
Disrupting EGFR–HER2 Transactivation by Pertuzumab in HER2-Positive Cancer: Quantitative Analysis Reveals EGFR Signal Input as Potential Predictor of Therapeutic Outcome

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
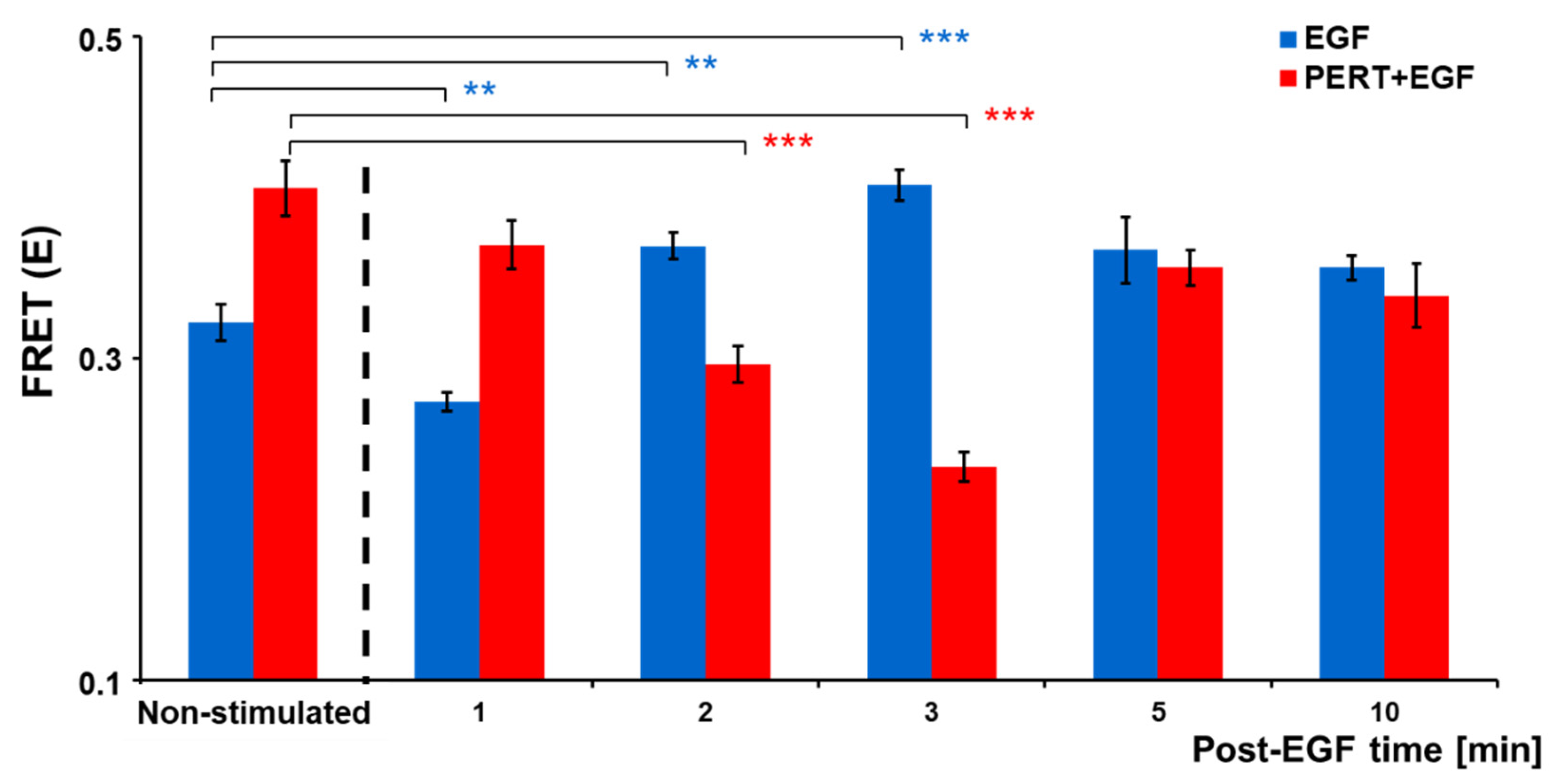
2.1. Molecular Interaction of EGFR and HER2 and Its Modulation by EGF and Pertuzumab
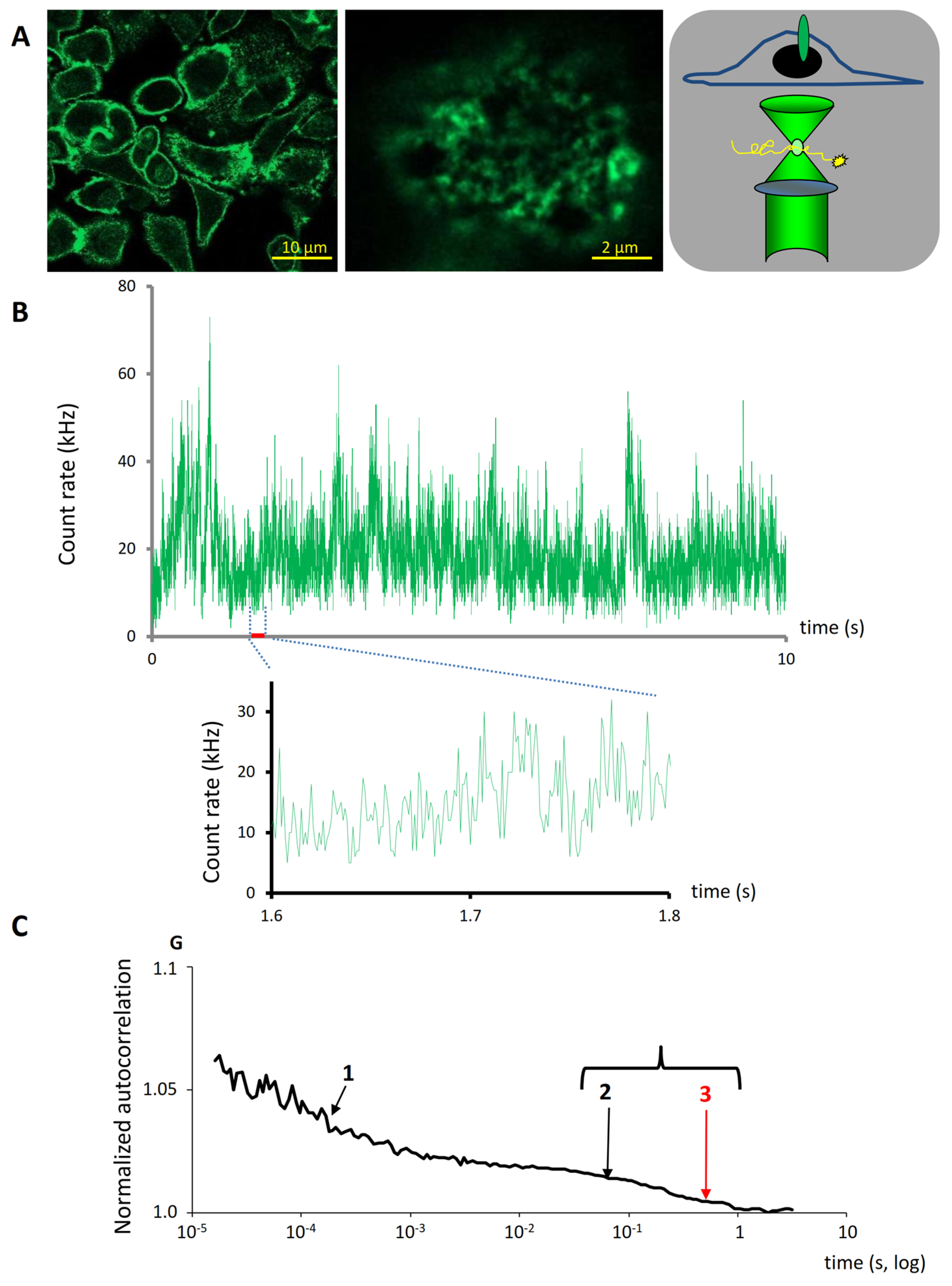
