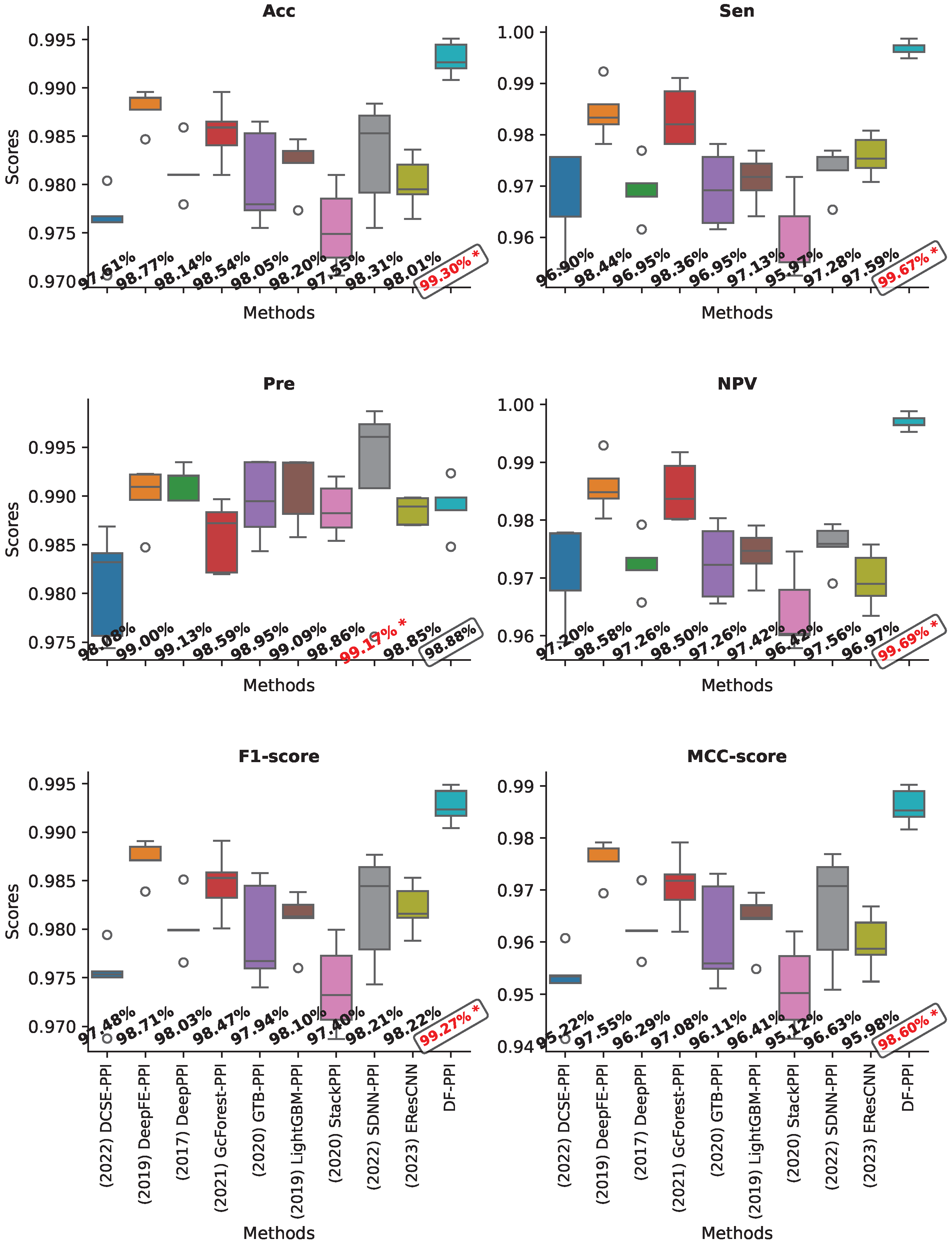
2.9. Validating on the Human Dataset
2.10. Testing on PPI Cross-Species Datasets
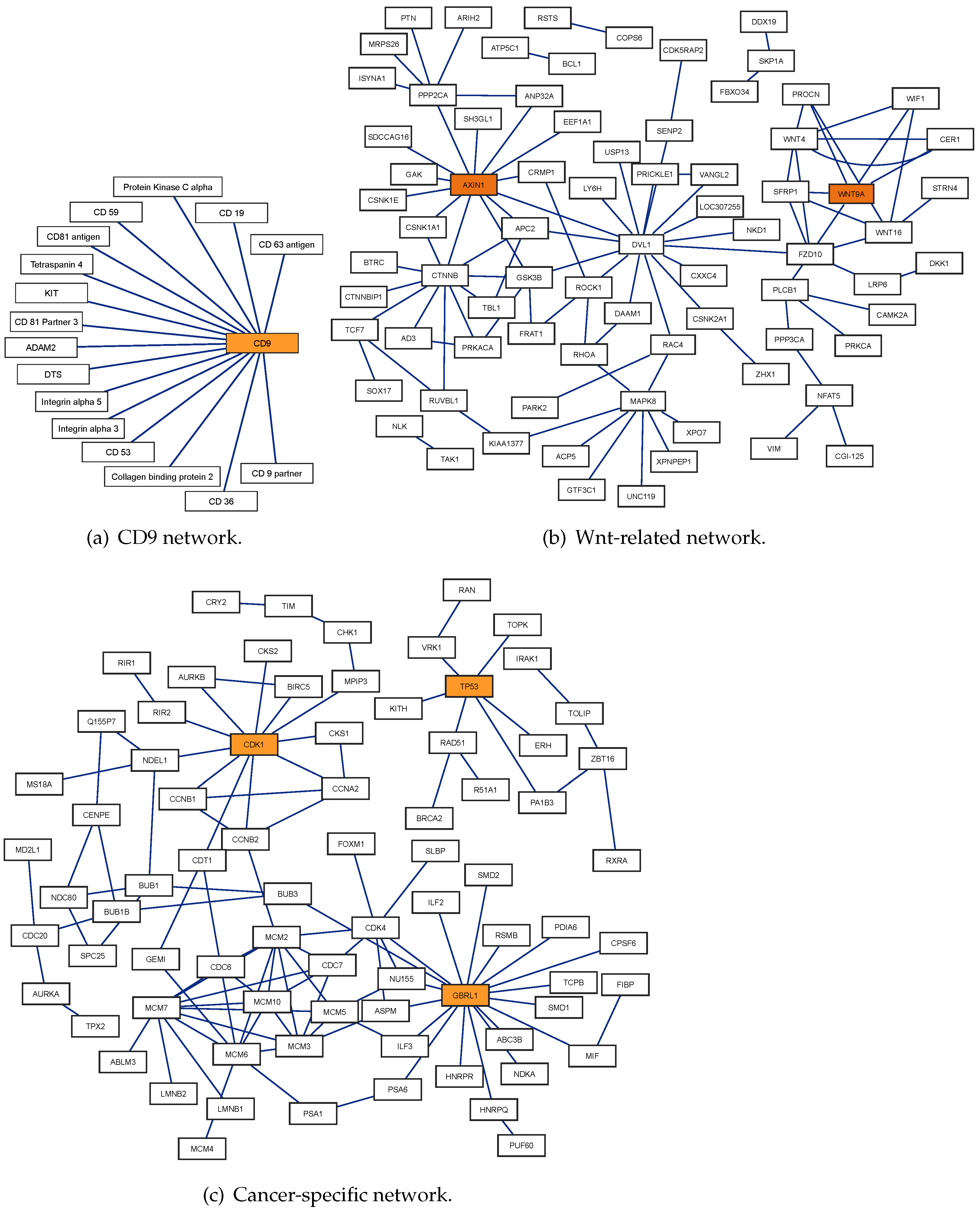
2.11. Testing on PPI Network Datasets
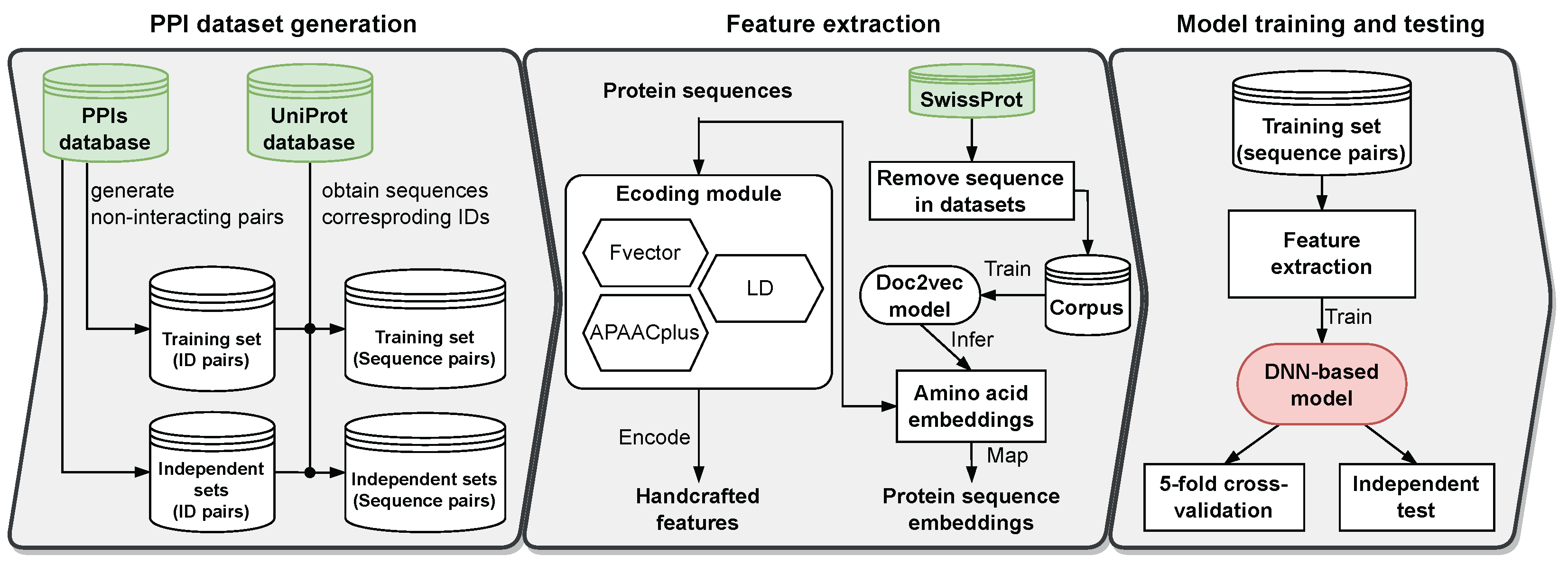
3. Materials and Methods
3.1. Handcrafted Features
3.1.1. Local Descriptor
3.1.2. F-Vector Descriptor
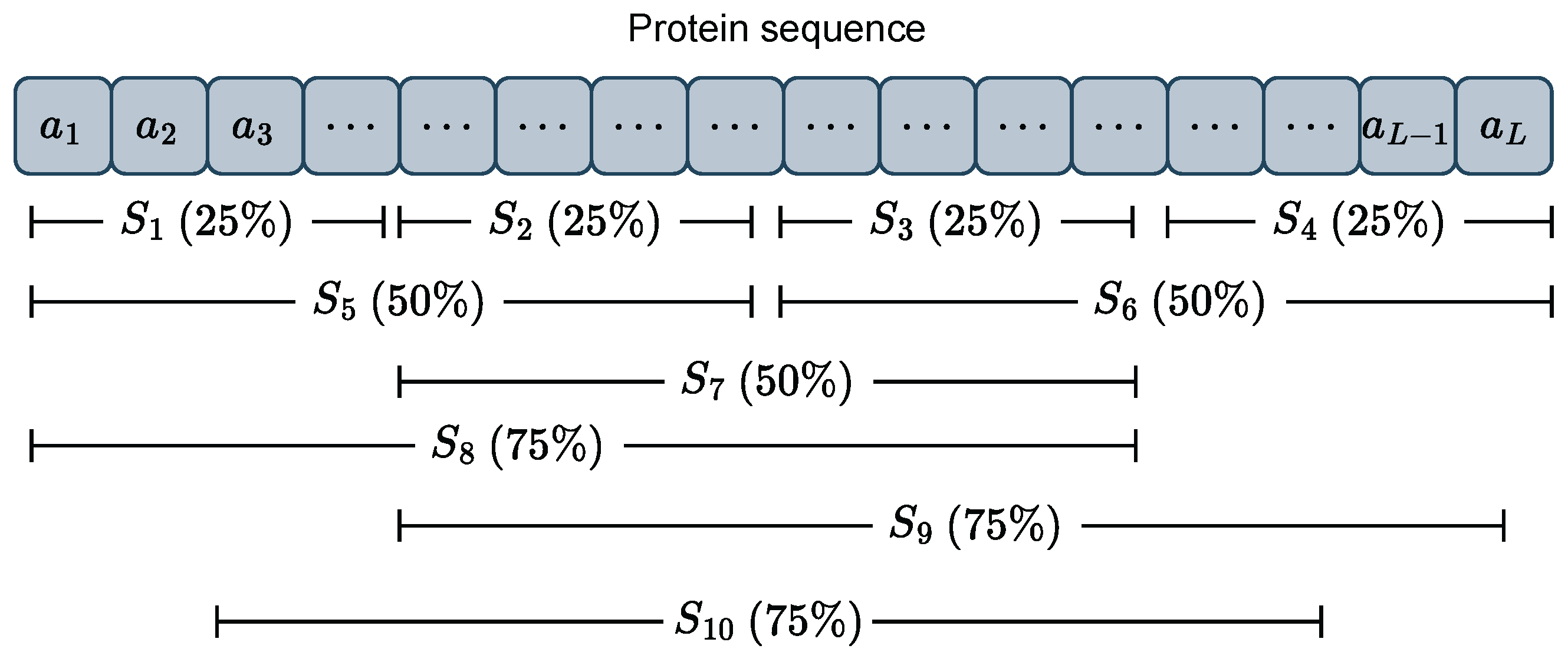
3.1.3. APAACplus Descriptor
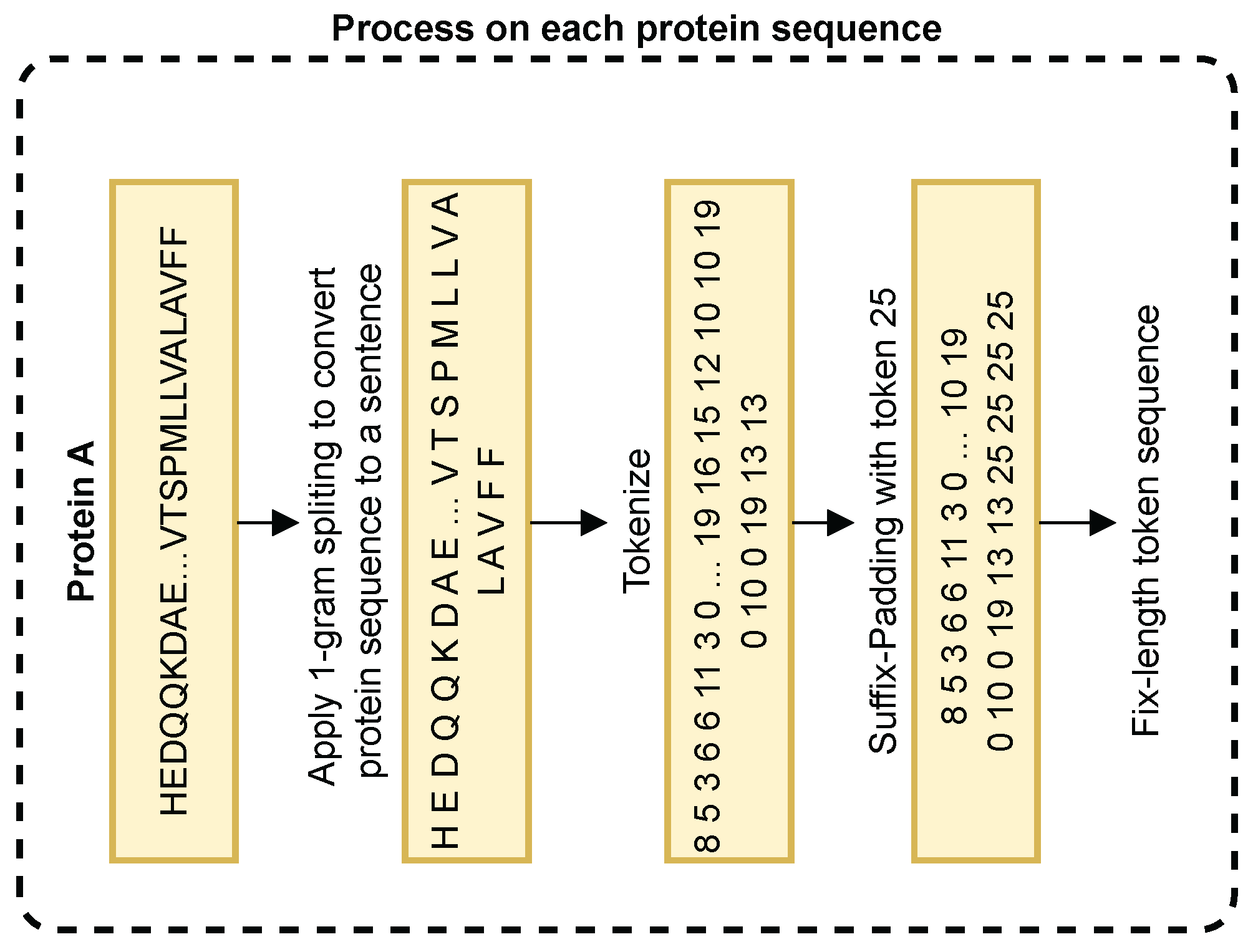
3.2. Protein Sequence Embedding
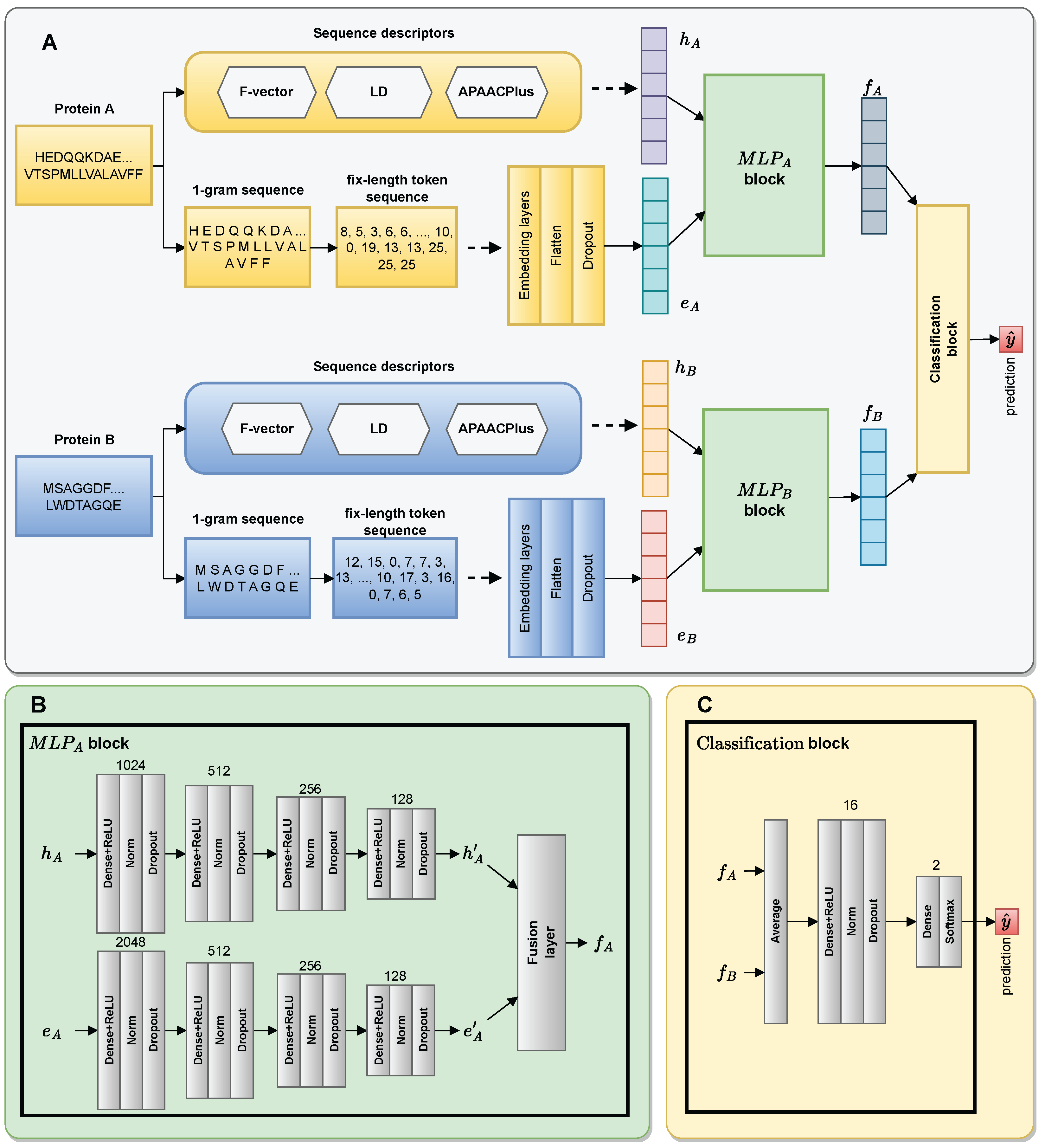
3.3. The Architecture of DF-PPI
3.3.1. Embedding Layer
3.3.2. MLP Blocks
3.3.3. Fusion Layer
3.3.4. Classification Block
3.4. Training the Model
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| PPI | Protein–Protein Interaction |
| APAAC | Amphiphilic Pseudo-Amino Acid Composition |
| LD | Local Descriptor |
| C, T, D | Composition, Transition, Distribution |
| PseAAC | Pseudo-Amino Acid Composition |
| PSSM | Position-Specific Score Matrix |
| ML | Machine Learning |
| DL | Deep Learning |
| CNN | Convolutional Neural Network |
| RNN | Recurrent Neural Network |
| GRU | Gated Recurrent Unit |
| MLP | Multilayer Perceptron |
| NLP | Natural Language Processing |
| SGD | Stochastic Gradient Descent |
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group Index | Amino Acid |
|---|---|
| Group 1 | Alanine (A), Glycine (G), Valine (V) |
| Group 2 | Cysteine (C) |
| Group 3 | Aspartic acid (D), Glutamic (E) |
| Group 4 | Phenylalanine (F), Proline (P), Isoleucine (I), Leucine (L) |
| Group 5 | Histidine (H), Glutamine (Q), Asparagine (N), Tryptophan (W) |
| Group 6 | Lysine (K), Arginine (R) |
| Group 7 | Methionine (M), Serine (S), Tyrosine (Y), Threonine (T) |
| Group Index | ||||
|---|---|---|---|---|
References
- Li, Y.; Golding, G.B.; Ilie, L. DELPHI: Accurate deep ensemble model for protein interaction sites prediction. Bioinformatics 2021, 37, 896–904. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Sun, S.; Hu, C.; Yao, Y.; Yan, Y.; Zhang, Y. DeepPPI: Boosting Prediction of Protein-Protein Interactions with Deep Neural Networks. J. Chem. Inf. Model. 2017, 57, 1499–1510. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Han, P.; Wang, G.; Chen, W.; Wang, S.; Song, T. SDNN-PPI: Self-attention with deep neural network effect on protein-protein interaction prediction. BMC Genom. 2022, 23, 474. [Google Scholar] [CrossRef]
