
Tailored Melatonin- and Donepezil-Based Hybrids Targeting Pathognomonic Changes in Alzheimer’s Disease: An In Vitro and In Vivo Investigation

 ,
,  ,
,  , ,
, , 
Abstract
1. Introduction
2. Results
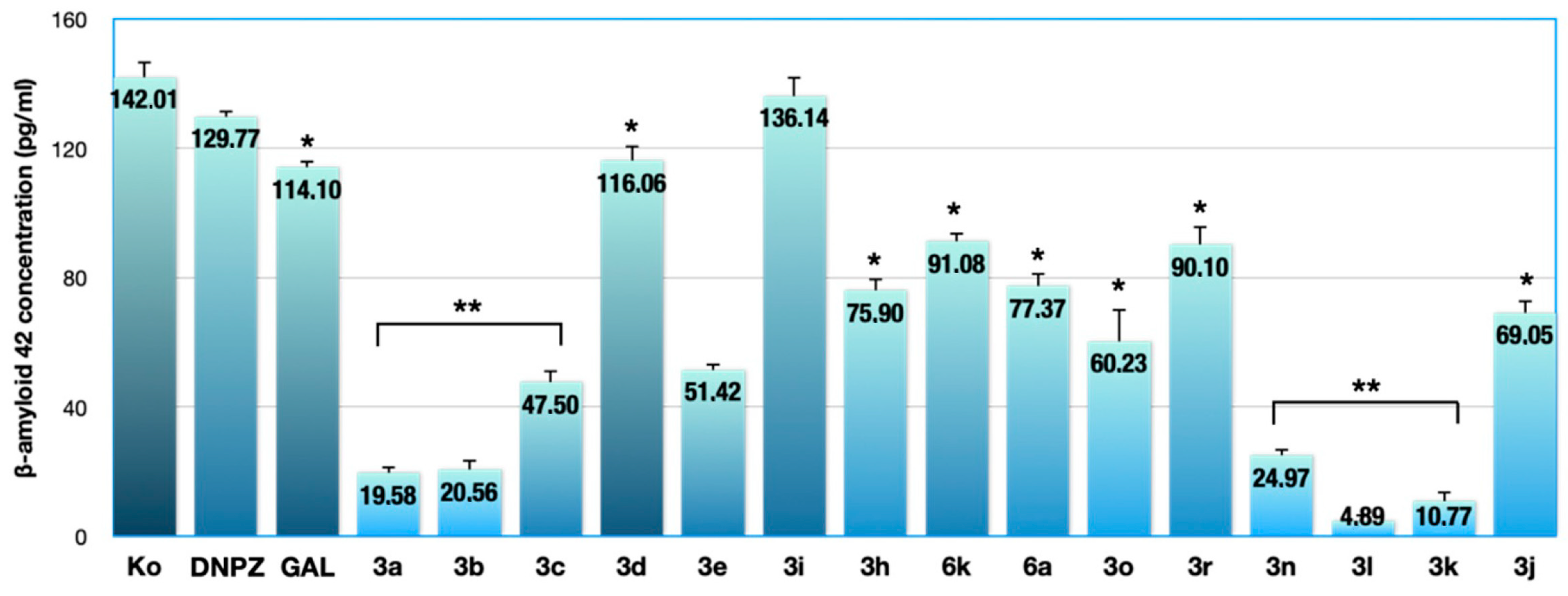
2.1. Results from the ELISA Assay
2.2. Results of the Acute Toxicity Study
2.3. Effects of the Tested Compounds on AChE Activity, MDA Quantity, and GSH Level in Mouse Brain Homogenate
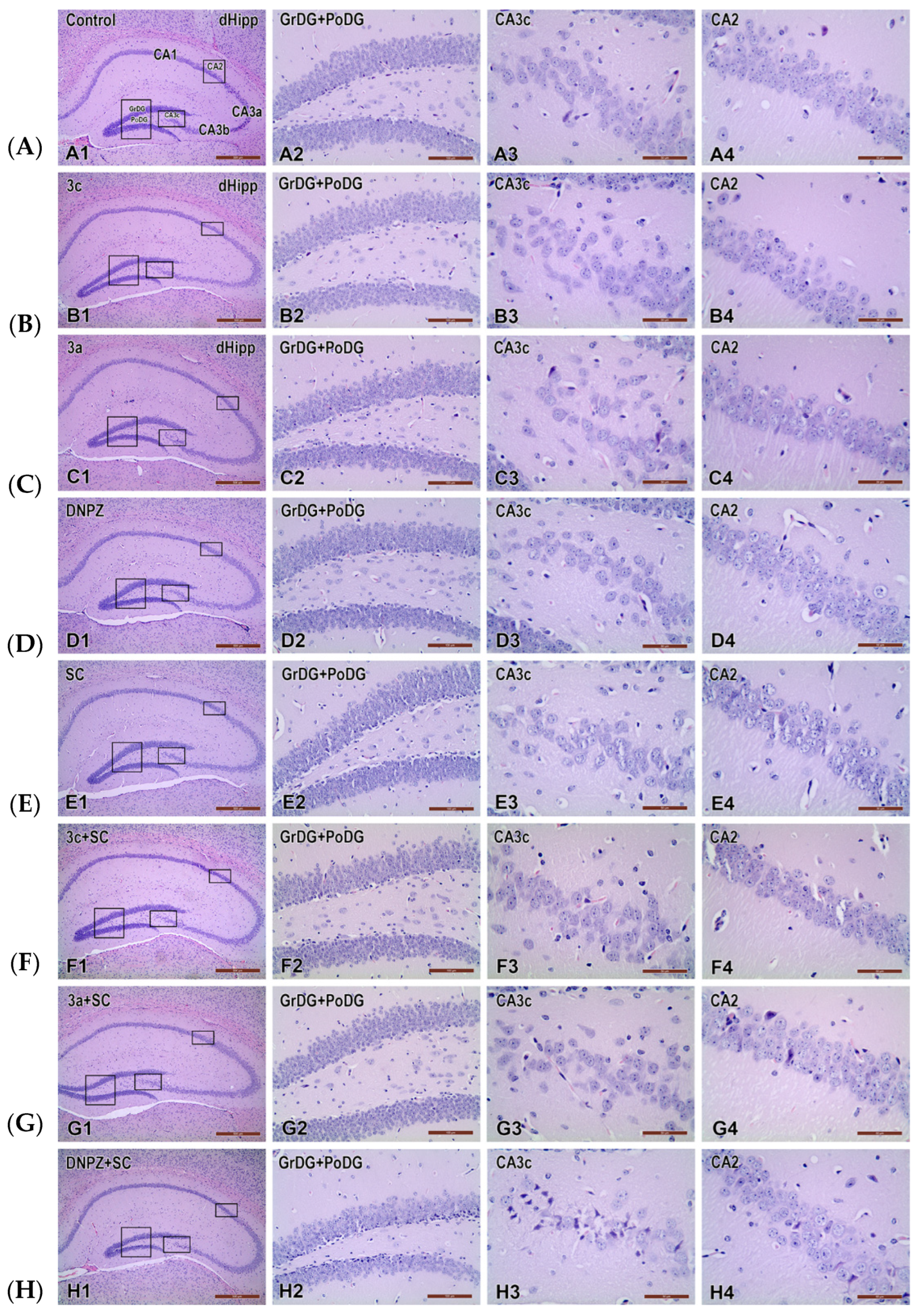
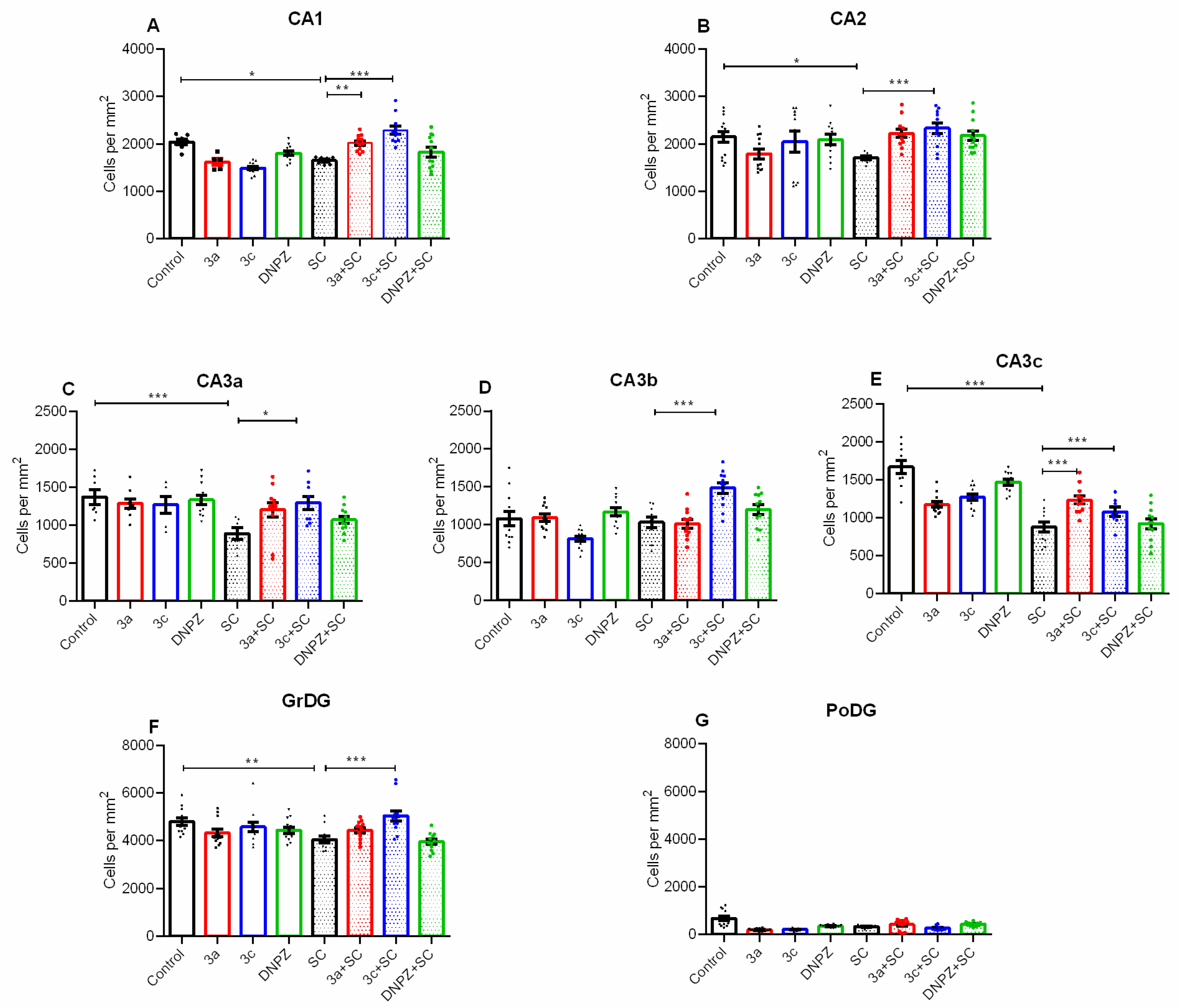
2.4. Effects of the Compounds Tested on Scopolamine-Induced Neuronal Damage in the Hippocampus of Mouse Brain Homogenate
3. Discussion
4. Materials and Methods
4.1. ELISA Assay
4.1.1. Cell Lines and Culture Conditions
4.1.2. Compounds
4.2. In Vivo Acute Toxicity Study
4.2.1. Experimental Animals
4.2.2. Drugs and Chemicals
4.2.3. Design of the In Vivo Experiment
- Group 1—control animals, treated with vehicle (0.9% saline i.p.) for 14 days;
- Group 2—animals treated with positive control DNPZ (1 mg/kg i.p.) for 14 days;
- Group 3—animals treated with the tested compound 3a alone at a dose 1/10 LD50 or 35 mg/kg i.p. for 14 days;
- Group 4—animals treated with the tested compound 3c alone at a dose 1/10 LD50 or 35 mg/kg i.p. for 14 days;
- Group 5—animals treated with SC alone (3 mg/kg i.p.) for 14 days;
- Group 6—animals treated with SC + DNPZ combination for 14 days;
- Group 7—animals treated with SC + 3a combination for 14 days;
- Group 8—animals treated with the combination of SC + 3c for 14 days.