2.2. Membrane Dynamics of HER2 Revealed by FCS
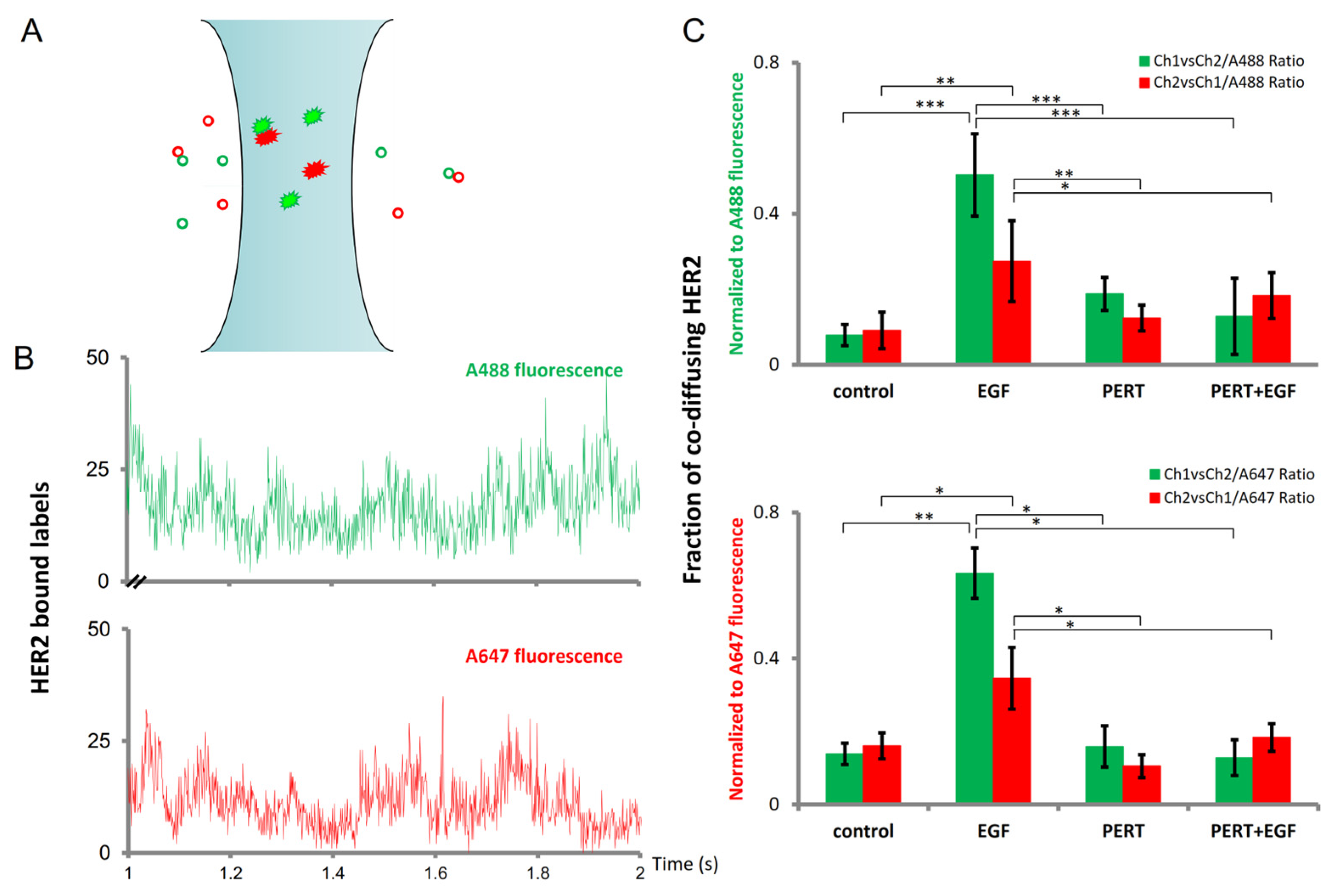
2.3. Fluorescence Cross-Correlation Spectroscopy Sensitively Detects the Fraction of Stably Co-Diffusing HER2 Molecules
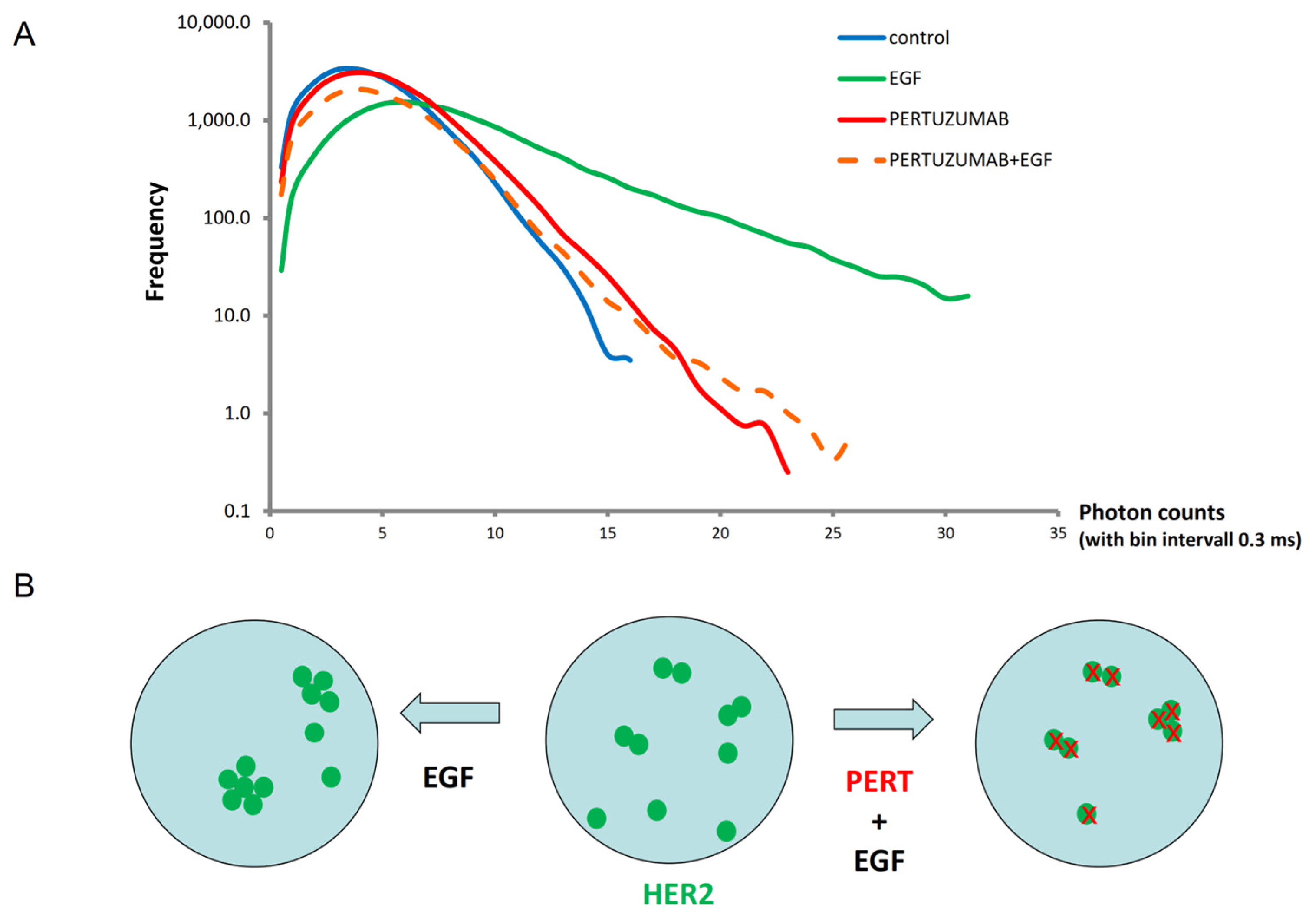
2.4. Photon-Count Histogram (PCH) Analysis of HER2 Oligomerization
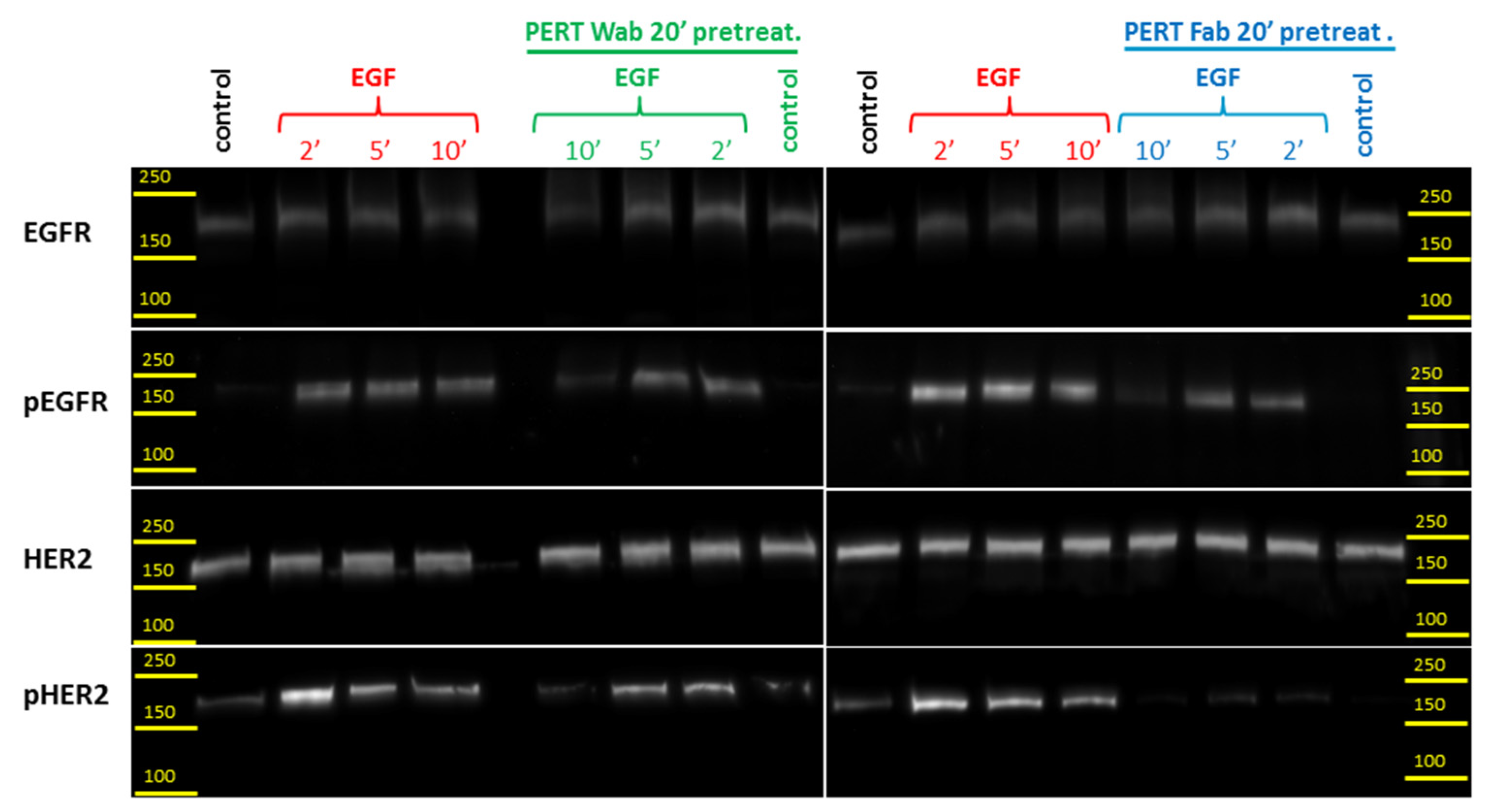
