
Patterns of the Predicted Mutation Burden in 19,778 Domesticated Barley Accessions Conserved Ex Situ


Abstract
:1. Introduction
2. Results
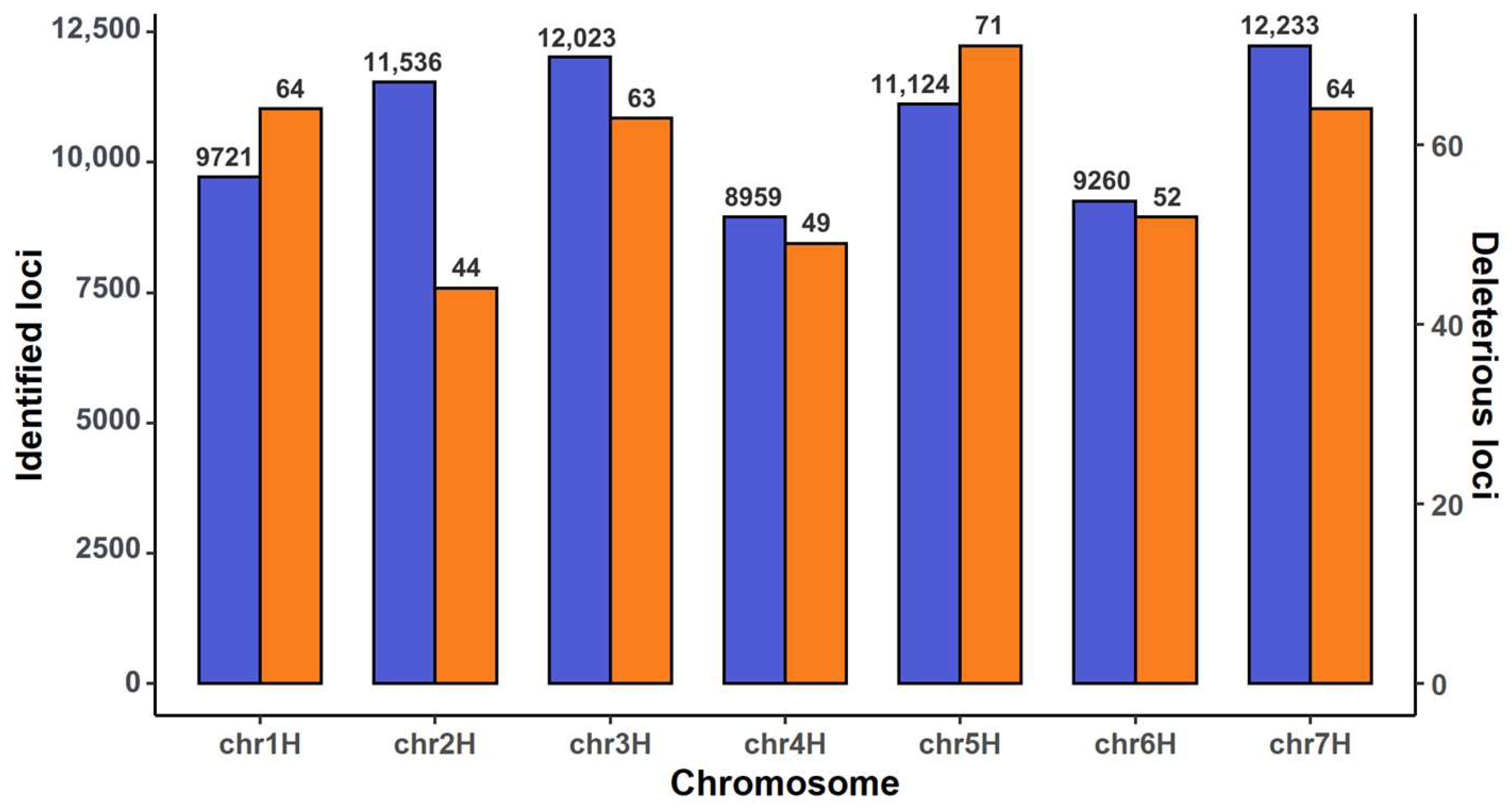
2.1. Annotating SNPs and Identifying Deleterious SNP
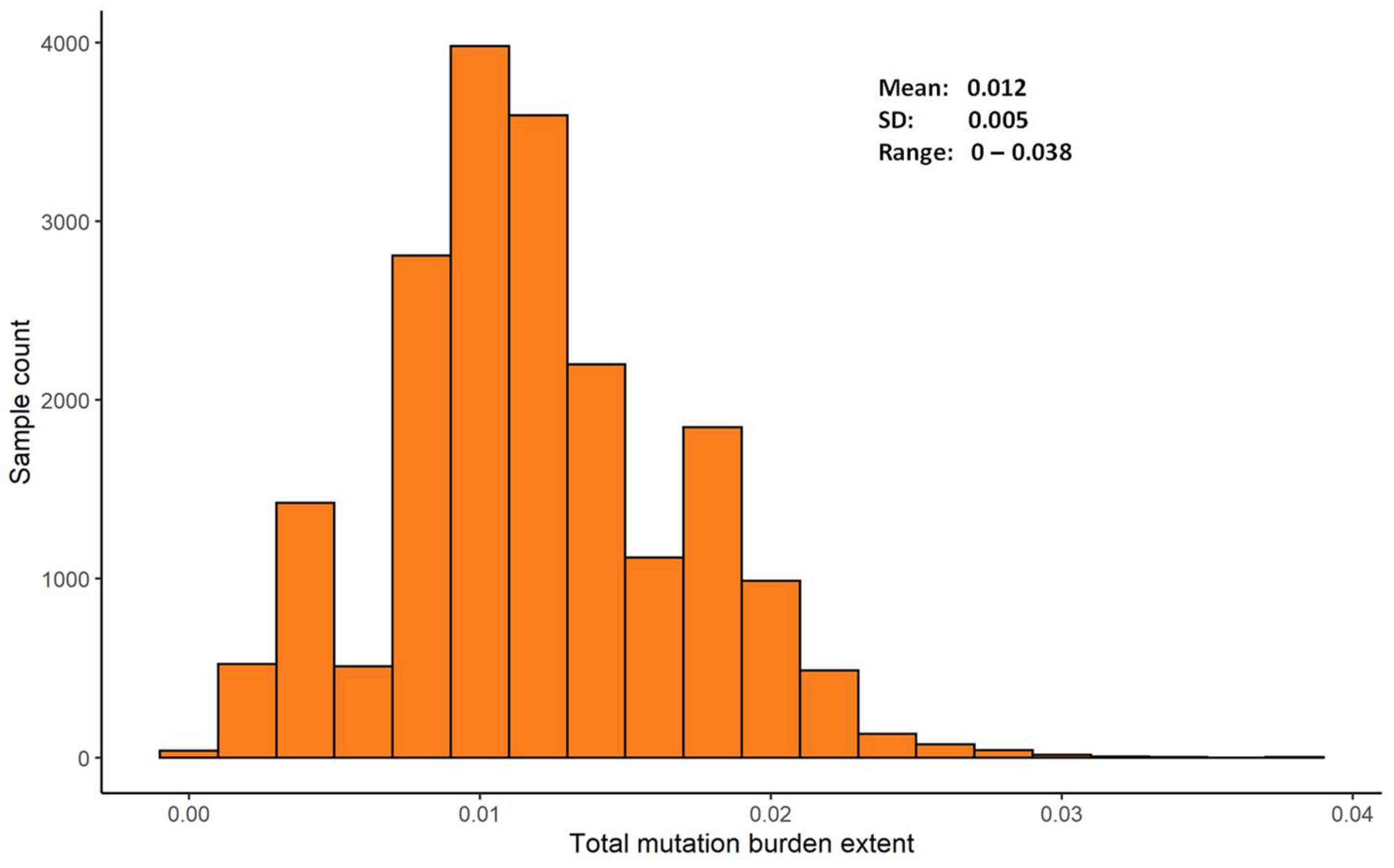
2.2. Estimating Mutation Burdens
2.3. Characterizing Mutation Burdens
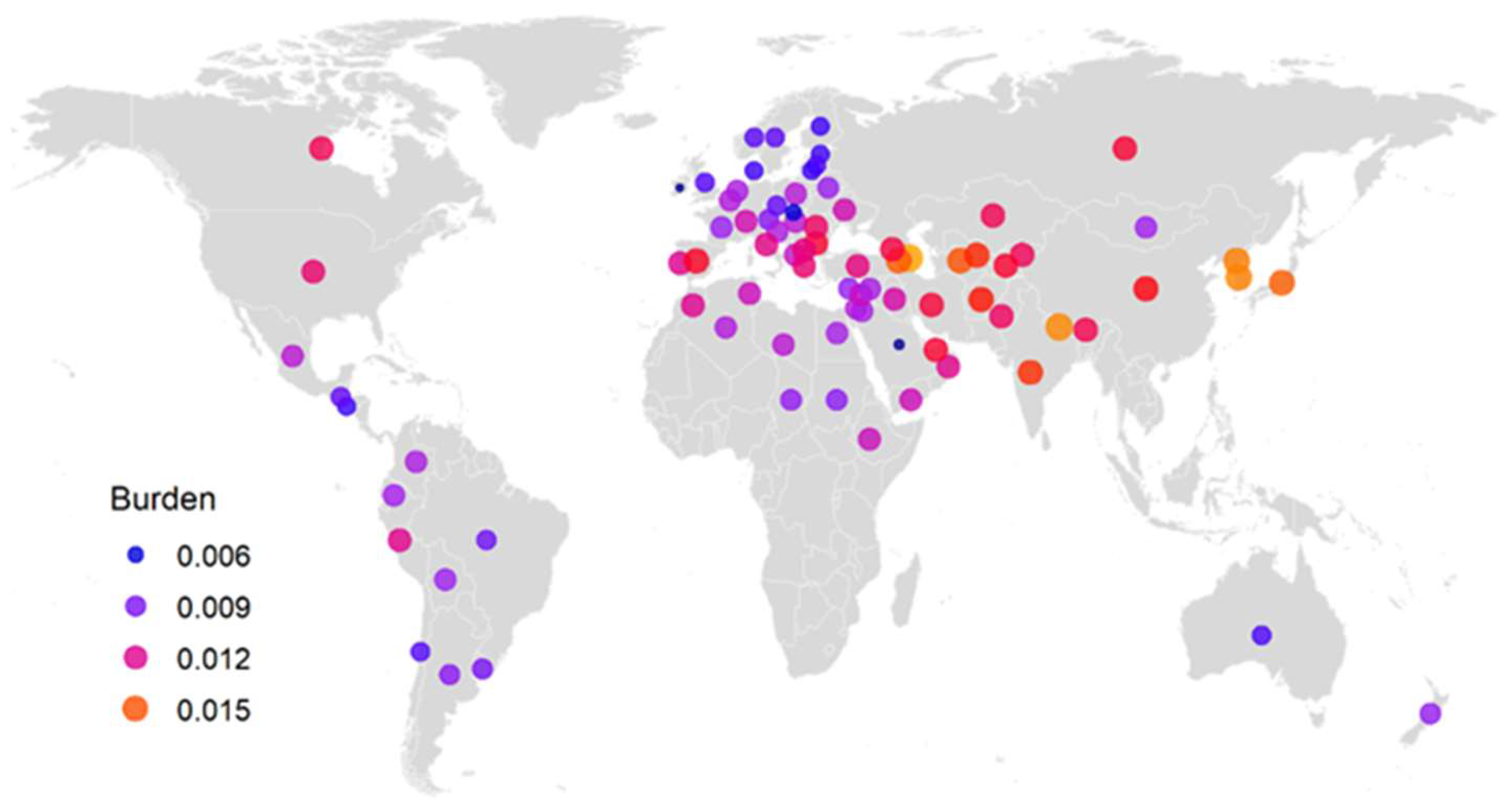
2.4. Correlating between Mutation Burdens and Averaged Pairwise Differences of Individual Samples
3. Discussion
4. Materials and Methods
5. Conclusions
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Food and Agriculture Organization of the United Nations. The Second Report on the State of the World’s Plant Genetic Resources for Food and Agriculture; FAO: Rome, Italy, 2010. [Google Scholar]
- Fu, Y.B. The vulnerability of plant genetic resources conserved ex situ. Crop Sci. 2017, 57, 2314–2328. [Google Scholar] [CrossRef]
- Schoen, D.J.; Brown, A.H. The conservation of wild plant species in seed banks. BioScience 2001, 51, 960–966. [Google Scholar] [CrossRef]
- Chebotar, S.; Röder, M.S.; Korzun, V.; Saal, B.; Weber, W.E.; Börner, A. Molecular studies on genetic integrity of open-pollinating species rye (Secale cereale L.) after long-term genebank maintenance. Theor. Appl. Genet. 2003, 107, 1469–1476. [Google Scholar] [CrossRef]
- Richards, C.M.; Lockwood, D.R.; Volk, G.M.; Walters, C. Modeling demographics and genetic diversity in ex situ collections during seed storage and regeneration. Crop Sci. 2010, 50, 2440–2447. [Google Scholar] [CrossRef]
- Krishnan, S.; Ranker, T.A.; Davis, A.P.; Rakotomalala, J.J. An assessment of the genetic integrity of ex situ germplasm collections of three endangered species of Coffea from Madagascar: Implications for the management of field germplasm collections. Genet. Resour. Crop Evol. 2013, 60, 1021–1036. [Google Scholar] [CrossRef]
- Dourado, A.M.; Roberts, E.H. Phenotypic mutations induced during storage in barley and pea seeds. Ann. Bot. 1984, 54, 781–790. [Google Scholar] [CrossRef]
- Fu, Y.B.; Peterson, G.W.; Horbach, C. Deleterious and adaptive mutations in plant germplasm conserved ex situ. Mol. Biol. Evol. 2023, 40, msad238. [Google Scholar] [CrossRef] [PubMed]
- Charlesworth, D.; Morgan, M.T.; Charlesworth, B. Mutation accumulation in finite outbreeding and inbreeding populations. Genet. Res. 1993, 61, 39–56. [Google Scholar] [CrossRef]
- Schoen, D.J.; David, J.L.; Bataillou, T.M. Deleterious mutation accumulation and the regeneration of genetic resources. Proc. Natl. Acad. Sci. USA 1998, 95, 394–399. [Google Scholar] [CrossRef]