4.2.4. Measurement of Acetylcholinesterase (AChE) Inhibition in Brain Homogenate
4.2.5. Measurement of Malondialdehyde (MDA) Levels in Brain Homogenate
4.2.6. Measurement of Glutathione (GSH) Levels in Brain Homogenate
4.2.7. Measurement of Hematological and Serum Biochemical Data
4.2.8. Pathomorphological Evaluation of Brain Tissue Specimens
4.2.9. Photodocumentation and Image Analysis
4.2.10. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Abubakar, M.B.; Sanusi, K.O.; Ugusman, A.; Mohamed, W.; Kamal, H.; Ibrahim, N.H.; Khoo, C.S.; Kumar, J. Alzheimer’s Disease: An Update and Insights into Pathophysiology. Front. Aging Neurosci. 2022, 14, 742408. [Google Scholar] [CrossRef]
- Alzheimer’s Association. Alzheimer’s Disease Facts and Figures; Alzheimer’s Association: Chicago, IL, USA, 2023; Volume 19, pp. 1598–1695. [Google Scholar]
- Tiwari, S.; Atluri, V.; Kaushik, A.; Yndart, A.; Nair, M. Alzheimer’s disease: Pathogenesis, diagnostics, and therapeutics. Int. J. Nanomed. 2019, 14, 5541–5554. [Google Scholar] [CrossRef] [PubMed]
- Danysz, W.; Parsons, C.G. Alzheimer’s disease, β-amyloid, glutamate, NMDA receptors and memantine—Searching for the connections. Br. J. Pharmacol. 2012, 167, 324–352. [Google Scholar] [CrossRef] [PubMed]
- Rostagno, A.A. Pathogenesis of Alzheimer’s Disease. Int. J. Mol. Sci. 2022, 24, 107. [Google Scholar] [CrossRef] [PubMed]
- Misrani, A.; Tabassum, S.; Yang, L. Mitochondrial Dysfunction and Oxidative Stress in Alzheimer’s Disease. Front. Aging Neurosci. 2021, 13, 617588. [Google Scholar] [CrossRef] [PubMed]
- Kocahan, S.; Doğan, Z. Mechanisms of Alzheimer’s Disease Pathogenesis and Prevention: The Brain, Neural Pathology, N-methyl-D-aspartate Receptors, Tau Protein and Other Risk Factors. Clin. Psychopharmacol. Neurosci. 2017, 15, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Guo, T.; Zhang, D.; Zeng, Y.; Huang, T.Y.; Xu, H.; Zhao, Y. Molecular and cellular mechanisms underlying the pathogenesis of Alzheimer’s disease. Mol. Neurodegener. 2020, 15, 40. [Google Scholar] [CrossRef]
- Kametani, F.; Hasegawa, M. Reconsideration of Amyloid Hypothesis and Tau Hypothesis in Alzheimer’s Disease. Front. Neurosci. 2018, 12, 25. [Google Scholar] [CrossRef]
- Dong, S.; Duan, Y.; Hu, Y.; Zhao, Z. Advances in the pathogenesis of Alzheimer’s disease: A re-evaluation of amyloid cascade hypothesis. Transl. Neurodegener. 2012, 1, 18. [Google Scholar] [CrossRef]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- Chen, G.F.; Xu, T.H.; Yan, Y.; Zhou, Y.R.; Jiang, Y.; Melcher, K.; Xu, H.E. Amyloid beta: Structure, biology and structure-based therapeutic development. Acta Pharmacol. Sin. 2017, 38, 1205–1235. [Google Scholar] [CrossRef] [PubMed]
- Barage, S.H.; Sonawane, K.D. Amyloid cascade hypothesis: Pathogenesis and therapeutic strategies in Alzheimer’s disease. Neuropeptides 2015, 52, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.-R.; Huang, J.-B.; Yang, S.-L.; Hong, F.-F. Role of Cholinergic Signaling in Alzheimer’s Disease. Molecules 2022, 27, 1816. [Google Scholar] [CrossRef] [PubMed]
- Ferreira-Vieira, T.H.; Guimaraes, I.M.; Silva, F.R.; Ribeiro, F.M. Alzheimer’s disease: Targeting the Cholinergic System. Curr. Neuropharmacol. 2016, 14, 101–115. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.-P.; Xie, Y.; Meng, X.-Y.; Kang, J.-S. History and progress of hypotheses and clinical trials for Alzheimer’s disease. Signal Transduct. Target. Ther. 2019, 4, 29. [Google Scholar] [CrossRef]