2.5. Western Blot Analysis of HER2 Transactivation by EGF Bound EGFR
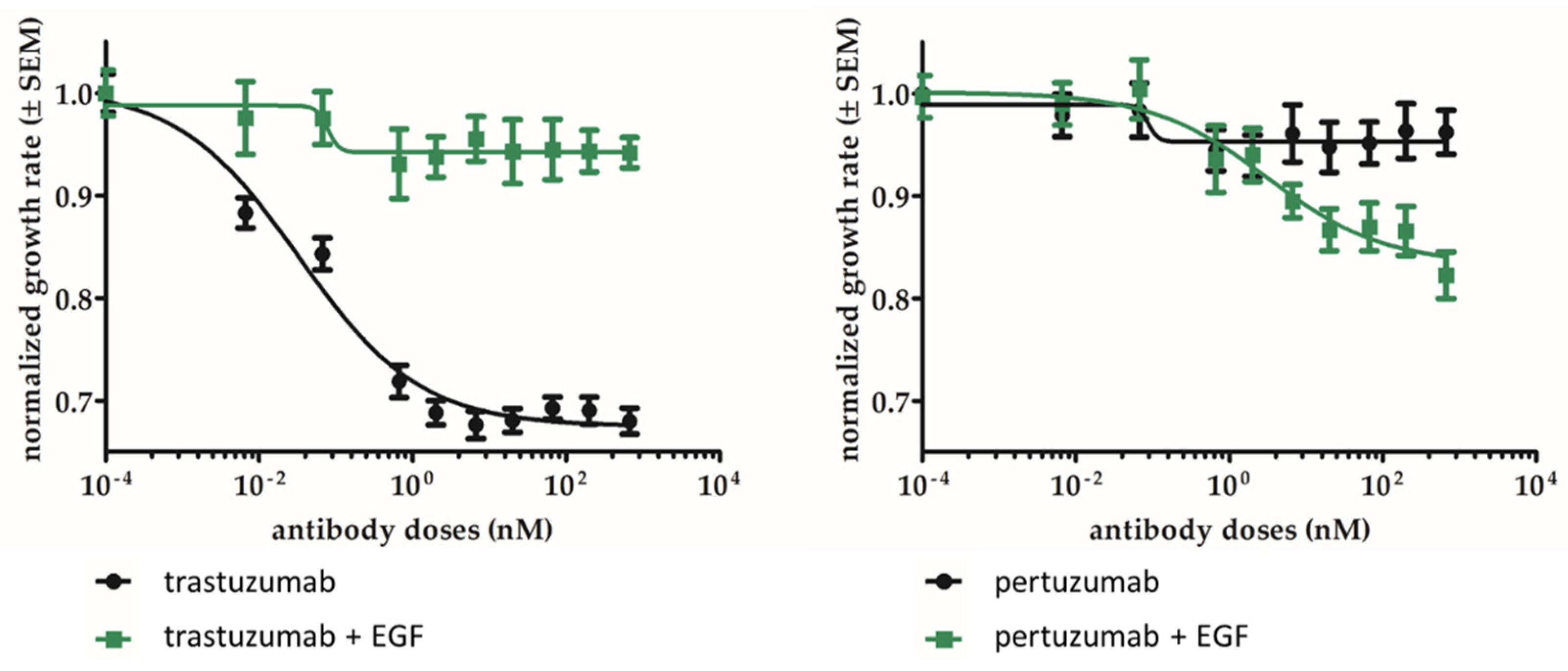
2.6. Pertuzumab Inhibits the Growth of EGFR and HER2 Double-Positive Cells In Vitro
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Concofal Microscopy Imaging and Spectrally Corrected Three-Filter (Ratiometric) FRET Imaging (riFRET)
4.3. Fluorescence Correlation and Cross-Correlation Spectroscopy
4.4. Analysis of FCS/FCCS Data
- -
- The fast two-dimensional membrane component is attributed to labeled monomers or smaller/more mobile oligomers in the membrane.
- -
- The slower but still two-dimensional membrane component is attributed to labeled higher oligomers/aggregates.
- -
- The model also contains a <10 ms time constant for the three-dimensional diffusion of non-bound Fab fragments.