- IWGSC; Appels, R.; Eversole, K.; Stein, N.; Feuillet, C.; Keller, B.; Rogers, J.; Pozniak, C.J.; Choulet, F.; Distelfeld, A.; et al. Shifting the limits in wheat research and breeding using a fully annotated reference genome. Science 2018, 361, eaar7191. [Google Scholar] [CrossRef]
- Cooper, G.M.; Stone, E.A.; Asimenos, G.; Green, E.D.; Batzoglou, S.; Sidow, A. Distribution and intensity of constraint in mammalian genomic sequence. Genome Res. 2005, 15, 901–913. [Google Scholar] [CrossRef]
- Henn, B.M.; Botigué, L.R.; Bustamante, C.D.; Clark, A.G.; Gravel, S. Estimating the mutation load in human genomes. Nat. Rev. Genet. 2015, 16, 333–343. [Google Scholar] [CrossRef] [PubMed]
- Ng, P.C.; Henikoff, S. SIFT: Predicting amino acid changes that affect protein function. Nucl. Acids Res. 2003, 31, 3812–3814. [Google Scholar] [CrossRef] [PubMed]
- Ramu, P.; Esuma, W.; Kawuki, R.; Rabbi, I.Y.; Egesi, C.; Bredeson, J.V.; Bart, R.S.; Verma, J.; Buckler, E.S.; Lu, F. Cassava haplotype map highlights fixation of deleterious mutations during clonal propagation. Nat. Genet. 2017, 49, 959–963. [Google Scholar] [CrossRef] [PubMed]
- Milner, S.G.; Jost, M.; Taketa, S.; Mazón, E.R.; Himmelbach, A.; Oppermann, M.; Weise, S.; Knüpffer, H.; Basterrechea, M.; König, P.; et al. Genebank genomics highlights the diversity of a global barley collection. Nat. Genet. 2019, 51, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Tang, T.; Tang, H.; Huang, J.; Shi, S.; Wu, C.I. The accumulation of deleterious mutations in rice genomes: A hypothesis on the cost of domestication. Trends Genet. 2006, 22, 126–131. [Google Scholar] [CrossRef] [PubMed]
- Günther, T.; Schmid, K.J. Deleterious amino acid polymorphisms in Arabidopsis thaliana and rice. Theor. Appl. Genet. 2010, 121, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Mezmouk, S.; Ross-Ibarra, J. The pattern and distribution of deleterious mutations in maize. G3 Genes Genomes Genet. 2014, 4, 163–171. [Google Scholar] [CrossRef]
- Renaut, S.; Rieseberg, L.H. The accumulation of deleterious mutations as a consequence of domestication and improvement in sunflowers and other Compositae crops. Mol. Biol. Evol. 2015, 32, 2273–2283. [Google Scholar] [CrossRef]
- Kono, T.J.; Fu, F.; Mohammadi, M.; Hoffman, P.J.; Liu, C.; Stupar, R.M.; Smith, K.P.; Tiffin, P.; Fay, J.C.; Morrell, P.L. The role of deleterious substitutions in crop genomes. Mol. Biol. Evol. 2016, 33, 2307–2317. [Google Scholar] [CrossRef]