- Chen, W.; Wang, S.; Song, T.; Li, X.; Han, P.; Gao, C. DCSE:Double-Channel-Siamese-Ensemble model for protein protein interaction prediction. BMC Genom. 2022, 23, 555. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Chen, C.; Li, S.; Wang, C.; Zhou, W.; Yu, B. Prediction of protein-protein interactions based on ensemble residual convolutional neural network. Comput. Biol. Med. 2023, 152, 106471. [Google Scholar] [CrossRef]
- Guo, Y.; Yu, L.; Wen, Z.; Li, M. Using support vector machine combined with auto covariance to predict protein–protein interactions from protein sequences. Nucleic Acids Res. 2008, 36, 3025–3030. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Xia, J.F.; Gui, J. Prediction of Protein-Protein Interactions from Protein Sequence Using Local Descriptors. Protein Pept. Lett. 2010, 17, 1085–1090. [Google Scholar] [CrossRef]
- You, Z.H.; Chan, K.C.; Hu, P. Predicting protein-protein interactions from primary protein sequences using a novel multi-scale local feature representation scheme and the random forest. PLoS ONE 2015, 10, e0125811. [Google Scholar] [CrossRef] [PubMed]
- You, Z.H.; Zhu, L.; Zheng, C.H.; Yu, H.J.; Deng, S.P.; Ji, Z. Prediction of protein-protein interactions from amino acid sequences using a novel multi-scale continuous and discontinuous feature set. BMC Bioinform. 2014, 15, S9. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Yu, H.; Ding, Y.; Guo, F.; Gong, X.J. Multi-scale encoding of amino acid sequences for predicting protein interactions using gradient boosting decision tree. PLoS ONE 2017, 12, 0181426. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Zhang, Q.; Ma, Q.; Yu, B. LightGBM-PPI: Predicting protein-protein interactions through LightGBM with multi-information fusion. Chemom. Intell. Lab. Syst. 2019, 191, 54–64. [Google Scholar] [CrossRef]
- Yu, B.; Chen, C.; Zhou, H.; Liu, B.; Ma, Q. GTB-PPI: Predict Protein–protein Interactions Based on L1-regularized Logistic Regression and Gradient Tree Boosting. Genom. Proteom. Bioinform. 2020, 18, 582–592. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.; Chen, C.; Wang, X.; Yu, Z.; Ma, A.; Liu, B. Prediction of protein–protein interactions based on elastic net and deep forest. Expert Syst. Appl. 2021, 176, 114876. [Google Scholar] [CrossRef]