4.5. Generation of the Photon-Count Histogram (PCH)
4.6. Western Blot Analysis of EGFR and HER2 Tyrosine Phosphorylation
4.7. Measurement of Cell Metabolic Activity
4.8. Statistical Evaluation of Data
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ADCC | Antibody dependent cellular cytotoxicity |
| EGF | Epidermal growth factor |
| EGFR | Epidermal growth factor receptor |
| FCS | Fluorescence correlation spectroscopy |
| FCCS | Fluorescence cross-correlation spectroscopy |
| FRET | Förster resonance energy transfer |
| HER2 | Human epidermal growth factor receptor-2 |
| PCH | Photon-count histogram |
| RiFRET | Ratiometric imaging FRET |
| TME | Tumor microenvironment |
References
- Yarden, Y.; Pines, G. The ERBB network: At last, cancer therapy meets systems biology. Nat. Rev. Cancer 2012, 12, 553–563. [Google Scholar] [CrossRef] [PubMed]
- Garrett, T.P.; McKern, N.M.; Lou, M.; Elleman, T.C.; Adams, T.E.; Lovrecz, G.O.; Kofler, M.; Jorissen, R.N.; Nice, E.C.; Burgess, A.W.; et al. The crystal structure of a truncated ErbB2 ectodomain reveals an active conformation, poised to interact with other ErbB receptors. Mol. Cell 2003, 11, 495–505. [Google Scholar] [CrossRef] [PubMed]
- Schneider, M.R.; Yarden, Y. Structure and function of epigen, the last EGFR ligand. Semin. Cell Dev. Biol. 2014, 28, 57–61. [Google Scholar] [CrossRef] [PubMed]
- Yarden, Y.; Sliwkowski, M.X. Untangling the ErbB signalling network. Nat. Rev. Mol. Cell Biol. 2001, 2, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Rubin, I.; Yarden, Y. The basic biology of HER2. Ann. Oncol. 2001, 12 (Suppl. 1), S3–S8. [Google Scholar] [CrossRef] [PubMed]
- Kato, S.; Tomson, B.N.; Buys, T.P.; Elkin, S.K.; Carter, J.L.; Kurzrock, R. Genomic Landscape of Malignant Mesotheliomas. Mol. Cancer Ther. 2016, 15, 2498–2507. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Xu, B. Targeted therapeutic options and future perspectives for HER2-positive breast cancer. Signal Transduct. Target. Ther. 2019, 4, 34. [Google Scholar] [CrossRef] [PubMed]
- Molnar, I.A.; Molnar, B.A.; Vizkeleti, L.; Fekete, K.; Tamas, J.; Deak, P.; Szundi, C.; Szekely, B.; Moldvay, J.; Vari-Kakas, S.; et al. Breast carcinoma subtypes show different patterns of metastatic behavior. Virchows Arch. 2017, 470, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Yan, M.; Parker, B.A.; Schwab, R.; Kurzrock, R. HER2 aberrations in cancer: Implications for therapy. Cancer Treat. Rev. 2014, 40, 770–780. [Google Scholar] [CrossRef] [PubMed]
- Ross, J.S.; Slodkowska, E.A.; Symmans, W.F.; Pusztai, L.; Ravdin, P.M.; Hortobagyi, G.N. The HER-2 receptor and breast cancer: Ten years of targeted anti-HER-2 therapy and personalized medicine. Oncologist 2009, 14, 320–368. [Google Scholar] [CrossRef] [PubMed]
- Baulida, J.; Kraus, M.H.; Alimandi, M.; Di Fiore, P.P.; Carpenter, G. All ErbB receptors other than the epidermal growth factor receptor are endocytosis impaired. J. Biol. Chem. 1996, 271, 5251–5257. [Google Scholar] [CrossRef] [PubMed]
- Waterman, H.; Sabanai, I.; Geiger, B.; Yarden, Y. Alternative intracellular routing of ErbB receptors may determine signaling potency. J. Biol. Chem. 1998, 273, 13819–13827. [Google Scholar] [CrossRef] [PubMed]
- Hendriks, B.S.; Wiley, H.S.; Lauffenburger, D. HER2-mediated effects on EGFR endosomal sorting: Analysis of biophysical mechanisms. Biophys. J. 2003, 85, 2732–2745. [Google Scholar] [CrossRef] [PubMed]
- Hendriks, B.S.; Opresko, L.K.; Wiley, H.S.; Lauffenburger, D. Quantitative analysis of HER2-mediated effects on HER2 and epidermal growth factor receptor endocytosis: Distribution of homo- and heterodimers depends on relative HER2 levels. J. Biol. Chem. 2003, 278, 23343–23351. [Google Scholar] [CrossRef]
- Lenferink, A.E.; Pinkas-Kramarski, R.; van de Poll, M.L.; van Vugt, M.J.; Klapper, L.N.; Tzahar, E.; Waterman, H.; Sela, M.; van Zoelen, E.J.; Yarden, Y. Differential endocytic routing of homo- and hetero-dimeric ErbB tyrosine kinases confers signaling superiority to receptor heterodimers. EMBO J. 1998, 17, 3385–3397. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.; Chantry, A.; Epstein, R.J. Overexpression of ErbB2 impairs ligand-dependent downregulation of epidermal growth factor receptors via a post-transcriptional mechanism. J. Cell. Biochem. 1999, 74, 23–30. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, L.; Yeung, T.K.; Chen, X. Endocytosis deficiency of epidermal growth factor (EGF) receptor-ErbB2 heterodimers in response to EGF stimulation. Mol. Biol. Cell 1999, 10, 1621–1636. [Google Scholar] [CrossRef] [PubMed]
- Friedlander, E.; Barok, M.; Szollosi, J.; Vereb, G. ErbB-directed immunotherapy: Antibodies in current practice and promising new agents. Immunol. Lett. 2008, 116, 126–140. [Google Scholar] [CrossRef] [PubMed]
- Szoor, A.; Toth, G.; Zsebik, B.; Szabo, V.; Eshhar, Z.; Abken, H.; Vereb, G. Trastuzumab derived HER2-specific CARs for the treatment of trastuzumab-resistant breast cancer: CAR T cells penetrate and eradicate tumors that are not accessible to antibodies. Cancer Lett. 2020, 484, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Albanell, J.; Baselga, J. Trastuzumab, a humanized anti-HER2 monoclonal antibody, for the treatment of breast cancer. Drugs Today 1999, 35, 931–946. [Google Scholar] [CrossRef]