- Liu, Q.; Zhou, Y.; Morrell, P.L.; Gaut, B.S. Deleterious variants in Asian rice and the potential cost of domestication. Mol. Biol. Evol. 2017, 34, 908–924. [Google Scholar] [CrossRef] [PubMed]
- Valluru, R.; Gazave, E.E.; Fernandes, S.B.; Ferguson, J.N.; Lozano, R.; Hirannaiah, P.; Zuo, T.; Brown, P.J.; Leakey, A.D.; Gore, M.A.; et al. Deleterious mutation burden and its association with complex traits in Sorghum (Sorghum bicolor). Genetics 2019, 211, 1075–1087. [Google Scholar] [CrossRef] [PubMed]
- Moyers, B.T.; Morrell, P.L.; McKay, J.K. Genetic costs of domestication and improvement. J. Hered. 2017, 109, 103–116. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.B. Variability in predicted deleterious mutations among barley accessions conserved ex situ. Crop Sci. 2024. acceptable upon a minor revison (14 May 2024). [Google Scholar]
- Vaser, R.; Adusumalli, S.; Leng, S.N.; Sikic, M.; Ng, P.C. SIFT missense predictions for genomes. Nat. Protoc. 2015, 11, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Davydov, E.V.; Goode, D.L.; Sirota, M.; Cooper, G.M.; Sidow, A.; Batzoglou, S. Identifying a high fraction of the human genome to be under selective constraint using GERP++. PLoS Comput. Biol. 2010, 6, e1001025. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.B. Assessing genetic distinctness and redundancy of plant germplasm conserved ex situ based on published genomic SNP data. Plants 2023, 12, 1476. [Google Scholar] [CrossRef] [PubMed]
- Kono, T.J.Y.; Liu, C.; Vonderharr, E.E.; Koenig, D.; Fay, J.C.; Smith, K.P.; Morrell, P.L. The fate of deleterious variants in a barley genomic prediction population. Genetics 2019, 213, 1531–1544. [Google Scholar] [CrossRef]
- Salse, J.; Bolot, S.; Throude, M.; Jouffe, V.; Piegu, B.; Quraishi, U.M.; Calcagno, T.; Cooke, R.; Delseny, M.; Feuillet, C. Identification and characterization of shared duplications between rice and wheat provide new insight into grass genome evolution. Plant Cell 2008, 20, 11–24. [Google Scholar] [CrossRef]
- Bikard, D.; Patel, D.; Le, M.; Giorgi, V.; Camilleri, C.; Bennett, M.J.; Loudet, O. Divergent evolution of duplicate genes leads to genetic incompatibilities within A. thaliana. Science 2009, 323, 623–626. [Google Scholar] [CrossRef]
- Mizuta, Y.; Harushima, Y.; Kurata, N. Rice pollen hybrid incompatibility caused by reciprocal gene loss of duplicated genes. Proc. Natl. Acad. Sci. USA 2010, 107, 20417–20422. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.B.; Horbach, C. Genetic diversity in a core subset of wild barley germplasm. Diversity 2012, 4, 239–257. [Google Scholar] [CrossRef]