- Hashemifar, S.; Neyshabur, B.; Khan, A.A.; Xu, J. Predicting protein-protein interactions through sequence-based deep learning. Bioinformatics 2018, 34, i802–i810. [Google Scholar] [CrossRef] [PubMed]
- Stringer, B.; de Ferrante, H.; Abeln, S.; Heringa, J.; Feenstra, K.A.; Haydarlou, R. PIPENN: Protein interface prediction from sequence with an ensemble of neural nets. Bioinformatics 2022, 38, 2111–2118. [Google Scholar] [CrossRef]
- Aybey, E.; Gümüş, Ö. SENSDeep: An Ensemble Deep Learning Method for Protein–Protein Interaction Sites Prediction. Interdiscip. Sci. Comput. Life Sci. 2022, 15, 55–87. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Wang, L.; Jia, H.; Tong, X.; Li, F. A Sequence-to-Sequence Deep Learning Architecture Based on Bidirectional GRU for Type Recognition and Time Location of Combined Power Quality Disturbance. IEEE Trans. Ind. Inform. 2019, 15, 4481–4493. [Google Scholar] [CrossRef]
- Jung, S.; Moon, J.; Park, S.; Hwang, E. An Attention-Based Multilayer GRU Model for Multistep-Ahead Short-Term Load Forecasting. Sensors 2021, 21, 1639. [Google Scholar] [CrossRef] [PubMed]
- Asgari, E.; Mofrad, M.R.K. Continuous Distributed Representation of Biological Sequences for Deep Proteomics and Genomics. PLoS ONE 2015, 10, e0141287. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Du, X.; Diao, Y.; Zhu, H. An integration of deep learning with feature embedding for protein–protein interaction prediction. PeerJ 2019, 7, e7126. [Google Scholar] [CrossRef] [PubMed]
- Mikolov, T.; Chen, K.; Corrado, G.; Dean, J. Efficient estimation of word representations in vector space. arXiv 2013, arXiv:1301.3781. [Google Scholar]
- Wang, Y.; You, Z.H.; Yang, S.; Li, X.; Jiang, T.H.; Zhou, X. A High Efficient Biological Language Model for Predicting Protein–Protein Interactions. Cells 2019, 8, 122. [Google Scholar] [CrossRef] [PubMed]
- Kudo, T. Subword Regularization: Improving Neural Network Translation Models with Multiple Subword Candidates; Association for Computational Linguistics: Stroudsburg, PA, USA, 2018; pp. 66–75. [Google Scholar] [CrossRef]