- Esteva, F.J.; Valero, V.; Booser, D.; Guerra, L.T.; Murray, J.L.; Pusztai, L.; Cristofanilli, M.; Arun, B.; Esmaeli, B.; Fritsche, H.A.; et al. Phase II study of weekly docetaxel and trastuzumab for patients with HER-2-overexpressing metastatic breast cancer. J. Clin. Oncol. 2002, 20, 1800–1808. [Google Scholar] [CrossRef] [PubMed]
- Cuello, M.; Ettenberg, S.A.; Clark, A.S.; Keane, M.M.; Posner, R.H.; Nau, M.M.; Dennis, P.A.; Lipkowitz, S. Down-regulation of the erbB-2 receptor by trastuzumab (herceptin) enhances tumor necrosis factor-related apoptosis-inducing ligand-mediated apoptosis in breast and ovarian cancer cell lines that overexpress erbB-2. Cancer Res. 2001, 61, 4892–4900. [Google Scholar] [PubMed]
- Nagata, Y.; Lan, K.H.; Zhou, X.; Tan, M.; Esteva, F.J.; Sahin, A.A.; Klos, K.S.; Li, P.; Monia, B.P.; Nguyen, N.T.; et al. PTEN activation contributes to tumor inhibition by trastuzumab, and loss of PTEN predicts trastuzumab resistance in patients. Cancer Cell 2004, 6, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Izumi, Y.; Xu, L.; di Tomaso, E.; Fukumura, D.; Jain, R.K. Tumour biology: Herceptin acts as an anti-angiogenic cocktail. Nature 2002, 416, 279–280. [Google Scholar] [CrossRef] [PubMed]
- Clynes, R.A.; Towers, T.L.; Presta, L.G.; Ravetch, J.V. Inhibitory Fc receptors modulate in vivo cytotoxicity against tumor targets. Nat. Med. 2000, 6, 443–446. [Google Scholar] [CrossRef] [PubMed]
- Barok, M.; Isola, J.; Palyi-Krekk, Z.; Nagy, P.; Juhasz, I.; Vereb, G.; Kauraniemi, P.; Kapanen, A.; Tanner, M.; Vereb, G.; et al. Trastuzumab causes antibody-dependent cellular cytotoxicity-mediated growth inhibition of submacroscopic JIMT-1 breast cancer xenografts despite intrinsic drug resistance. Mol. Cancer Ther. 2007, 6, 2065–2072. [Google Scholar] [CrossRef] [PubMed]
- Barok, M.; Balazs, M.; Nagy, P.; Rakosy, Z.; Treszl, A.; Toth, E.; Juhasz, I.; Park, J.W.; Isola, J.; Vereb, G.; et al. Trastuzumab decreases the number of circulating and disseminated tumor cells despite trastuzumab resistance of the primary tumor. Cancer Lett. 2008, 260, 198–208. [Google Scholar] [CrossRef] [PubMed]
- Capelan, M.; Pugliano, L.; De Azambuja, E.; Bozovic, I.; Saini, K.S.; Sotiriou, C.; Loi, S.; Piccart-Gebhart, M.J. Pertuzumab: New hope for patients with HER2-positive breast cancer. Ann. Oncol. 2013, 24, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Sakai, K.; Yokote, H.; Murakami-Murofushi, K.; Tamura, T.; Saijo, N.; Nishio, K. Pertuzumab, a novel HER dimerization inhibitor, inhibits the growth of human lung cancer cells mediated by the HER3 signaling pathway. Cancer Sci. 2007, 98, 1498–1503. [Google Scholar] [CrossRef] [PubMed]
- Mann, M.; Sheng, H.; Shao, J.; Williams, C.S.; Pisacane, P.I.; Sliwkowski, M.X.; DuBois, R.N. Targeting cyclooxygenase 2 and HER-2/neu pathways inhibits colorectal carcinoma growth. Gastroenterology 2001, 120, 1713–1719. [Google Scholar] [CrossRef] [PubMed]
- Mendoza, N.; Phillips, G.L.; Silva, J.; Schwall, R.; Wickramasinghe, D. Inhibition of ligand-mediated HER2 activation in androgen-independent prostate cancer. Cancer Res. 2002, 62, 5485–5488. [Google Scholar] [PubMed]
- Mullen, P.; Cameron, D.A.; Hasmann, M.; Smyth, J.F.; Langdon, S.P. Sensitivity to pertuzumab (2C4) in ovarian cancer models: Cross-talk with estrogen receptor signaling. Mol. Cancer Ther. 2007, 6, 93–100. [Google Scholar] [CrossRef] [PubMed]
- DeFazio-Eli, L.; Strommen, K.; Dao-Pick, T.; Parry, G.; Goodman, L.; Winslow, J. Quantitative assays for the measurement of HER1-HER2 heterodimerization and phosphorylation in cell lines and breast tumors: Applications for diagnostics and targeted drug mechanism of action. Breast Cancer Res. 2011, 13, R44. [Google Scholar] [CrossRef] [PubMed]
- Swain, S.M.; Kim, S.B.; Cortes, J.; Ro, J.; Semiglazov, V.; Campone, M.; Ciruelos, E.; Ferrero, J.M.; Schneeweiss, A.; Knott, A.; et al. Pertuzumab, trastuzumab, and docetaxel for HER2-positive metastatic breast cancer (CLEOPATRA study): Overall survival results from a randomised, double-blind, placebo-controlled, phase 3 study. Lancet Oncol. 2013, 14, 461–471. [Google Scholar] [CrossRef] [PubMed]
- Howie, L.J.; Scher, N.S.; Amiri-Kordestani, L.; Zhang, L.; King-Kallimanis, B.L.; Choudhry, Y.; Schroeder, J.; Goldberg, K.B.; Kluetz, P.G.; Ibrahim, A.; et al. FDA Approval Summary: Pertuzumab for Adjuvant Treatment of HER2-Positive Early Breast Cancer. Clin. Cancer Res. 2019, 25, 2949–2955. [Google Scholar] [CrossRef] [PubMed]
- Toth, G.; Szoor, A.; Simon, L.; Yarden, Y.; Szollosi, J.; Vereb, G. The combination of trastuzumab and pertuzumab administered at approved doses may delay development of trastuzumab resistance by additively enhancing antibody-dependent cell-mediated cytotoxicity. MAbs 2016, 8, 1361–1370. [Google Scholar] [CrossRef]
- von Minckwitz, G.; Procter, M.; de Azambuja, E.; Zardavas, D.; Benyunes, M.; Viale, G.; Suter, T.; Arahmani, A.; Rouchet, N.; Clark, E.; et al. Adjuvant Pertuzumab and Trastuzumab in Early HER2-Positive Breast Cancer. N. Engl. J. Med. 2017, 377, 122–131. [Google Scholar] [CrossRef]