- Gaut, B.S.; Díez, C.M.; Morrell, P.L. Genomics and the contrasting dynamics of annual and perennial domestication. Trends Genet. 2015, 31, 709–719. [Google Scholar] [CrossRef] [PubMed]
- Visioni, A.; Basile, B.; Amri, A.; Sanchez-Garcia, M.; Corrado, G. Advancing the conservation and utilization of barley genetic resources: Insights into germplasm management and breeding for sustainable agriculture. Plants 2023, 12, 3186. [Google Scholar] [CrossRef] [PubMed]
- Hay, F.R.; Whitehouse, K.J.; Ellis, R.H.; Sackville Hamilton, N.R.; Lusty, C.; Ndjiondjop, M.N.; Tia, D.; Wenzel, P.; Santos, L.G.; Yazbek, M.; et al. CGIAR genebank viability data reveal inconsistencies in seed collection management. Glob. Food Sec. 2021, 30, 100557. [Google Scholar] [CrossRef]
- Food and Agriculture Organization of the United Nations. Genebank Standards for Plant Genetic Resources for Food and Agriculture, Revised ed.; FAO: Rome, Italy, 2014. [Google Scholar]
- Food and Agriculture Organization of the United Nations. Practical Guide for the Application of the Genebank Standards for Plant Genetic Resources for Food and Agriculture: Conservation of Orthodox Seeds in Seed Genebanks; FAO: Rome, Italy, 2022. [Google Scholar] [CrossRef]
- Roles, A.J.; Conner, J.K. Fitness effects of mutation accumulation in a natural outbred population of wild radish (Raphanus raphanistrum): Comparison of field and greenhouse environments. Evolution 2008, 62, 1066–1075. [Google Scholar] [CrossRef] [PubMed]
- Pourkheirandish, M.; Hensel, G.; Kilian, B.; Senthil, N.; Chen, G.; Sameri, M.; Azhaguvel, P.; Sakuma, S.; Dhanagond, S.; Sharma, R.; et al. Evolution of the grain dispersal system in barley. Cell 2015, 162, 527–539. [Google Scholar] [CrossRef] [PubMed]
- Mascher, M.; Gundlach, H.; Himmelbach, A.; Beier, S.; Twardziok, S.O.; Wicker, T.; Radchuk, V.; Dockter, C.; Hedley, P.E.; Russell, J.; et al. A chromosome conformation capture ordered sequence of the barley genome. Nature 2017, 544, 427–433. [Google Scholar] [CrossRef]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl variant effect predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef]
- Naithani, S.; Geniza, M.; Jaiswal, P. Variant effect prediction analysis using resources available at Gramene Database. In Plant Genomics Databases: Methods and Protocols; van Dijk, A., Ed.; Humana Press: New York, NY, USA, 2017; pp. 270–297. [Google Scholar] [CrossRef]