- Yoshiyama, Y.; Kojima, A.; Itoh, K.; Isose, S.; Koide, M.; Hori, K.; Arai, K. Does Anticholinergic Activity Affect Neuropathology Implication of Neuroinflammation in Alzheimer’s Disease. Neurodegener. Dis. 2015, 15, 140–148. [Google Scholar] [CrossRef] [PubMed]
- Stéphan, A.; Phillips, A.G. A case for a non-transgenic animal model of Alzheimer’s disease. Genes Brain Behav. 2005, 4, 157–172. [Google Scholar] [CrossRef]
- Kar, S.; Slowikowski SP, M.; Westaway, D.; Mount, H.T.J. Interactions between beta-amyloid and central cholinergic neurons: Implications for Alzheimer’s disease. J. Psychiatry Neurosci. JPN 2004, 29, 427–441. [Google Scholar]
- Lin, L.; Huang, Q.-X.; Yang, S.-S.; Chu, J.; Wang, J.-Z.; Tian, Q. Melatonin in Alzheimer’s Disease. Int. J. Mol. Sci. 2013, 14, 14575–14593. [Google Scholar] [CrossRef]
- Cardinali, D.P.; Furio, A.M.; Brusco, L.I. Clinical Aspects of Melatonin Intervention in Alzheimers Disease Progression. Curr. Neuropharmacol. 2010, 8, 218–227. [Google Scholar] [CrossRef]
- Nous, A.; Engelborghs, S.; Smolders, I. Melatonin levels in the Alzheimer’s disease continuum: A systematic review. Alzheimer’s Res. Ther. 2021, 13, 52. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, A.R.; Barbosa, D.J.; Remião, F.; Silva, R. Alzheimer’s disease: Insights and new prospects in disease pathophysiology, biomarkers and disease-modifying drugs. Biochem. Pharmacol. 2023, 211, 115522. [Google Scholar] [CrossRef] [PubMed]
- Sumsuzzman, D.M.; Choi, J.; Jin, Y.; Hong, Y. Neurocognitive effects of melatonin treatment in healthy adults and individuals with Alzheimer’s disease and insomnia: A systematic review and meta-analysis of randomized controlled trials. Neurosci. Biobehav. Rev. 2021, 127, 459–473. [Google Scholar] [CrossRef] [PubMed]
- Cardinali, D.; Vigo, D.; Olivar, N.; Vidal, M.; Brusco, L. Melatonin Therapy in Patients with Alzheimer’s Disease. Antioxidants 2014, 3, 245–277. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, J.; Wan, J.; Liu, A.; Sun, J. Melatonin regulates Aβ production/clearance balance and Aβ neurotoxicity: A potential therapeutic molecule for Alzheimer’s disease. Biomed. Pharmacother. 2020, 132, 110887. [Google Scholar] [CrossRef]
- Wu, Y.-H.; Zhou, J.-N.; Van Heerikhuize, J.; Jockers, R.; Swaab, D.F. Decreased MT1 melatonin receptor expression in the suprachiasmatic nucleus in aging and Alzheimer’s disease. Neurobiol. Aging 2007, 28, 1239–1247. [Google Scholar] [CrossRef] [PubMed]
- Roy, J.; Wong, K.Y.; Aquili, L.; Uddin, M.S.; Heng, B.C.; Tipoe, G.L.; Wong, K.H.; Fung, M.L.; Lim, L.W. Role of melatonin in Alzheimer’s disease: From preclinical studies to novel melatonin-based therapies. Front. Neuroendocrinol. 2022, 65, 100986. [Google Scholar] [CrossRef] [PubMed]
- Pappolla, M.; Reiter, R.; Bryant-Thomas, T.; Poeggeler, B. Oxidative Mediated Neurodegeneration in Alzheimers Disease: Melatonin and Related Indoles as Neuroprotective Agents. Curr. Med. Chem.-Immunol. Endocr. Metab. Agents 2003, 3, 33–46. [Google Scholar] [CrossRef]
- Bendheim, P.E.; Poeggeler, B.; Neria, E.; Ziv, V.; Pappolla, M.A.; Chain, D.G. Development of indole-3-propionic acid (OXIGONTM) for alzheimer’s disease. J. Mol. Neurosci. 2002, 19, 213–217. [Google Scholar] [CrossRef]