- Roszik, J.; Toth, G.; Szollosi, J.; Vereb, G. Validating pharmacological disruption of protein-protein interactions by acceptor photobleaching FRET imaging. Methods Mol. Biol. 2013, 986, 165–178. [Google Scholar] [CrossRef] [PubMed]
- Schwille, P.; Haupts, U.; Maiti, S.; Webb, W.W. Molecular dynamics in living cells observed by fluorescence correlation spectroscopy with one- and two-photon excitation. Biophys. J. 1999, 77, 2251–2265. [Google Scholar] [CrossRef] [PubMed]
- Widengren, J.; Thyberg, P. FCS cell surface measurements--photophysical limitations and consequences on molecular ensembles with heterogenic mobilities. Cytom. A 2005, 68, 101–112. [Google Scholar] [CrossRef]
- Vamosi, G.; Baudendistel, N.; von der Lieth, C.W.; Szaloki, N.; Mocsar, G.; Muller, G.; Brazda, P.; Waldeck, W.; Damjanovich, S.; Langowski, J.; et al. Conformation of the c-Fos/c-Jun complex in vivo: A combined FRET, FCCS, and MD-modeling study. Biophys. J. 2008, 94, 2859–2868. [Google Scholar] [CrossRef] [PubMed]
- Vamosi, G.; Friedlander-Brock, E.; Ibrahim, S.M.; Brock, R.; Szollosi, J.; Vereb, G. EGF Receptor Stalls upon Activation as Evidenced by Complementary Fluorescence Correlation Spectroscopy and Fluorescence Recovery after Photobleaching Measurements. Int. J. Mol. Sci. 2019, 20, 3370. [Google Scholar] [CrossRef] [PubMed]
- Mezosi-Csaplar, M.; Szoor, A.; Vereb, G. CD28 and 41BB Costimulatory Domains Alone or in Combination Differentially Influence Cell Surface Dynamics and Organization of Chimeric Antigen Receptors and Early Activation of CAR T Cells. Cancers 2023, 15, 3081. [Google Scholar] [CrossRef] [PubMed]
- Reho, B.; Fadel, L.; Brazda, P.; Benziane, A.; Hegedus, E.; Sen, P.; Gadella, T.W.J.; Toth, K.; Nagy, L.; Vamosi, G. Agonist-controlled competition of RAR and VDR nuclear receptors for heterodimerization with RXR is manifested in their DNA binding. J. Biol. Chem. 2023, 299, 102896. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Muller, J.D.; Berland, K.M.; Gratton, E. Fluorescence fluctuation spectroscopy. Methods 1999, 19, 234–252. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Muller, J.D.; So, P.T.; Gratton, E. The photon counting histogram in fluorescence fluctuation spectroscopy. Biophys. J. 1999, 77, 553–567. [Google Scholar] [CrossRef] [PubMed]
- Ujlaky-Nagy, L.; Nagy, P.; Szollosi, J.; Vereb, G. Flow Cytometric FRET Analysis of Protein Interactions. Methods Mol. Biol. 2018, 1678, 393–419. [Google Scholar] [CrossRef] [PubMed]
- Vereb, G.; Nagy, P.; Szollosi, J. Flow cytometric FRET analysis of protein interaction. Methods Mol. Biol. 2011, 699, 371–392. [Google Scholar] [CrossRef] [PubMed]
- Roszik, J.; Lisboa, D.; Szollosi, J.; Vereb, G. Evaluation of intensity-based ratiometric FRET in image cytometry—Approaches and a software solution. Cytom. A 2009, 75, 761–767. [Google Scholar] [CrossRef] [PubMed]
- Muller, J.D.; Chen, Y.; Gratton, E. Resolving heterogeneity on the single molecular level with the photon-counting histogram. Biophys. J. 2000, 78, 474–486. [Google Scholar] [CrossRef]
- Kask, P.; Palo, K.; Ullmann, D.; Gall, K. Fluorescence-intensity distribution analysis and its application in biomolecular detection technology. Proc. Natl. Acad. Sci. USA 1999, 96, 13756–13761. [Google Scholar] [CrossRef] [PubMed]
- Thompson, N.L.; Lieto, A.M.; Allen, N.W. Recent advances in fluorescence correlation spectroscopy. Curr. Opin. Struct. Biol. 2002, 12, 634–641. [Google Scholar] [CrossRef] [PubMed]
- Thatte, H.S.; Bridges, K.R.; Golan, D.E. Microtubule inhibitors differentially affect translational movement, cell surface expression, and endocytosis of transferrin receptors in K562 cells. J. Cell. Physiol. 1994, 160, 345–357. [Google Scholar] [CrossRef] [PubMed]
- Diermeier-Daucher, S.; Hasmann, M.; Brockhoff, G. Flow cytometric FRET analysis of erbB receptor interaction on a cell-by-cell basis. Ann. N. Y. Acad. Sci. 2008, 1130, 280–286. [Google Scholar] [CrossRef] [PubMed]
- Diermeier-Daucher, S.; Breindl, S.; Buchholz, S.; Ortmann, O.; Brockhoff, G. Modular anti-EGFR and anti-Her2 targeting of SK-BR-3 and BT474 breast cancer cell lines in the presence of ErbB receptor-specific growth factors. Cytometry Part A 2011, 79, 684–693. [Google Scholar] [CrossRef] [PubMed]
- Colavito, S.A. AXL as a Target in Breast Cancer Therapy. J. Oncol. 2020, 2020, 5291952. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, H.; Yamada, T.; Wang, R.; Tanimura, K.; Adachi, Y.; Nishiyama, A.; Tanimoto, A.; Takeuchi, S.; Araujo, L.H.; Boroni, M.; et al. AXL confers intrinsic resistance to osimertinib and advances the emergence of tolerant cells. Nat. Commun. 2019, 10, 259. [Google Scholar] [CrossRef] [PubMed]
- Geng, K.; Kumar, S.; Kimani, S.G.; Kholodovych, V.; Kasikara, C.; Mizuno, K.; Sandiford, O.; Rameshwar, P.; Kotenko, S.V.; Birge, R.B. Requirement of Gamma-Carboxyglutamic Acid Modification and Phosphatidylserine Binding for the Activation of Tyro3, Axl, and Mertk Receptors by Growth Arrest-Specific 6. Front. Immunol. 2017, 8, 1521. [Google Scholar] [CrossRef] [PubMed]