- Wang, L.; Beissinger, T.M.; Lorant, A.; Ross-Ibarra, C.; Ross-Ibarra, J.; Hufford, M.B. The interplay of demography and selection during maize domestication and expansion. Genome Biol. 2017, 18, 215. [Google Scholar] [CrossRef] [PubMed]
- Knüpffer, H.; Terentyeva, I.; Hammer, K.; Kovaleva, O.; Sato, K.; Yasuda, S. Ecogeographical diversity—A Vavilovian approach. In Diversity in Barley (Hordeum vulgare); von Bothmer, R., van Hintum, T.J., Knüpffer, H., Sato, K., Eds.; Eslevier: Amsterdam, The Netherlands, 2003; pp. 53–76. [Google Scholar]
- Fu, Y.B. Redundancy and distinctness in flax germplasm as revealed by RAPD dissimilarity. Plant Gen. Res. 2006, 4, 117–124. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SNP Annotation | Count |
|---|---|
| VEP consequence type | 74,856 |
| missense_variant | 12,783 |
| splice_region_variant | 854 |
| stop_gained | 356 |
| splice_donor_variant | 134 |
| start_lost | 118 |
| splice_acceptor_variant | 95 |
| stop_lost | 53 |
| stop_retained_variant | 16 |
| synonymous_variant | 8544 |
| 5_prime_UTR_variant | 4752 |
| 3_prime_UTR_variant | 4644 |
| non_coding_transcript_exon_variant | 1350 |
| intron_variant | 9726 |
| upstream_gene_variant | 6585 |
| downstream_gene_variant | 5645 |
| intergenic_variant | 19,201 |
| SIFT prediction | 64,898 |
| deleterious | 19,839 |
| deleterious_low_confidence | 5265 |
| tolerated | 33,699 |
| tolerated_low_confidence | 6095 |
| Group | Count | Mean | SD | Min | Max | ANOVA |
|---|---|---|---|---|---|---|
| Germplasm_panel | p = 0.9504 | |||||
| IPK_Genebank | 18,714 | 0.0119 | 0.0049 | 0.0000 | 0.0376 | |
| Chinese_Genebank | 257 | 0.0158 | 0.0071 | 0.0000 | 0.0323 | |
| Swiss_Genebank | 669 | 0.0113 | 0.0038 | 0.0025 | 0.0205 | |
| Pourkheirandish_et_al_2015 | 138 | 0.0113 | 0.0042 | 0.0025 | 0.0223 | |
| Material_type | p < 0.00001 | |||||
| Landrace | 12,792 | 0.0122 | 0.0044 | 0.0000 | 0.0323 | |
| Cultivar | 5118 | 0.0115 | 0.0056 | 0.0000 | 0.0376 | |
| Breeding_material | 1864 | 0.0112 | 0.0054 | 0.0000 | 0.0273 | |
| Unknown | 4 | 0.0105 | 0.0012 | 0.0099 | 0.0123 | |
| Growth_habit | p < 0.00001 | |||||
| Spring | 14,643 | 0.0110 | 0.0046 | 0.0000 | 0.0323 | |
| Intermediate | 152 | 0.0146 | 0.0045 | 0.0049 | 0.0253 | |
| Winter | 3920 | 0.0151 | 0.0048 | 0.0000 | 0.0376 | |
| Unknown | 1063 | 0.0115 | 0.0040 | 0.0025 | 0.0272 | |
| Row_type | p < 0.00001 | |||||
| Two-rowed | 3819 | 0.0092 | 0.0044 | 0.0000 | 0.0250 | |
| Intermedium | 347 | 0.0162 | 0.0049 | 0.0024 | 0.0272 | |
| Six-rowed | 7821 | 0.0134 | 0.0046 | 0.0000 | 0.0376 | |
