
Fatty Acid Synthase as Interacting Anticancer Target of the Terpenoid Myrianthic Acid Disclosed by MS-Based Proteomics Approaches

 ,
,  ,
,  ,
,  ,
, 
Abstract
:1. Introduction
2. Results
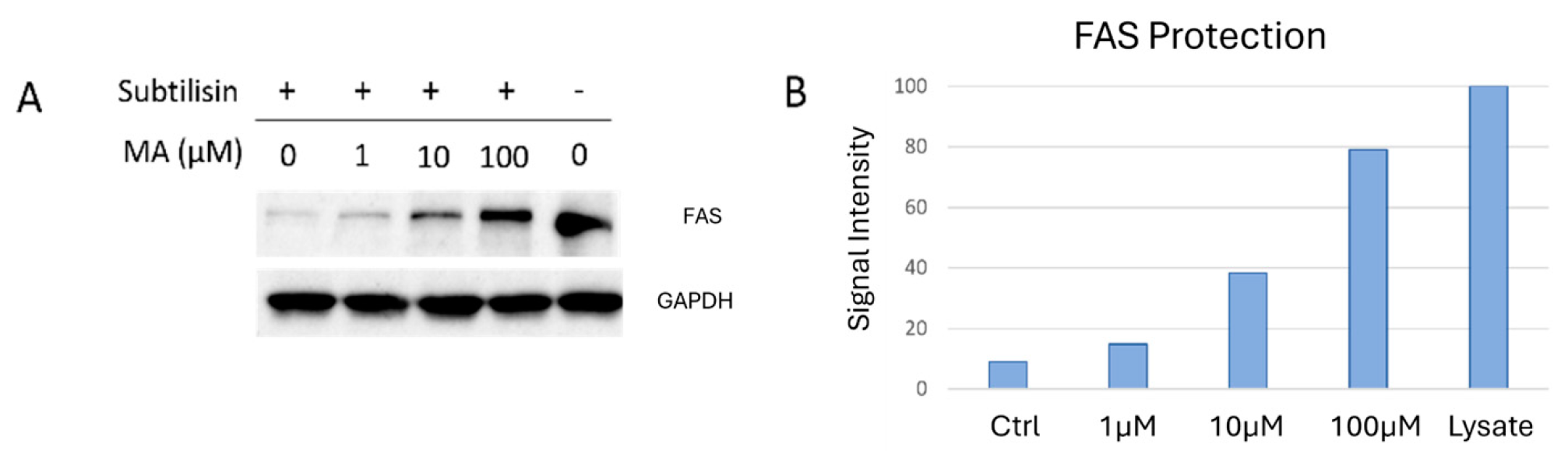
2.1. DARTS for Myrianthic Acid Target(s) Identification
2.2. Analysis of MA/FAS Interaction Features through t-LiP
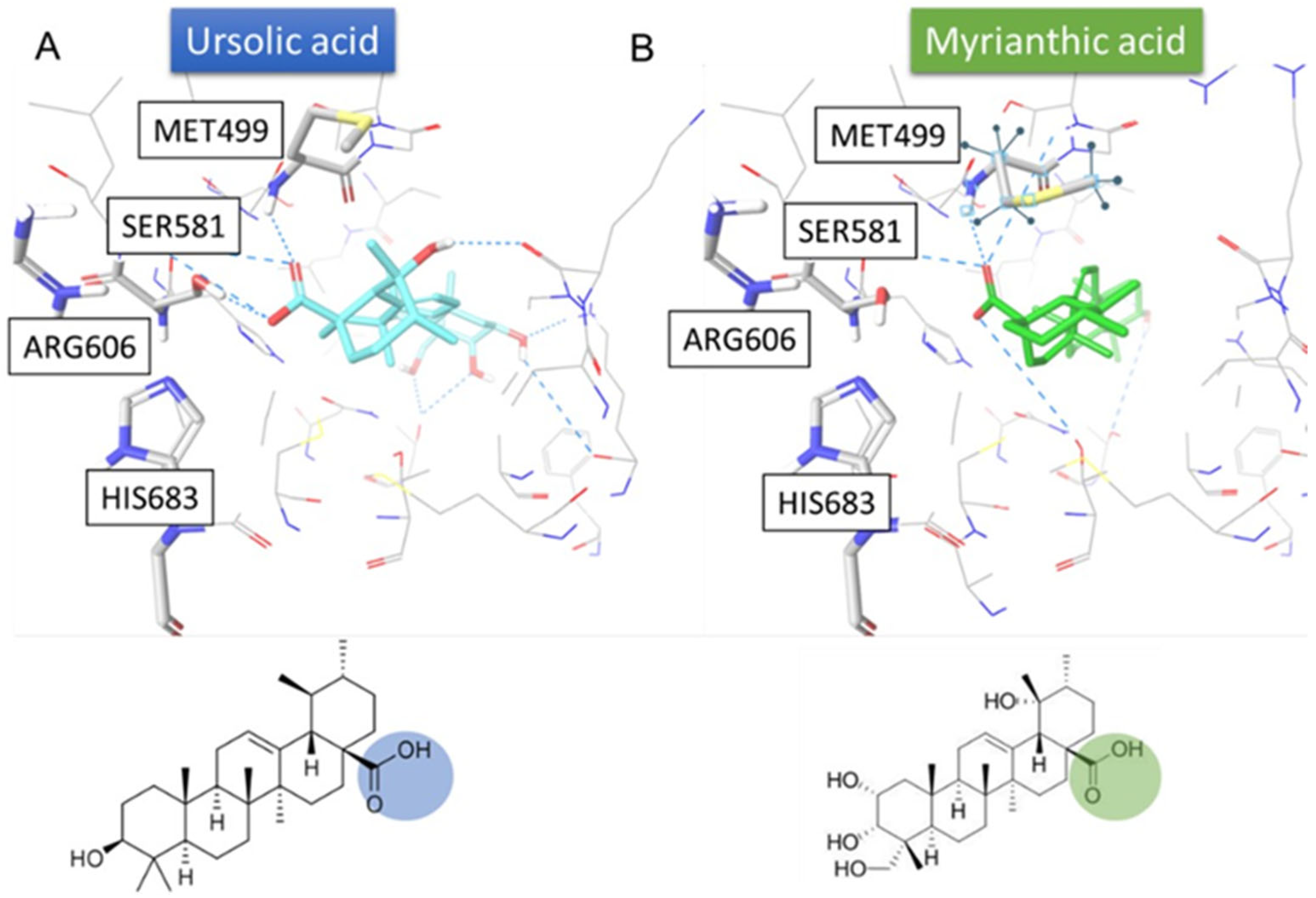
2.3. MA/FAS Complex: Molecular Docking
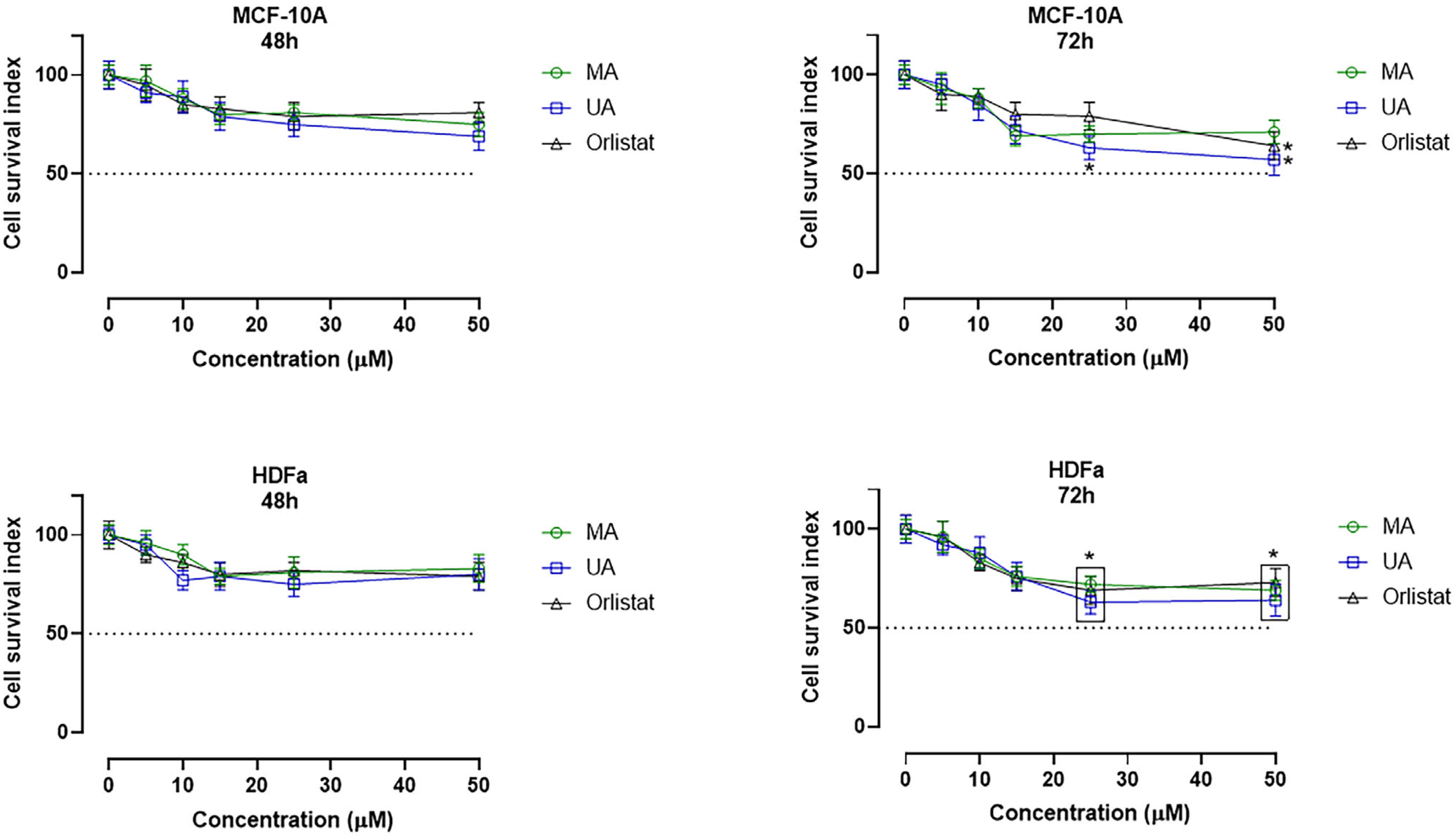
2.4. In Vitro Bioscreens and IC50 Evaluation
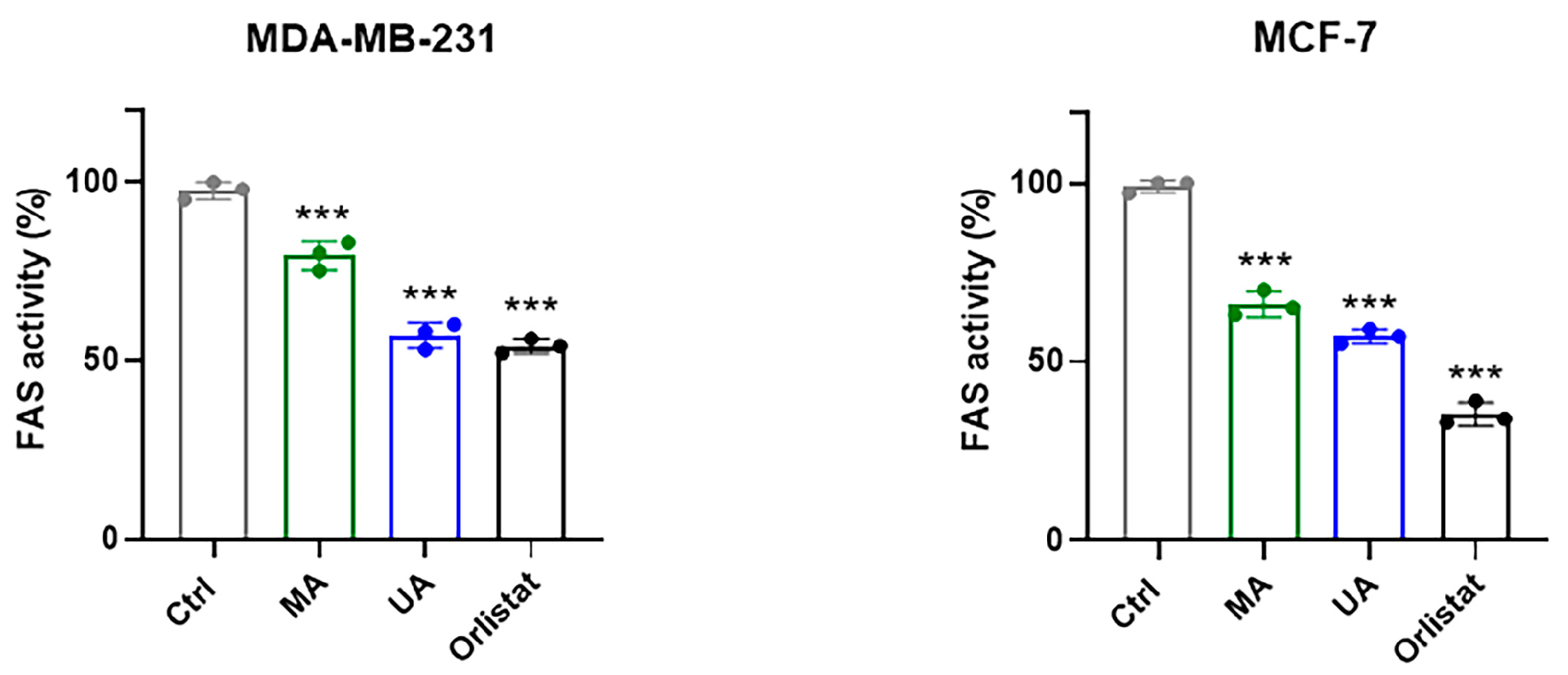
2.5. Regulation of FAS Activity by Myrianthic Acid
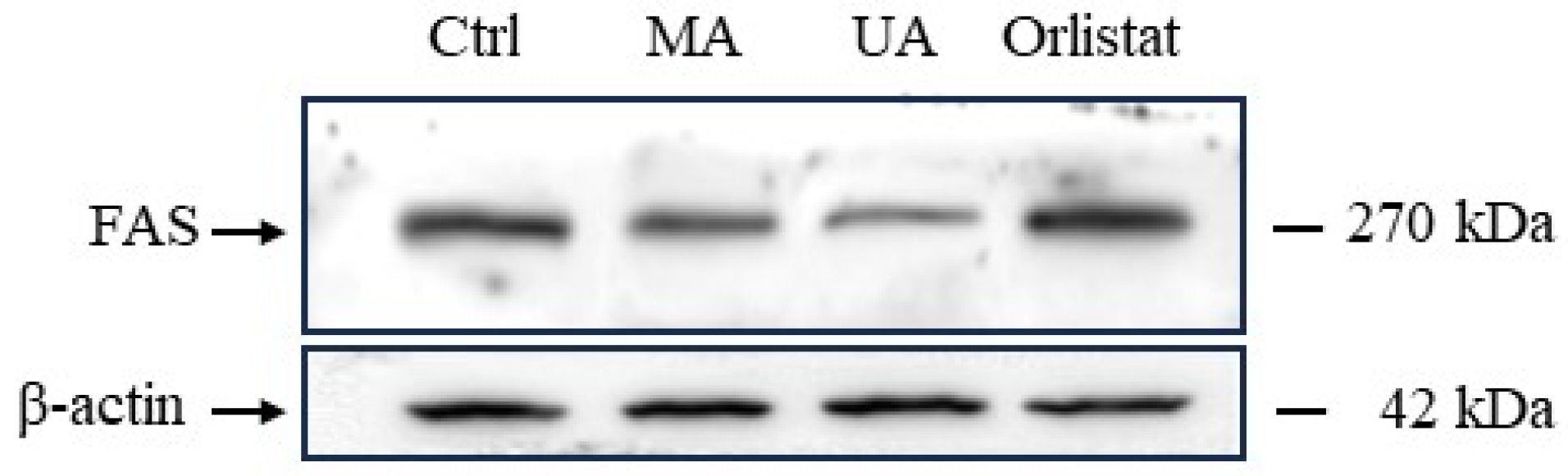
2.6. Regulation of FAS Expression by Myrianthic Acid
3. Discussion
4. Materials and Methods
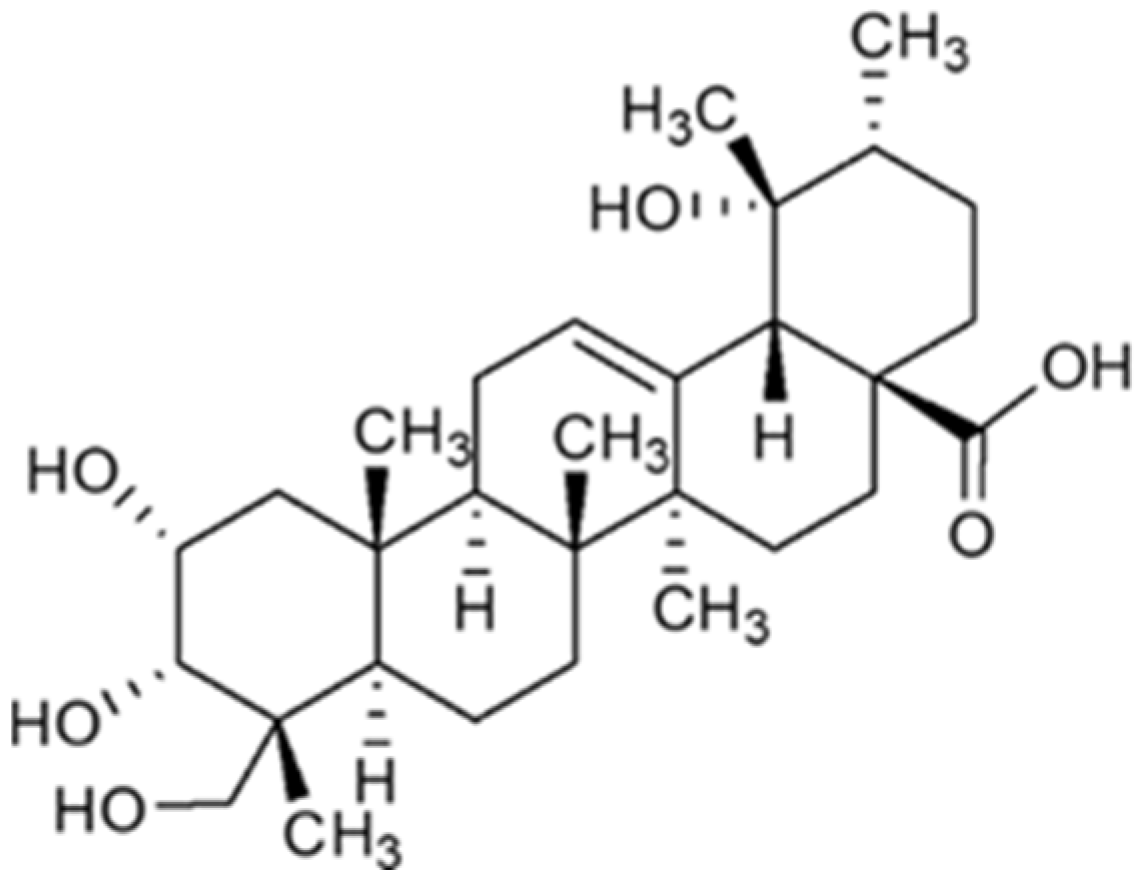
4.1. Myrianthic Acid
4.2. MA Target Identification through DARTS
4.3. Western Blotting for DARTS Validation
4.4. MRM Methods Fine-Tuning
4.5. T-LiP-MRM-MS for the Interactions Study of the Complexes MA-FAS
4.6. Molecular Docking
4.7. Biological Evaluations
4.7.1. Cancer Cell Cultures
4.7.2. Healthy Cell Cultures
4.7.3. Bioscreens In Vitro and IC50 Evaluation
4.7.4. Preparation of Cellular Extracts
4.7.5. Evaluation of FAS Activity in Protein Extracts
4.7.6. Evaluation of FAS Activity in Cells
4.7.7. Western Blot Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Terstappen, G.C.; Schlüpen, C.; Raggiaschi, R.; Gaviraghi, G. Target deconvolution strategies in drug discovery. Nat. Rev. Drug Discov. 2007, 6, 891–903. [Google Scholar] [CrossRef] [PubMed]
- Raida, M. Drug target deconvolution by chemical proteomics. Curr. Opin. Chem. Biol. 2011, 15, 570–575. [Google Scholar] [CrossRef] [PubMed]
- Saxena, C.; Zhen, E.; Higgs, R.E.; Hale, J.E. An Immuno-Chemo-Proteomics Method for Drug Target Deconvolution. J. Proteome Res. 2008, 7, 3490–3497. [Google Scholar] [CrossRef] [PubMed]