- Angelova, V.T.; Georgiev, B.; Pencheva, T.; Pajeva, I.; Rangelov, M.; Todorova, N.; Zheleva-Dimitrova, D.; Kalcheva-Yovkova, E.; Valkova, I.V.; Vassilev, N.; et al. Design, Synthesis, In Silico Studies and In Vitro Evaluation of New Indole- and/or Donepezil-like Hybrids as Multitarget-Directed Agents for Alzheimer’s Disease. Pharmaceuticals 2023, 16, 1194. [Google Scholar] [CrossRef]
- Serfilippi, L.M.; Pallman, D.R.S.; Russell, B. Serum clinical chemistry and hematology reference values in outbred stocks of albino mice from three commonly used vendors and two inbred strains of albino mice. Contemp. Top. Lab. Anim. Sci. 2003, 42, 46–52. [Google Scholar] [PubMed]
- Kurtz, D.M.; Travlos, G.S. The Clinical Chemistry of Laboratory Animals; CRC Press: Boca Raton, FL, USA, 2018. [Google Scholar]
- Wquimby, F.; Hluong, R. Clinical Chemistry of the Laboratory Mouse. In The Mouse in Biomedical Research; Elsevier: Amsterdam, The Netherlands, 2007; Volume 3, pp. 171–216. [Google Scholar]
- Watanabe, T.; Tomioka, N.H.; Watanabe, S.; Tsuchiya, M.; Hosoyamada, M. False In Vitro and In Vivo Elevations of Uric Acid Levels in Mouse Blood. Nucleosides Nucleotides Nucleic Acids 2014, 33, 192–198. [Google Scholar] [CrossRef] [PubMed]
- Tchekalarova, J.; Ivanova, P.; Krushovlieva, D.; Kortenska, L.; Angelova, V.T. Protective Effect of the Novel Melatonin Analogue Containing Donepezil Fragment on Memory Impairment via MT/ERK/CREB Signaling in the Hippocampus in a Rat Model of Pinealectomy and Subsequent Aβ1-42 Infusion. Int. J. Mol. Sci. 2024, 25, 1867. [Google Scholar] [CrossRef] [PubMed]
- Hampel, H.; Hardy, J.; Blennow, K.; Chen, C.; Perry, G.; Kim, S.H.; Vergallo, A. The amyloid-β pathway in Alzheimer’s disease. Mol. Psychiatry 2021, 26, 5481–5503. [Google Scholar] [CrossRef] [PubMed]
- Marshall, K.E.; Vadukul, D.M.; Staras, K.; Serpell, L.C. Misfolded amyloid-β-42 impairs the endosomal–lysosomal pathway. Cell Mol. Life Sci. 2020, 77, 5031–5043. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Kienlen-Campard, P.; Ahmed, M.; Liu, W.; Li, H.; Elliott, J.I.; Smith, S.O. Inhibitors of amyloid toxicity based on β-sheet packing of Aβ40 and Aβ42. Biochemistry 2006, 45, 5503–5516. [Google Scholar] [CrossRef] [PubMed]
- LaFerla, F.M.; Green, K.N.; Oddo, S. Intracellular amyloid-β in Alzheimer’s disease. Nat. Rev. Neurosci. 2007, 8, 499–509. [Google Scholar] [CrossRef] [PubMed]
- Cheignon, C.; Tomas, M.; Bonnefont-Rousselot, D.; Faller, P.; Hureau, C.; Collin, F. Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox Biol. 2018, 14, 450–464. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Mi, J.; Li, S.; Liu, Z.; Yang, J.; Chen, R.; Sang, Z. Design, synthesis and evaluation of quinoline-O-carbamate derivatives as multifunctional agents for the treatment of Alzheimer’s disease. J. Enzym. Inhib. Med. Chem. 2023, 38, 2169682. [Google Scholar] [CrossRef]
- Gomaa, A.A.; Makboul, R.M.; El-Mokhtar, M.A.; Abdel-Rahman, E.A.; Ahmed, E.A.; Nicola, M.A. Evaluation of the neuroprotective effect of donepezil in type 2 diabetic rats. Fundam. Clin. Pharmacol. 2021, 35, 97–112. [Google Scholar] [CrossRef] [PubMed]
- Hodge, A.; Sterner, B. Toxicity Classes; Canadian Center for Occupational Health and Safety: Hamilton, ON, Canada, 2005. [Google Scholar]
- Umukoro, S.; Adewole, F.; Eduviere, A.; Aderibigbe, A.; Onwuchekwa, C. Free Radical Scavenging Effect of Donepezil as the Possible Contribution to its Memory Enhancing Activity in Mice. Drug Res. 2013, 64, 236–239. [Google Scholar] [CrossRef] [PubMed]