| Deficiens | 696 | 0.0117 | 0.0032 | 0.0025 | 0.0209 | |
| Labile | 242 | 0.0120 | 0.0022 | 0.0074 | 0.0201 | |
| Unknown | 6853 | 0.0115 | 0.0049 | 0.0000 | 0.0278 | |
| 1000-accession_core_set | p < 0.00001 | |||||
| Core | 998 | 0.0126 | 0.0051 | 0.0000 | 0.0280 | |
| Non-core | 18,780 | 0.0119 | 0.0049 | 0.0000 | 0.0376 | |
| Region | p < 0.00001 | |||||
| Near Eastern Centre | 1850 | 0.0122 | 0.0042 | 0.0024 | 0.0294 | |
| Mediterranean Centre | 2327 | 0.0129 | 0.0046 | 0.0000 | 0.0321 | |
| Middle Asian Centre | 857 | 0.0138 | 0.0049 | 0.0025 | 0.0296 | |
| East Asiatic Centre | 2490 | 0.0152 | 0.0052 | 0.0000 | 0.0376 | |
| European-Siberian Centre | 5021 | 0.0105 | 0.0050 | 0.0000 | 0.0297 | |
| Ethiopian Centre | 3952 | 0.0113 | 0.0031 | 0.0000 | 0.0271 | |
| New World Centre | 1024 | 0.0120 | 0.0052 | 0.0000 | 0.0271 | |
| Unknown | 2257 | 0.0105 | 0.0052 | 0.0000 | 0.0271 |
| Country | Region | Count | Mean | SD | Min | Max |
|---|---|---|---|---|---|---|
| Ireland | ES | 5 | 0.00346 | 0.00136 | 0.00246 | 0.00498 |
| Saudi Arabia | M | 2 | 0.00369 | 0.00173 | 0.00246 | 0.00491 |
| Slovakia | ES | 162 | 0.00574 | 0.00224 | 0.00000 | 0.01605 |
| El Salvador | NW | 2 | 0.00744 | 0.00341 | 0.00503 | 0.00985 |
| Lithuania | ES | 3 | 0.00747 | 0.00260 | 0.00493 | 0.01013 |
| Estonia | ES | 4 | 0.00748 | 0.00205 | 0.00498 | 0.01000 |
| Denmark | ES | 123 | 0.00753 | 0.00369 | 0.00246 | 0.02010 |
| Finland | ES | 59 | 0.00753 | 0.00229 | 0.00491 | 0.01478 |
| Costa Rica | NW | 1 | 0.00756 | NA | 0.00756 | 0.00756 |
| Australia | NW | 48 | 0.00777 | 0.00324 | 0.00246 | 0.01489 |
| Latvia | ES | 5 | 0.00790 | 0.00269 | 0.00493 | 0.01232 |
| UK | ES | 211 | 0.00796 | 0.00516 | 0.00000 | 0.02970 |
| Chile | NW | 12 | 0.00798 | 0.00458 | 0.00491 | 0.01609 |
| Norway | ES | 10 | 0.00813 | 0.00234 | 0.00491 | 0.01238 |
| Sweden | ES | 196 | 0.00819 | 0.00407 | 0.00000 | 0.02100 |
| Guatemala | NW | 8 | 0.00832 | 0.00127 | 0.00737 | 0.00990 |
| Czechia | ES | 29 | 0.00835 | 0.00397 | 0.00246 | 0.02020 |
| Brazil | NW | 2 | 0.00867 | 0.00184 | 0.00737 | 0.00998 |
| Uruguay | NW | 27 | 0.00904 | 0.00353 | 0.00246 | 0.01737 |
| Austria | ES | 252 | 0.00949 | 0.00423 | 0.00246 | 0.02222 |
| Argentina | NW | 25 | 0.00959 | 0.00432 | 0.00248 | 0.02057 |
| Sudan (the) | E | 11 | 0.00971 | 0.00224 | 0.00503 | 0.01250 |
| South Africa | E | 1 | 0.00993 | NA | 0.00993 | 0.00993 |
| Chad | E | 20 | 0.00993 | 0.00295 | 0.00250 | 0.01746 |
| Cyprus | M | 6 | 0.01005 | 0.00489 | 0.00495 | 0.01728 |