- Boldi, A.M. Libraries from natural product-like scaffolds. Curr. Opin. Chem. Biol. 2004, 8, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Davison, E.K.; Brimble, M.A. Natural product derived privileged scaffolds in drug discovery. Curr. Opin. Chem. Biol. 2019, 52, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Aly, S.H.; Elbadry, A.M.M.; Doghish, A.S.; El-Nashar, H.A.S. Unveiling the pharmacological potential of plant triterpenoids in breast cancer management: An updated review. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2024. [Google Scholar] [CrossRef]
- Kazakova, O.; Șoica, C.; Babaev, M.; Petrova, A.; Khusnutdinova, E.; Poptsov, A.; Macașoi, I.; Drăghici, G.; Avram, Ș.; Vlaia, L.; et al. 3-Pyridinylidene Derivatives of Chemically Modified Lupane and Ursane Triterpenes as Promising Anticancer Agents by Targeting Apoptosis. Int. J. Mol. Sci. 2021, 22, 10695. [Google Scholar] [CrossRef] [PubMed]
- Ojinnaka, C.M.; Okogun, J.I.; Okorie, D.A. Myrianthic acid: A triterpene acid from the rootwood of Myrianthus arboreus. Phytochemistry 1984, 23, 1125–1127. [Google Scholar] [CrossRef]
- Festa, C.; D’Auria, M.V.; Sepe, V.; Ilaš, J.; Leick, A.; N’Gom, S.; De Marino, S. Triterpenoid profile and bioactivity study of Oenothera maritima. Phytochem. Lett. 2015, 13, 324–329. [Google Scholar] [CrossRef]
- Alqahtani, A.; Hamid, K.; Kam, A.; Wong, K.H.; Abdelhak, Z.; Razmovski-Naumovski, V.; Chan, K.; Li, K.M.; Groundwater, P.W.; Li, G.Q. The pentacyclic triterpenoids in herbal medicines and their pharmacological activities in diabetes and diabetic complications. Curr. Med. Chem. 2013, 20, 908–931. [Google Scholar]
- Laszczyk, M.N. Pentacyclic Triterpenes of the Lupane, Oleanane and Ursane Group as Tools in Cancer Therapy. Planta Medica 2009, 75, 1549–1560. [Google Scholar] [CrossRef] [PubMed]
- Lomenick, B.; Hao, R.; Jonai, N.; Chin, R.M.; Aghajan, M.; Warburton, S.; Wang, J.; Wu, R.P.; Gomez, F.; Loo, J.A.; et al. Target identification using drug affinity responsive target stability (DARTS). Proc. Natl. Acad. Sci. USA 2009, 106, 21984–21989. [Google Scholar] [CrossRef] [PubMed]
- Lomenick, B.; Jung, G.; Wohlschlegel, J.A.; Huang, J. Target Identification Using Drug Affinity Responsive Target Stability (DARTS). Curr. Protoc. Chem. Biol. 2011, 3, 163–180. [Google Scholar] [CrossRef] [PubMed]