- Simeonova, R.; Atanasova, M.; Stavrakov, G.; Philipova, I.; Doytchinova, I. Ex Vivo Antioxidant and Cholinesterase Inhibiting Effects of a Novel Galantamine–Curcumin Hybrid on Scopolamine-Induced Neurotoxicity in Mice. Int. J. Mol. Sci. 2022, 23, 14843. [Google Scholar] [CrossRef] [PubMed]
- Valgimigli, L. Lipid peroxidation and antioxidant protection. Biomolecules 2023, 13, 1291. [Google Scholar] [CrossRef]
- Chen, W.N.; Yeong, K.Y. Scopolamine, a toxin-induced experimental model used to study Alzheimer’s disease. CNS Neurol. Disord. Drug Targets 2020, 19, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Chen, J.S.; Li, S.; Zhang, F.; Deng, J.; Zeng, L.H.; Tan, J. Amyloid precursor protein: A regulatory hub in Alzheimer’s disease. Aging Dis. 2024, 15, 201. [Google Scholar]
- Takada-Takatori, Y.; Nakagawa, S.; Kimata, R.; Nao, Y.; Mizukawa, Y.; Urushidani, T.; Kume, T. DNPZ modulates amyloid precursor protein endocytosis and reduction by upregulating SNX33 expression in primary cortical neurons. Sci. Rep. 2019, 9, 11922. [Google Scholar] [CrossRef]
- Ongnok, B.; Khuanjing, T.; Chunchai, T.; Kerdphoo, S.; Jaiwongkam, T.; Chattipakorn, N.; Chattipakorn, S.C. Donepezil provides neuroprotective effects against brain injury and Alzheimer’s pathology under conditions of cardiac ischemia/reperfusion injury. Biochim. Biophys. Acta Mol. Basis Dis. 2021, 1867, 165975. [Google Scholar] [CrossRef]
- Jeong, J.H.; Choi, B.Y.; Kho, A.R.; Lee, S.H.; Hong, D.K.; Lee, S.H.; Lee, S.Y.; Song, H.K.; Choi, H.C.; Suh, S.W. Diverse Effects of an Acetylcholinesterase Inhibitor, Donepezil, on Hippocampal Neuronal Death after Pilocarpine-Induced Seizure. Int. J. Mol. Sci. 2017, 18, 2311. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.-J.; Park, J.-H.; Jeong, Y.J.; Hwang, J.-W.; Lee, S.; Lee, H.; Seol, E.; Kim, I.-W.; Cha, B.-Y.; Seo, J.; et al. Donepezil ameliorates Aβ pathology but not tau pathology in 5xFAD mice. Mol. Brain 2022, 15, 63. [Google Scholar] [CrossRef]
- Simeonova, R.; Vitcheva, V.; Kostadinova, I.; Valkova, I.; Philipova, I.; Stavrakov, G.; Danchev, N.; Doytchinova, I. Biochemical Studies on a Novel Potent Acetylcholinesterase Inhibitor with Dual site Binding for Treatment of Alzheimer’s Disease. C. R. Acad. Bulg. Sci. 2021, 74, 219–225. [Google Scholar]
- Lorke, D. A new approach to practical acute toxicity testing. Arch. Toxicol. 1983, 54, 275–287. [Google Scholar] [CrossRef] [PubMed]
- Ellman, G.L.; Courtney, K.D.; Andres, V.; Featherstone, R.M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Deby, C.; Goutier, R. New perspectives on the biochemistry of superoxide anion and the efficiency of superoxide dismutases. Biochem. Pharmacol. 1990, 39, 399–405. [Google Scholar] [CrossRef]
- Bump, E.A.; Taylor, Y.C.; Brown, J.M. Role of glutathione in the hypoxic cell cytotoxicity of misonidazole. Cancer Res. 1983, 43, 997–1002. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Hematology | Controls | 3a (35 mg/kg, i.p.) | 3c (35 mg/kg, i.p.) | DNPZ (1 mg/kg, i.p.) | SC (3 mg/kg, i.p.) | SC (3 mg/kg, i.p.) + DNPZ (1 mg/kg, i.p.) | SC (3 mg/kg, i.p.) + 3a (35 mg/kg, i.p.) | SC (3 mg/kg, i.p.) + 3c (35 mg/kg, i.p.) | Ref. Range (mice) |
|---|---|---|---|---|---|---|---|---|---|
| WBC ×103/µL | 9.75 ± 2.3 | 7.65 ± 2.27 | 8.63 ± 1.03 | 8.25 ± 1.21 | 10.63 ± 3.46 | 12.03 ± 2.62 * | 8.7 ± 1.42 | 9.12 ± 1.85 | 2.9–15.3 |
| RBC ×106/µL | 9.11 ± 0.17 | 9.00 ± 0.37 | 8.69 ± 0.69 | 9.05 ± 0.17 | 8.98 ± 0.22 | 8.58 ± 0.42 | 8.00 ± 0.64 | 8.55 ± 0.37 | 5.6–10.4 |