| New Zealand | NW | 2 | 0.01010 | 0.01428 | 0.00000 | 0.02020 |
| Germany | ES | 1415 | 0.01026 | 0.00560 | 0.00000 | 0.02519 |
| Belarus | ES | 7 | 0.01029 | 0.00327 | 0.00495 | 0.01478 |
| France | ES | 267 | 0.01038 | 0.00507 | 0.00000 | 0.02793 |
| Mongolia | EA | 33 | 0.01045 | 0.00334 | 0.00491 | 0.01733 |
| Bolivia | NW | 35 | 0.01048 | 0.00424 | 0.00246 | 0.01852 |
| Ecuador | NW | 14 | 0.01057 | 0.00355 | 0.00246 | 0.01724 |
| Egypt | M | 32 | 0.01062 | 0.00364 | 0.00491 | 0.01970 |
| Syria | NE | 149 | 0.01066 | 0.00365 | 0.00248 | 0.02020 |
| Jordan | NE | 26 | 0.01068 | 0.00345 | 0.00318 | 0.01605 |
| Colombia | NW | 22 | 0.01071 | 0.00376 | 0.00493 | 0.01985 |
| Netherlands | ES | 88 | 0.01071 | 0.00574 | 0.00246 | 0.02256 |
| Croatia | ES | 7 | 0.01073 | 0.00383 | 0.00491 | 0.01519 |
| Algeria | M | 49 | 0.01081 | 0.00359 | 0.00246 | 0.01980 |
| Belgium | ES | 27 | 0.01082 | 0.00606 | 0.00249 | 0.02211 |
| Israel | NE | 51 | 0.01086 | 0.00515 | 0.00246 | 0.02239 |
| Poland | ES | 166 | 0.01090 | 0.00573 | 0.00246 | 0.02356 |
| Hungary | ES | 84 | 0.01094 | 0.00471 | 0.00254 | 0.02228 |
| Mexico | NW | 26 | 0.01096 | 0.00363 | 0.00254 | 0.01500 |
| Libya | M | 162 | 0.01098 | 0.00348 | 0.00248 | 0.02036 |
| Albania | ES | 52 | 0.01124 | 0.00390 | 0.00249 | 0.01852 |
| Lebanon | NE | 10 | 0.01128 | 0.00123 | 0.00983 | 0.01244 |
| Ethiopia | E | 3919 | 0.01135 | 0.00312 | 0.00000 | 0.02709 |
| Tunisia | M | 56 | 0.01136 | 0.00385 | 0.00493 | 0.02015 |
| Yemen | M | 56 | 0.01140 | 0.00210 | 0.00493 | 0.01720 |
| Ukraine | ES | 100 | 0.01151 | 0.00342 | 0.00493 | 0.01980 |
| Switzerland | ES | 786 | 0.01151 | 0.00395 | 0.00246 | 0.02513 |
| Iraq | NE | 71 | 0.01171 | 0.00439 | 0.00249 | 0.02681 |
| Morocco | M | 111 | 0.01202 | 0.00348 | 0.00248 | 0.02073 |
| Portugal | M | 17 | 0.01206 | 0.00517 | 0.00491 | 0.01857 |
| Peru | NW | 29 | 0.01210 | 0.00316 | 0.00739 | 0.01741 |
| Oman | M | 10 | 0.01210 | 0.00337 | 0.00494 | 0.01481 |
| Italy | M | 324 | 0.01223 | 0.00467 | 0.00000 | 0.02730 |
| Turkey | NE | 1383 | 0.01234 | 0.00414 | 0.00240 | 0.02941 |
| Eritrea | E | 1 | 0.01235 | NA | 0.01235 | 0.01235 |
| Macedonia | M | 2 | 0.01236 | 0.00002 | 0.01235 | 0.01238 |
| Greece | M | 328 | 0.01255 | 0.00419 | 0.00262 | 0.02463 |
| USA | NW | 605 | 0.01259 | 0.00509 | 0.00000 | 0.02709 |
| Romania | ES | 105 | 0.01276 | 0.00534 | 0.00247 | 0.02519 |
| Pakistan | M | 321 | 0.01282 | 0.00466 | 0.00372 | 0.02757 |
| Kyrgyzstan | MA | 4 | 0.01296 | 0.00548 | 0.00491 | 0.01720 |