- Lomenick, B.; Olsen, R.W.; Huang, J. Identification of Direct Protein Targets of Small Molecules. ACS Chem. Biol. 2010, 6, 34–46. [Google Scholar] [CrossRef] [PubMed]
- Pai, M.Y.; Lomenick, B.; Hwang, H.; Schiestl, R.; McBride, W.; Loo, J.A.; Huang, J. Drug affinity responsive target stability (DARTS) for small-molecule target identification. In Methods in Molecular Biology; Humana Press: New York, NY, USA, 2015; Volume 1263, pp. 287–298. [Google Scholar] [CrossRef]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Proteomics. Tissue-Based Map of the Human Proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef] [PubMed]
- Buckley, D.; Duke, G.; Heuer, T.S.; O’Farrell, M.; Wagman, A.S.; McCulloch, W.; Kemble, G. Fatty acid synthase—Modern tumor cell biology insights into a classical oncology target. Pharmacol. Ther. 2017, 177, 23–31. [Google Scholar] [CrossRef]
- Shevchenko, A.; Wilm, M.; Vorm, O.; Mann, M. Mass Spectrometric Sequencing of Proteins from Silver-Stained Polyacrylamide Gels. Anal. Chem. 1996, 68, 850–858. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; De Franceschi, G.; Kahraman, A.; Soste, M.; Melnik, A.; Boersema, P.J.; De Laureto, P.P.; Nikolaev, Y.; Oliveira, A.P.; Picotti, P. Global analysis of protein structural changes in complex proteomes. Nat. Biotechnol. 2014, 32, 1036–1044. [Google Scholar] [CrossRef] [PubMed]
- Fontana, A.; De Laureto, P.P.; Spolaore, B.; Frare, E.; Picotti, P.; Zambonin, M. Probing protein structure by limited proteolysis. Acta Biochim. Pol. 2004, 51, 299–321. [Google Scholar] [CrossRef]
- Piazza, I.; Beaton, N.; Bruderer, R.; Knobloch, T.; Barbisan, C.; Chandat, L.; Sudau, A.; Siepe, I.; Rinner, O.; de Souza, N.; et al. A machine learning-based chemoproteomic approach to identify drug targets and binding sites in complex proteomes. Nat. Commun. 2020, 11, 4200. [Google Scholar] [CrossRef]