- Yang, X.; Yang, S.; Li, Q.; Wuchty, S.; Zhang, Z. Prediction of human-virus protein-protein interactions through a sequence embedding-based machine learning method. Comput. Struct. Biotechnol. J. 2020, 18, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Le, Q.V.; Mikolov, T. Distributed Representations of Sentences and Documents. In Proceedings of the International Conference on Machine Learning, Beijing, China, 21–26 June 2014. [Google Scholar]
- Brandes, N.; Ofer, D.; Peleg, Y.; Rappoport, N.; Linial, M. ProteinBERT: A universal deep-learning model of protein sequence and function. Bioinformatics 2022, 38, 2102–2110. [Google Scholar] [CrossRef] [PubMed]
- Kong, M.; Zhang, Y.; Xu, D.; Chen, W.; Dehmer, M. FCTP-WSRC: Protein–Protein Interactions Prediction via Weighted Sparse Representation Based Classification. Front. Genet. 2020, 11, 18. [Google Scholar] [CrossRef] [PubMed]
- Chou, K.C. Using amphiphilic pseudo amino acid composition to predict enzyme subfamily classes. Bioinformatics 2005, 21, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Zhang, J.; Luo, X.; Zhu, W.; Yu, K.; Chen, K.; Li, Y.; Jiang, H. Predicting protein-protein interactions based only on sequences information. Nucleic Acids Res. 2007, 104, 4337–4341. [Google Scholar] [CrossRef] [PubMed]
- Xenarios, I. DIP, the Database of Interacting Proteins: A research tool for studying cellular networks of protein interactions. Nucleic Acids Res. 2002, 30, 303–305. [Google Scholar] [CrossRef]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.A.; You, Z.H.; Gao, X.; Wong, L.; Wang, L. Using Weighted Sparse Representation Model Combined with Discrete Cosine Transformation to Predict Protein-Protein Interactions from Protein Sequence. BioMed Res. Int. 2015, 2015, 902198. [Google Scholar] [CrossRef]
- Le, N.Q.K.; Do, D.T.; Nguyen, T.T.D.; Le, Q.A. A sequence-based prediction of Kruppel-like factors proteins using XGBoost and optimized features. Gene 2021, 787, 145643. [Google Scholar] [CrossRef] [PubMed]
- Kha, Q.H.; Le, V.H.; Hung, T.N.K.; Nguyen, N.T.K.; Le, N.Q.K. Development and Validation of an Explainable Machine Learning-Based Prediction Model for Drug–Food Interactions from Chemical Structures. Sensors 2023, 23, 3962. [Google Scholar] [CrossRef] [PubMed]
- Kudo, T.; Richardson, J. SentencePiece: A simple and language independent subword tokenizer and detokenizer for Neural Text Processing. arXiv 2018, arXiv:1808.06226. [Google Scholar]