| Hgb g/L | 153 ± 8.87 | 157 ± 6.88 | 154.8 ± 6.1 | 153.5 ± 4.8 | 159.5 ± 8.1 | 151.3 ± 6.2 | 133 ± 4.5 | 144.5 ± 6.8 | 120–160 |
| HCT % | 43.15 ± 2.6 | 44.03 ± 1.95 | 44.83 ± 2.95 | 41.1 ± 1.21 | 45.33 ± 3.59 | 41.65 ± 1.15 | 40.15 ± 4.94 | 41.63 ± 1.62 | 36–52 |
| PLT ×103/µL | 437 ± 30.1 | 487.3 ± 41.2 | 395.8 ± 24.5 | 618 ± 32.2 * | 426.8 ± 48.4 | 435.5 ± 24.5 | 618.5 ± 30.1 * | 430 ± 37.6 | 127–939 |
| Blood Biochemistry | Controls | 3a (35 mg/kg, i.p.) | 3c (35 mg/kg, i.p.) | DNPZ (1 mg/kg, i.p.) | SC (3 mg/kg, i.p.) | SC (3 mg/kg, i.p.) + DNPZ (1 mg/kg, i.p.) | SC (3 mg/kg, i.p.) + 3a (35 mg/kg, i.p.) | SC (3 mg/kg, i.p.) + 3c (35 mg/kg, i.p.) | Ref. Range (mice) |
|---|---|---|---|---|---|---|---|---|---|
| GLU mmol/L | 8.95 ± 0.82 | 9.41 ± 0.77 | 8.95 ± 0.76 | 10.3 ± 0.84 | 10.96 ± 0.9 | 9.86 ± 0.79 | 10.33 ± 0.68 | 9.21 ± 0.56 | 4.2–11.6 [32] |
| UREA mmol/L | 12.01 ± 0.32 | 9.73 ± 0.36 | 10.41 ± 0.28 | 10.5 ± 0.22 | 11.37 ± 0.4 | 8.33 ± 0.33 | 10.58 ± 0.45 | 7.59 ± 0.52 | 3.8–12.3 [32] |
| CREAT µmol/L | 34.9 ± 2.3 | 31.8 ± 28.8 | 28.6 ± 6.6 | 32.6 ± 5.6 | 34.4 ± 4.8 | 33.6 ± 3.2 | 31.4 ± 3.6 | 41.5 ± 3.5 | 35–53 [32] |
| TP g/L | 58.1 ± 2.2 | 40.7 ± 3.1 | 59.6 ± 2.6 | 59.1 ± 3.6 | 58.5 ± 3.8 | 61 ± 5.6 | 60.2 ± 6.3 | 60.8 ± 5.7 | 53–63 [32] |
| ALB g/L | 35.8 ± 1.8 | 35.2 ± 1.7 | 33.4 ± 2.2 | 35.8 ± 3.1 | 37.6 ± 2.2 | 36.6 ± 2.8 | 36.2 ± 3.2 | 34.8 ± 2.6 | 26–39 [33] |
| ASAT U/L | 313 ± 4.5 | 383.7 ± 5.2 *+ | 382.3 ± 3.6 *+ | 321 ± 4.1 | 320.5 ± 6.8 | 368.8 ± 7.2 *+ | 365.7 ± 6.3 *+ | 369 ± 5.8 *+ | 57–329 [33] |
| ALAT U/L | 103.8 ± 2.2 | 70.5 ± 3.1 | 116.8 ± 3.3 | 89.2 ± 3.4 | 107 ± 4.5 | 87 ± 6.3 | 83.1 ± 7.2 | 94.8 ± 7.7 | 7–227 [34] |
| AMYL U/L | 3363 ± 42.3 | 1464 ± 38.3 | 1224 ± 48.3 | 1909 ± 28 | 1386 ± 22 | 1321 ± 42.3 | 1501 ± 52.1 | 2155 ± 55.2 | 1512–3084 [34] |
| Uric acid µmol/L | 83.6 ± 4.4 | 72.3 ± 2.3 | 177 ± 3.7 * | 120 ± 6.7 * | 88.3 ± 3.3 | 119 ± 7.8 * | 115 ± 8.2 * | 68.2 ± 4.4 | 0.1–760 [35] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mihaylova, R.; Angelova, V.T.; Tchekalarova, J.; Atanasova, D.; Ivanova, P.; Simeonova, R. Tailored Melatonin- and Donepezil-Based Hybrids Targeting Pathognomonic Changes in Alzheimer’s Disease: An In Vitro and In Vivo Investigation. Int. J. Mol. Sci. 2024, 25, 5969. https://doi.org/10.3390/ijms25115969
Mihaylova R, Angelova VT, Tchekalarova J, Atanasova D, Ivanova P, Simeonova R. Tailored Melatonin- and Donepezil-Based Hybrids Targeting Pathognomonic Changes in Alzheimer’s Disease: An In Vitro and In Vivo Investigation. International Journal of Molecular Sciences. 2024; 25(11):5969. https://doi.org/10.3390/ijms25115969
Chicago/Turabian StyleMihaylova, Rositsa, Violina T. Angelova, Jana Tchekalarova, Dimitrinka Atanasova, Petja Ivanova, and Rumyana Simeonova. 2024. "Tailored Melatonin- and Donepezil-Based Hybrids Targeting Pathognomonic Changes in Alzheimer’s Disease: An In Vitro and In Vivo Investigation" International Journal of Molecular Sciences 25, no. 11: 5969. https://doi.org/10.3390/ijms25115969
APA StyleMihaylova, R., Angelova, V. T., Tchekalarova, J., Atanasova, D., Ivanova, P., & Simeonova, R. (2024). Tailored Melatonin- and Donepezil-Based Hybrids Targeting Pathognomonic Changes in Alzheimer’s Disease: An In Vitro and In Vivo Investigation. International Journal of Molecular Sciences, 25(11), 5969. https://doi.org/10.3390/ijms25115969