- Picotti, P.; Aebersold, R. Selected reaction monitoring–based proteomics: Workflows, potential, pitfalls and future directions. Nat. Methods 2012, 9, 555–566. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Tian, W.; Ma, X.; Ding, W. Evaluation of inhibition of fatty acid synthase by ursolic acid: Positive cooperation mechanism. Biochem. Biophys. Res. Commun. 2010, 392, 386–390. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.-S.; Lei, J.-P.; Wei, G.-Q.; Chen, H.; Ma, C.-Y.; Jiang, H.-Z. Natural fatty acid synthase inhibitors as potent therapeutic agents for cancers: A review. Pharm. Biol. 2016, 54, 1919–1925. [Google Scholar] [CrossRef] [PubMed]
- Maier, T.; Leibundgut, M.; Ban, N. The Crystal Structure of a Mammalian Fatty Acid Synthase. Science 2008, 321, 1315–1322. [Google Scholar] [CrossRef] [PubMed]
- Rittner, A.; Paithankar, K.S.; Himmler, A.; Grininger, M. Type I fatty acid synthase trapped in the octanoyl-bound state. Protein Sci. 2020, 29, 589–605. [Google Scholar] [CrossRef] [PubMed]
- Schiffer, A.; Fritz, G.; Kroneck, P.M.H.; Ermler, U. Reaction Mechanism of the Iron−Sulfur Flavoenzyme Adenosine-5′-Phosphosulfate Reductase Based on the Structural Characterization of Different Enzymatic States. Biochemistry 2006, 45, 2960–2967. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Bae, J.; Kim, D.; Choi, Y.; Im, Y.J.; Koh, S.; Kim, J.S.; Kim, M.-K.; Kang, G.B.; Hong, S.-I.; et al. Stereoselectivity of fructose-1,6-bisphosphate aldolase in Thermus caldophilus. Biochem. Biophys. Res. Commun. 2006, 347, 616–625. [Google Scholar] [CrossRef] [PubMed]
- Pappenberger, G.; Benz, J.; Gsell, B.; Hennig, M.; Ruf, A.; Stihle, M.; Thoma, R.; Rudolph, M.G. Structure of the Human Fatty Acid Synthase KS–MAT Didomain as a Framework for Inhibitor Design. J. Mol. Biol. 2010, 397, 508–519. [Google Scholar] [CrossRef] [PubMed]
- Rittner, A.; Paithankar, K.S.; Huu, K.V.K.V.; Grininger, M. Characterization of the Polyspecific Transferase of Murine Type I Fatty Acid Synthase (FAS) and Implications for Polyketide Synthase (PKS) Engineering. ACS Chem. Biol. 2018, 13, 723–732. [Google Scholar] [CrossRef]
- Yin, R.; Li, T.; Tian, J.X.; Xi, P.; Liu, R.H. Ursolic acid, a potential anticancer compound for breast cancer therapy. Crit. Rev. Food Sci. Nutr. 2017, 58, 568–574. [Google Scholar] [CrossRef]