- Chen, C.; Zhang, Q.; Yu, B.; Yu, Z.; Lawrence, P.J.; Ma, Q.; Zhang, Y. Improving protein-protein interactions prediction accuracy using XGBoost feature selection and stacked ensemble classifier. Comput. Biol. Med. 2020, 123, 103899. [Google Scholar] [CrossRef] [PubMed]
- Rapposelli, S.; Gaudio, E.; Bertozzi, F.; Gul, S. Editorial: Protein–Protein Interactions: Drug Discovery for the Future. Front. Chem. 2021, 9, 811190. [Google Scholar] [CrossRef] [PubMed]
- Dimitrakopoulos, G.N.; Vrahatis, A.G.; Exarchos, T.P.; Krokidis, M.G.; Vlamos, P. Drug and Protein Interaction Network Construction for Drug Repurposing in Alzheimer’s Disease. Future Pharmacol. 2023, 3, 731–741. [Google Scholar] [CrossRef]
- Frolikova, M.; Manaskova-Postlerova, P.; Cerny, J.; Jankovicova, J.; Simonik, O.; Pohlova, A.; Secova, P.; Antalikova, J.; Dvorakova-Hortova, K. CD9 and CD81 Interactions and Their Structural Modelling in Sperm Prior to Fertilization. Int. J. Mol. Sci. 2018, 19, 1236. [Google Scholar] [CrossRef] [PubMed]
- Nie, X.; Liu, H.; Liu, L.; Wang, Y.D.; Chen, W.D. Emerging Roles of Wnt Ligands in Human Colorectal Cancer. Front. Oncol. 2020, 10, 01341. [Google Scholar] [CrossRef] [PubMed]
- Qiu, L.; Sun, Y.; Ning, H.; Chen, G.; Zhao, W.; Gao, Y. The scaffold protein AXIN1: Gene ontology, signal network, and physiological function. Cell Commun. Signal. 2024, 22, 77. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, X. Targeting the Wnt/β-catenin signaling pathway in cancer. J. Hematol. Oncol. 2020, 13, 165. [Google Scholar] [CrossRef] [PubMed]
- Enserink, J.M.; Kolodner, R.D. An overview of Cdk1-controlled targets and processes. Cell Div. 2010, 5, 11. [Google Scholar] [CrossRef] [PubMed]
- Marei, H.E.; Althani, A.; Afifi, N.; Hasan, A.; Caceci, T.; Pozzoli, G.; Morrione, A.; Giordano, A.; Cenciarelli, C. p53 signaling in cancer progression and therapy. Cancer Cell Int. 2021, 21, 703. [Google Scholar] [CrossRef] [PubMed]
- The UniProt Consortium. UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2016, 45, D158–D169. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.B.; Chen, C.; Li, Z.C.; Zou, X.Y. Using Chou’s amphiphilic pseudo-amino acid composition and support vector machine for prediction of enzyme subfamily classes. J. Theor. Biol. 2007, 248, 546–551. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.Y.; You, Z.H.; Li, S.; Zhu, Z. Using Chou’s amphiphilic Pseudo-Amino Acid Composition and Extreme Learning Machine for prediction of Protein-protein interactions. In Proceedings of the 2014 International Joint Conference on Neural Networks, Beijing, China, 6–11 July 2014; IEEE: Piscataway, NJ, USA, 2014; pp. 2952–2956. [Google Scholar] [CrossRef]
- Chou, K.C.; Cai, Y.D. Prediction of Membrane Protein Types by Incorporating Amphipathic Effects. J. Chem. Inf. Model. 2005, 45, 407–413. [Google Scholar] [CrossRef]
- Rehurek, R.; Sojka, P. Gensim–Python Framework for Vector Space Modelling; NLP Centre, Faculty of Informatics, Masaryk University: Brno, Czech Republic, 2011; Volume 3. [Google Scholar]
- Shrestha, A.; Mahmood, A. Review of deep learning algorithms and architectures. IEEE Access 2019, 22, 53040–53065. [Google Scholar] [CrossRef]
- Tran, H.N.; Nguyen, P.X.Q.; Peng, X.; Wang, J. An integration of deep learning with feature fusion for protein-protein interaction prediction. In Proceedings of the 2022 IEEE International Conference on Bioinformatics and Biomedicine (BIBM), Las Vegas, NV, USA, 6–8 December 2022. [Google Scholar] [CrossRef]
- Ioffe, S.; Szegedy, C. Batch normalization: Accelerating deep network training by reducing internal covariate shift. In Proceedings of the International Conference on Machine Learning, Lille, France, 7–9 July 2015; Volume 1. [Google Scholar]
- Garbin, C.; Zhu, X.; Marques, O. Dropout vs. batch normalization: An empirical study of their impact to deep learning. Multimed. Tools Appl. 2020, 79, 12777–12815. [Google Scholar] [CrossRef]
- Goodfellow, I.; Bengio, Y.; Courville, A. Deep Learning; MIT Press: Cambridge, MA, USA, 2016; Available online: http://www.deeplearningbook.org (accessed on 20 February 2024).
- Kingma, D.P.; Ba, J.L. Adam: A method for stochastic optimization. arXiv 2015, arXiv:1412.6980. [Google Scholar]
- Park, J.; Yi, D.; Ji, S. A Novel Learning Rate Schedule in Optimization for Neural Networks and It’s Convergence. Symmetry 2020, 12, 660. [Google Scholar] [CrossRef]

| Dimension | Acc | Sen | Pre | NPV | F1 | MCC | AUC | AUPR |
|---|---|---|---|---|---|---|---|---|
| 8 | 95.90 ± 0.23 | 94.05 ± 0.39 | 97.66± 0.29 | 94.26 ± 0.35 | 95.82 ± 0.24 | 91.86 ± 0.46 | 98.70 ± 0.07 | 99.04 ± 0.04 |