| Kazakhstan | MA | 7 | 0.01303 | 0.00341 | 0.00739 | 0.01728 |
| Canada | NW | 166 | 0.01303 | 0.00616 | 0.00246 | 0.02604 |
| Bhutan | EA | 10 | 0.01312 | 0.00160 | 0.01013 | 0.01538 |
| Georgia | NE | 125 | 0.01327 | 0.00379 | 0.00256 | 0.02217 |
| Iran | MA | 422 | 0.01345 | 0.00487 | 0.00246 | 0.02672 |
| Russia | ES | 217 | 0.01354 | 0.00399 | 0.00246 | 0.02481 |
| United Arab Emirates | M | 2 | 0.01357 | 0.00519 | 0.00990 | 0.01724 |
| Bulgaria | ES | 143 | 0.01358 | 0.00396 | 0.00493 | 0.02239 |
| Tajikistan | MA | 10 | 0.01361 | 0.00456 | 0.00985 | 0.02233 |
| Spain | M | 170 | 0.01373 | 0.00418 | 0.00491 | 0.02302 |
| China | EA | 806 | 0.01396 | 0.00584 | 0.00000 | 0.03226 |
| Afghanistan | MA | 345 | 0.01410 | 0.00501 | 0.00248 | 0.02778 |
| Uzbekistan | MA | 38 | 0.01424 | 0.00414 | 0.00493 | 0.02463 |
| India | M | 679 | 0.01433 | 0.00510 | 0.00000 | 0.03210 |
| Turkmenistan | MA | 31 | 0.01501 | 0.00602 | 0.00249 | 0.02965 |
| Armenia | NE | 22 | 0.01516 | 0.00423 | 0.00761 | 0.02006 |
| Japan | EA | 722 | 0.01531 | 0.00502 | 0.00000 | 0.03756 |
| North Korea | EA | 116 | 0.01619 | 0.00405 | 0.00000 | 0.02799 |
| South Korea | EA | 249 | 0.01637 | 0.00353 | 0.00513 | 0.02744 |
| Nepal | EA | 554 | 0.01652 | 0.00472 | 0.00293 | 0.02750 |
| Azerbaijan | NE | 13 | 0.01754 | 0.00510 | 0.00739 | 0.02228 |
| Moldova | ES | 1 | 0.02211 | NA | 0.02211 | 0.02211 |
| Unknown | 1888 | 0.01064 | 0.00522 | 0.00000 | 0.02709 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
Crown Copyright: @ His Majesty the King in Right of Canada, 2024. Submitted for possible open access publication under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fu, Y.-B. Patterns of the Predicted Mutation Burden in 19,778 Domesticated Barley Accessions Conserved Ex Situ. Int. J. Mol. Sci. 2024, 25, 5930. https://doi.org/10.3390/ijms25115930
Fu Y-B. Patterns of the Predicted Mutation Burden in 19,778 Domesticated Barley Accessions Conserved Ex Situ. International Journal of Molecular Sciences. 2024; 25(11):5930. https://doi.org/10.3390/ijms25115930
Chicago/Turabian StyleFu, Yong-Bi. 2024. "Patterns of the Predicted Mutation Burden in 19,778 Domesticated Barley Accessions Conserved Ex Situ" International Journal of Molecular Sciences 25, no. 11: 5930. https://doi.org/10.3390/ijms25115930
APA StyleFu, Y.-B. (2024). Patterns of the Predicted Mutation Burden in 19,778 Domesticated Barley Accessions Conserved Ex Situ. International Journal of Molecular Sciences, 25(11), 5930. https://doi.org/10.3390/ijms25115930