- Seguin, F.; Carvalho, M.A.; Bastos, D.C.; Agostini, M.; Zecchin, K.G.; Alvarez-Flores, M.P.; Chudzinski-Tavassi, A.M.; Coletta, R.D.; Graner, E. The fatty acid synthase inhibitor orlistat reduces experimental metastases and angiogenesis in B16-F10 melanomas. Br. J. Cancer 2012, 107, 977–987. [Google Scholar] [CrossRef] [PubMed]
- Schcolnik-Cabrera, A.; Chavez-Blanco, A.; Dominguez-Gomez, G.; Taja-Chayeb, L.; Morales-Barcenas, R.; Trejo-Becerril, C.; Perez-Cardenas, E.; Gonzalez-Fierro, A.; Duenas-Gonzalez, A. Orlistat as a FASN inhibitor and multitargeted agent for cancer therapy. Expert Opin. Investig. Drugs 2018, 27, 475–489. [Google Scholar] [CrossRef] [PubMed]
- Khwaza, V.; Oyedeji, O.O.; Aderibigbe, B.A. Ursolic Acid-Based Derivatives as Potential Anti-Cancer Agents: An Update. Int. J. Mol. Sci. 2020, 21, 5920. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Yang, Y.; Xiong, H.; Dong, G. The implications of FASN in immune cell biology and related diseases. Cell Death Dis. 2024, 15, 88. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, J.; Abbasi, B.A.; Ahmad, R.; Mahmood, T.; Kanwal, S.; Ali, B.; Khalil, A.T.; Shah, S.A.; Alam, M.M.; Badshah, H. Ursolic acid a promising candidate in the therapeutics of breast cancer: Current status and future implications. Biomed. Pharmacother. 2018, 108, 752–756. [Google Scholar] [CrossRef] [PubMed]
- Vanauberg, D.; Schulz, C.; Lefebvre, T. Involvement of the pro-oncogenic enzyme fatty acid synthase in the hallmarks of cancer: A promising target in anti-cancer therapies. Oncogenesis 2023, 12, 16. [Google Scholar] [CrossRef] [PubMed]
- Fhu, C.W.; Ali, A. Fatty Acid Synthase: An Emerging Target in Cancer. Molecules 2020, 25, 3935. [Google Scholar] [CrossRef] [PubMed]
- Ferraro, M.G.; Bocchetti, M.; Riccardi, C.; Trifuoggi, M.; Paduano, L.; Montesarchio, D.; Misso, G.; Santamaria, R.; Piccolo, M.; Irace, C. Triple Negative Breast Cancer Preclinical Therapeutic Management by a Cationic Ruthenium-Based Nucleolipid Nanosystem. Int. J. Mol. Sci. 2023, 24, 6473. [Google Scholar] [CrossRef] [PubMed]