| 16 | 96.17 ± 0.28 | 94.71 ± 0.49 | 97.55 ± 0.22 | 94.86 ± 0.45 | 96.11 ± 0.29 | 92.37 ± 0.54 | 98.86 ± 0.06 | 99.15 ± 0.06 |
| 32 | 96.34± 0.34 | 95.05± 0.58 | 97.56 ± 0.39 | 95.18± 0.54 | 96.29± 0.35 | 92.70± 0.67 | 98.87± 0.08 | 99.16± 0.04 |
| 64 | 95.91 ± 0.27 | 94.28 ± 0.43 | 97.45 ± 0.43 | 94.46 ± 0.38 | 95.84 ± 0.27 | 91.86 ± 0.53 | 98.65 ± 0.07 | 99.03 ± 0.05 |
| 128 | 96.09 ± 0.21 | 94.53 ± 0.25 | 97.57 ± 0.29 | 94.70 ± 0.23 | 96.02 ± 0.21 | 92.22 ± 0.42 | 98.73 ± 0.05 | 99.07 ± 0.05 |
| Acc | Sen | Pre | NPV | F1 | MCC | AUROC | AUPRC | |
|---|---|---|---|---|---|---|---|---|
| APAAC [28] | 96.10 ± 0.20 | 94.76 ± 0.48 | 97.38 ± 0.45 | 94.90 ± 0.43 | 96.05 ± 0.21 | 92.24 ± 0.41 | 98.88 ± 0.07 | 99.16 ± 0.05 |
| APAACplus (Ours) | 96.34 ± 0.34 | 95.05 ± 0.58 | 97.56 ± 0.39 | 95.18 ± 0.54 | 96.29 ± 0.35 | 92.70 ± 0.67 | 98.87 ± 0.08 | 99.16 ± 0.04 |
| Metrics | Acc | Pre | Sen | NPV | F1 | MCC | AUROC | AUPRC | |
|---|---|---|---|---|---|---|---|---|---|
| Yeast core | H | 94.33 | 97.25 | 91.26 | 91.78 | 94.15 | 88.85 | 98.04 | 98.51 |
| E | 95.41 | 96.60 | 94.15 | 94.30 | 95.36 | 90.86 | 98.46 | 98.87 | |
| H + E | 96.34 | 97.56 | 95.05 | 95.18 | 96.29 | 92.70 | 98.87 | 99.16 | |
| Human | H | 96.99 | 98.88 | 94.77 | 95.42 | 96.77 | 94.04 | 99.25 | 99.02 |
| E | 99.11 | 98.90 | 99.23 | 99.29 | 99.07 | 98.21 | 99.66 | 99.42 | |
| H + E | 99.30 | 98.88 | 99.67 | 99.69 | 99.27 | 98.60 | 99.75 | 99.57 | |
| Acc | Sen | Pre | NPV | F1 | MCC | |
|---|---|---|---|---|---|---|
| 0.1 | 96.07 ± 0.20 | 94.51 ± 0.46 | 97.55 ± 0.23 | 94.68 ± 0.41 | 96.00 ± 0.21 | 92.18 ± 0.38 |
| 0.3 | 96.22 ± 0.24 | 94.82 ± 0.24 | 97.56 ± 0.47 | 94.96 ± 0.22 | 96.17 ± 0.24 | 92.48 ± 0.48 |
| 0.5 | 96.34 ± 0.34 | 95.05 ± 0.58 | 97.56 ± 0.39 | 95.18 ± 0.54 | 96.29 ± 0.35 | 92.70 ± 0.67 |
| 0.7 | 96.08 ± 0.17 | 94.73 ± 0.22 | 97.35 ± 0.23 | 94.87 ± 0.20 | 96.02 ± 0.17 | 92.19 ± 0.33 |
| 0.9 | 96.07 ± 0.32 | 94.83 ± 0.69 | 97.23 ± 0.29 | 94.96 ± 0.63 | 96.02 ± 0.33 | 92.17 ± 0.62 |
| Word | Sentence/Document | Embedding Vector Dimension | Training Corpus | |
|---|---|---|---|---|
| Bio2vec [22] | Unigram 1 | Protein sequence | 32 | SwissProt |
| Res2vec [20] | 1-gram | Protein sequence | 20 | SwissProt |
| ProtVec [19] | 3-gram | Protein sequence | 100 | UniRef50 |