- Pastena, P.; Perera, H.; Martinino, A.; Kartsonis, W.; Giovinazzo, F. Unraveling Biomarker Signatures in Triple-Negative Breast Cancer: A Systematic Review for Targeted Approaches. Int. J. Mol. Sci. 2024, 25, 2559. [Google Scholar] [CrossRef]
- Kuhajda, F.P.; Jenner, K.; Wood, F.D.; Hennigar, R.A.; Jacobs, L.B.; Dick, J.D.; Pasternack, G.R. Fatty acid synthesis: A potential selective target for antineoplastic therapy. Proc. Natl. Acad. Sci. USA 1994, 91, 6379–6383. [Google Scholar] [CrossRef]
- Cheng, J.; Liu, Y.; Liu, Y.; Liu, D.; Liu, Y.; Guo, Y.; Wu, Z.; Li, H.; Wang, H. Ursolic acid alleviates lipid accumulation by activating the AMPK signaling pathwayin vivoandin vitro. J. Food Sci. 2020, 85, 3998–4008. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.-N.; Wang, C.C.N.; Chang, H.-Y.; Chu, F.-Y.; Hsu, Y.-A.; Cheng, W.-K.; Ma, W.-C.; Chen, C.-J.; Wan, L.; Lim, Y.-P. Ursolic Acid, a Novel Liver X Receptor α (LXRα) Antagonist Inhibiting Ligand-Induced Nonalcoholic Fatty Liver and Drug-Induced Lipogenesis. J. Agric. Food Chem. 2018, 66, 11647–11662. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Bhuiyan, M.J.H.; Jun, H.-J.; Lee, J.H.; Hoang, M.H.; Lee, H.-J.; Kim, N.; Lee, D.; Hwang, K.Y.; Hwang, B.Y.; et al. Ursolic acid is a PPAR-α agonist that regulates hepatic lipid metabolism. Bioorg. Med. Chem. Lett. 2011, 21, 5876–5880. [Google Scholar] [CrossRef] [PubMed]
- Shevchenko, A.; Tomas, H.; Havlis, J.; Olsen, J.V.; Mann, M. In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat. Protoc. 2006, 1, 2856–2860. [Google Scholar] [CrossRef] [PubMed]
- Perez-Riverol, Y.; Bai, J.; Bandla, C.; Garcia-Seisdedos, D.; Hewapathirana, S.; Kamatchinathan, S.; Kundu, D.J.; Prakash, A.; Frericks-Zipper, A.; Eisenacher, M.; et al. The PRIDE database resources in 2022: A hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res. 2021, 50, D543–D552. [Google Scholar] [CrossRef] [PubMed]
- Capuano, A.; D’Urso, G.; Aliberti, M.; Ruggiero, D.; Terracciano, S.; Festa, C.; Tosco, A.; Chini, M.G.; Lauro, G.; Bifulco, G.; et al. Chemoproteomics Reveals USP5 (Ubiquitin Carboxyl-Terminal Hydrolase 5) as Promising Target of the Marine Polyketide Gracilioether A. Mar. Drugs 2024, 22, 41. [Google Scholar] [CrossRef] [PubMed]
- Piccolo, M.; Misso, G.; Ferraro, M.G.; Riccardi, C.; Capuozzo, A.; Zarone, M.R.; Maione, F.; Trifuoggi, M.; Stiuso, P.; D’Errico, G.; et al. Exploring cellular uptake, accumulation and mechanism of action of a cationic Ru-based nanosystem in human preclinical models of breast cancer. Sci. Rep. 2019, 9, 7006. [Google Scholar] [CrossRef] [PubMed]
- Irace, C.; Misso, G.; Capuozzo, A.; Piccolo, M.; Riccardi, C.; Luchini, A.; Caraglia, M.; Paduano, L.; Montesarchio, D.; Santamaria, R. Antiproliferative effects of ruthenium-based nucleolipidic nanoaggregates in human models of breast cancer in vitro: Insights into their mode of action. Sci. Rep. 2017, 7, 45236. [Google Scholar] [CrossRef]
- Fiorito, F.; Irace, C.; Nocera, F.P.; Piccolo, M.; Ferraro, M.G.; Ciampaglia, R.; Tenore, G.C.; Santamaria, R.; De Martino, L. MG-132 interferes with iron cellular homeostasis and alters virulence of bovine herpesvirus 1. Res. Vet. Sci. 2021, 137, 1–8. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fatty Acid Synthase Abundance | ||||
|---|---|---|---|---|
| 1 µM/ctrl | 10 µM/ctrl | 100 µM/ctrl | Lysate/ctrl | |
| Replicate A | 1.829 | 2.594 | 3.962 | 15.79 |
| Replicate B | 0.431 | 0.518 | 2.114 | 5.966 |
| Replicate C | 2.517 | 3.368 | 5.443 | 23.426 |
| Treated | Lysate | |||||
|---|---|---|---|---|---|---|
| Q1_mz | Q3_mz | ID | Fc | p-Value | Fc | p-Value |
| 808.11 | 215.14 | T[2483-2505]R | 6.46 | 3.18 × 10−3 | 52.85 | 4.12 × 10−4 |
| 718.34 | 851.9 | E[612-631]K | 1.79 | 5.00 × 10−2 | 8.47 | 1.36 × 10−2 |
| 623.94 | 504.28 | F[1117-1131]R | 1.70 | 3.05 × 10−2 | 10.72 | 1.02 × 10−2 |
| 610.99 | 495.3 | F[1772-1787]K | 2.49 | 1.55 × 10−2 | 6.04 | 1.07 × 10−2 |
| 546.26 | 637.29 | C[1828-1841]R | 1.75 | 1.56 × 10−3 | 1.65 | 4.53 × 10−2 |
| 807.42 | 515.33 | E[2207-2220]R | 3.04 | 7.20 × 10−3 | 3.11 | 1.93 × 10−2 |
| 775.40 | 291.17 | A[943-957]K | 1.67 | 3.58 × 10−3 | 5.79 | 1.64 × 10−3 |
| 703.41 | 708.45 | D[2126-2138]R | 7.56 | 3.93 × 10−3 | 24.72 | 1.03 × 10−2 |
| 596.79 | 691.31 | L[203-213]K | 2.29 | 1.76 × 10−2 | 14.82 | 2.55 × 10−3 |