| Yang’s work [24] | 5-gram | Protein sequence | 32 | SwissProt |
| ProteinBERT [26] | 1-gram | Protein sequence | 128 | UniRef90 |
| Approaches | Yeast Core | Human | ||||
|---|---|---|---|---|---|---|
| Acc | F1 | MCC | Acc | F1 | MCC | |
| Bio2Vec (2019) [22] | 95.47 ± 0.36 | 95.35 ± 0.37 | 91.07 ± 0.72 | 98.64 ± 0.14 | 98.57 ± 0.14 | 97.28 ± 0.27 |
| Res2vec (2019) [20] | 95.86 ± 0.23 | 95.79 ± 0.24 | 91.78 ± 0.46 | 99.25 ± 0.17 | 99.22 ± 0.18 | 98.50 ± 0.35 |
| ProtVec (2020) [19] | 96.02 ± 0.25 | 95.94 ± 0.27 | 92.11 ± 0.49 | 99.14 ± 0.13 | 99.10 ± 0.14 | 98.28 ± 0.27 |
| Yang’s work (2020) [24] | 95.84 ± 0.54 | 95.75 ± 0.57 | 91.77 ± 1.04 | 99.15 ± 0.15 | 99.12 ± 0.15 | 98.31 ± 0.29 |
| ProteinBERT (2022) [26] | 96.07 ± 0.24 | 95.99 ± 0.27 | 92.21 ± 0.45 | 99.08 ± 0.20 | 99.04 ± 0.21 | 98.16 ± 0.40 |
| Doc2vec (ours) | 96.34 ± 0.16 | 96.29 ± 0.35 | 92.70 ± 0.67 | 99.30 ± 0.16 | 99.27 ± 0.16 | 98.60 ± 0.32 |
| DF-PPI (Ours) | LightGBM-PPI (2020) | GTB-PPI (2020) | GcForest-PPI (2021) | SDNN-PPI (2022) | EResCNN (2023) | |
|---|---|---|---|---|---|---|
| CD9 | 16/16 | 16/16 | 15/16 | 16/16 | 16/16 | 16/16 |
| Wnt | 96/96 | 89/96 | 92/96 | 94/96 | 96/96 | 90/96 |
| Cancer | 108/108 | None | None | 108/108 | 108/108 | 107/108 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tran, H.-N.; Nguyen, P.-X.-Q.; Guo, F.; Wang, J. Prediction of Protein–Protein Interactions Based on Integrating Deep Learning and Feature Fusion. Int. J. Mol. Sci. 2024, 25, 5820. https://doi.org/10.3390/ijms25115820
Tran H-N, Nguyen P-X-Q, Guo F, Wang J. Prediction of Protein–Protein Interactions Based on Integrating Deep Learning and Feature Fusion. International Journal of Molecular Sciences. 2024; 25(11):5820. https://doi.org/10.3390/ijms25115820
Chicago/Turabian StyleTran, Hoai-Nhan, Phuc-Xuan-Quynh Nguyen, Fei Guo, and Jianxin Wang. 2024. "Prediction of Protein–Protein Interactions Based on Integrating Deep Learning and Feature Fusion" International Journal of Molecular Sciences 25, no. 11: 5820. https://doi.org/10.3390/ijms25115820
APA StyleTran, H.-N., Nguyen, P.-X.-Q., Guo, F., & Wang, J. (2024). Prediction of Protein–Protein Interactions Based on Integrating Deep Learning and Feature Fusion. International Journal of Molecular Sciences, 25(11), 5820. https://doi.org/10.3390/ijms25115820