| 449.26 | 204.13 | L[1583-1591]K | 1.78 | 1.66 × 10−3 | 2.06 | 4.42 × 10−3 |
| IC50 (µM)—48 h | ||||
|---|---|---|---|---|
| MDA-MB-231 | MCF-7 | MCF-10A | HDFa | |
| Myrianthic acid | 50 ± 1 | 46 ± 2 | >50 | >50 |
| Ursolic acid | 23 ± 5 | 24 ± 3 | >50 | >50 |
| Orlistat | 19 ± 3 | 13 ± 4 | >50 | >50 |
| IC50 (µM)—72 h | ||||
|---|---|---|---|---|
| MDA-MB-231 | MCF-7 | MCF-10A | HDFa | |
| Myrianthic acid | 25 ± 3 | 21 ± 6 | >50 | >50 |
| Ursolic acid | 11 ± 3 | 9 ± 2 | >50 | >50 |
| Orlistat | 4 ± 2 | 5 ± 1 | >50 | >50 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Capuano, A.; D’Urso, G.; Gazzillo, E.; Lauro, G.; Chini, M.G.; D’Auria, M.V.; Ferraro, M.G.; Iazzetti, F.; Irace, C.; Bifulco, G.; et al. Fatty Acid Synthase as Interacting Anticancer Target of the Terpenoid Myrianthic Acid Disclosed by MS-Based Proteomics Approaches. Int. J. Mol. Sci. 2024, 25, 5918. https://doi.org/10.3390/ijms25115918
Capuano A, D’Urso G, Gazzillo E, Lauro G, Chini MG, D’Auria MV, Ferraro MG, Iazzetti F, Irace C, Bifulco G, et al. Fatty Acid Synthase as Interacting Anticancer Target of the Terpenoid Myrianthic Acid Disclosed by MS-Based Proteomics Approaches. International Journal of Molecular Sciences. 2024; 25(11):5918. https://doi.org/10.3390/ijms25115918
Chicago/Turabian StyleCapuano, Alessandra, Gilda D’Urso, Erica Gazzillo, Gianluigi Lauro, Maria Giovanna Chini, Maria Valeria D’Auria, Maria Grazia Ferraro, Federica Iazzetti, Carlo Irace, Giuseppe Bifulco, and et al. 2024. "Fatty Acid Synthase as Interacting Anticancer Target of the Terpenoid Myrianthic Acid Disclosed by MS-Based Proteomics Approaches" International Journal of Molecular Sciences 25, no. 11: 5918. https://doi.org/10.3390/ijms25115918
APA StyleCapuano, A., D’Urso, G., Gazzillo, E., Lauro, G., Chini, M. G., D’Auria, M. V., Ferraro, M. G., Iazzetti, F., Irace, C., Bifulco, G., & Casapullo, A. (2024). Fatty Acid Synthase as Interacting Anticancer Target of the Terpenoid Myrianthic Acid Disclosed by MS-Based Proteomics Approaches. International Journal of Molecular Sciences, 25(11), 5918. https://doi.org/10.3390/ijms25115918