- Birge, R.B.; Boeltz, S.; Kumar, S.; Carlson, J.; Wanderley, J.; Calianese, D.; Barcinski, M.; Brekken, R.A.; Huang, X.; Hutchins, J.T.; et al. Phosphatidylserine is a global immunosuppressive signal in efferocytosis, infectious disease, and cancer. Cell Death Differ. 2016, 23, 962–978. [Google Scholar] [CrossRef]
- Goyette, M.A.; Duhamel, S.; Aubert, L.; Pelletier, A.; Savage, P.; Thibault, M.P.; Johnson, R.M.; Carmeliet, P.; Basik, M.; Gaboury, L.; et al. The Receptor Tyrosine Kinase AXL Is Required at Multiple Steps of the Metastatic Cascade during HER2-Positive Breast Cancer Progression. Cell Rep. 2018, 23, 1476–1490. [Google Scholar] [CrossRef]
- Noronha, A.; Belugali Nataraj, N.; Lee, J.S.; Zhitomirsky, B.; Oren, Y.; Oster, S.; Lindzen, M.; Mukherjee, S.; Will, R.; Ghosh, S.; et al. AXL and Error-Prone DNA Replication Confer Drug Resistance and Offer Strategies to Treat EGFR-Mutant Lung Cancer. Cancer Discov. 2022, 12, 2666–2683. [Google Scholar] [CrossRef] [PubMed]
- Mollard, A.; Warner, S.L.; Call, L.T.; Wade, M.L.; Bearss, J.J.; Verma, A.; Sharma, S.; Vankayalapati, H.; Bearss, D.J. Design, Synthesis and Biological Evaluation of a Series of Novel Axl Kinase Inhibitors. ACS Med. Chem. Lett. 2011, 2, 907–912. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Liao, M.; Li, M.; Sun, M.; Xi, N.; Zeng, Y. Structure-based discovery of potent inhibitors of Axl: Design, synthesis, and biological evaluation. RSC Med. Chem. 2022, 13, 1246–1264. [Google Scholar] [CrossRef] [PubMed]
- Fang, F.; Dai, Y.; Wang, H.; Ji, Y.; Liang, X.; Peng, X.; Li, J.; Zhao, Y.; Li, C.; Wang, D.; et al. Structure-based drug discovery of novel fused-pyrazolone carboxamide derivatives as potent and selective AXL inhibitors. Acta Pharm. Sin. B 2023, 13, 4918–4933. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Sun, D.; Wang, W.; Peng, X.; Zhan, Z.; Ji, Y.; Shen, Y.; Geng, M.; Ai, J.; Duan, W. Discovery of pyrrolo [2,3-d]pyrimidine derivatives as potent Axl inhibitors: Design, synthesis and biological evaluation. Eur. J. Med. Chem. 2021, 220, 113497. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.; Zhang, Y.; Wang, J.; Yu, Q.; Peng, X.; Zou, J.; Zhou, L.; Tan, L.; Duan, Y.; Zhou, Y.; et al. Discovery of 3-Aminopyrazole Derivatives as New Potent and Orally Bioavailable AXL Inhibitors. J. Med. Chem. 2022, 65, 15374–15390. [Google Scholar] [CrossRef]
- Holland, S.J.; Pan, A.; Franci, C.; Hu, Y.; Chang, B.; Li, W.; Duan, M.; Torneros, A.; Yu, J.; Heckrodt, T.J.; et al. R428, a selective small molecule inhibitor of Axl kinase, blocks tumor spread and prolongs survival in models of metastatic breast cancer. Cancer Res. 2010, 70, 1544–1554. [Google Scholar] [CrossRef] [PubMed]
- Okura, N.; Nishioka, N.; Yamada, T.; Taniguchi, H.; Tanimura, K.; Katayama, Y.; Yoshimura, A.; Watanabe, S.; Kikuchi, T.; Shiotsu, S.; et al. ONO-7475, a Novel AXL Inhibitor, Suppresses the Adaptive Resistance to Initial EGFR-TKI Treatment in EGFR-Mutated Non-Small Cell Lung Cancer. Clin. Cancer Res. 2020, 26, 2244–2256. [Google Scholar] [CrossRef] [PubMed]
- McDaniel, N.K.; Cummings, C.T.; Iida, M.; Hulse, J.; Pearson, H.E.; Vasileiadi, E.; Parker, R.E.; Orbuch, R.A.; Ondracek, O.J.; Welke, N.B.; et al. MERTK Mediates Intrinsic and Adaptive Resistance to AXL-targeting Agents. Mol. Cancer Ther. 2018, 17, 2297–2308. [Google Scholar] [CrossRef] [PubMed]
- Bhalla, S.; Fattah, F.J.; Ahn, C.; Williams, J.; Macchiaroli, A.; Padro, J.; Pogue, M.; Dowell, J.E.; Putnam, W.C.; McCracken, N.; et al. Phase 1 trial of bemcentinib (BGB324), a first-in-class, selective AXL inhibitor, with docetaxel in patients with previously treated advanced non-small cell lung cancer. Lung Cancer 2023, 182, 107291. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.W.; Liu, Y.C.; Chang, Y.H.; Lin, C.C.; Huang, P.M.; Hua, K.T.; Lee, J.M.; Hsieh, M.S. Cabozantinib (XL184) and R428 (BGB324) Inhibit the Growth of Esophageal Squamous Cell Carcinoma (ESCC). Front. Oncol. 2019, 9, 1138. [Google Scholar] [CrossRef] [PubMed]
- Aveic, S.; Corallo, D.; Porcu, E.; Pantile, M.; Boso, D.; Zanon, C.; Viola, G.; Sidarovich, V.; Mariotto, E.; Quattrone, A.; et al. TP-0903 inhibits neuroblastoma cell growth and enhances the sensitivity to conventional chemotherapy. Eur. J. Pharmacol. 2018, 818, 435–448. [Google Scholar] [CrossRef] [PubMed]
- Sinha, S.; Boysen, J.C.; Chaffee, K.G.; Kabat, B.F.; Slager, S.L.; Parikh, S.A.; Secreto, C.R.; Call, T.; Shanafelt, T.D.; Leis, J.F.; et al. Chronic lymphocytic leukemia cells from ibrutinib treated patients are sensitive to Axl receptor tyrosine kinase inhibitor therapy. Oncotarget 2018, 9, 37173–37184. [Google Scholar] [CrossRef] [PubMed]
- Park, I.K.; Mundy-Bosse, B.; Whitman, S.P.; Zhang, X.; Warner, S.L.; Bearss, D.J.; Blum, W.; Marcucci, G.; Caligiuri, M.A. Receptor tyrosine kinase Axl is required for resistance of leukemic cells to FLT3-targeted therapy in acute myeloid leukemia. Leukemia 2015, 29, 2382–2389. [Google Scholar] [CrossRef]
- Gao, J.; Li, H.R.; Jin, C.; Jiang, J.H.; Ding, J.Y. Strategies to overcome acquired resistance to EGFR TKI in the treatment of non-small cell lung cancer. Clin. Transl. Oncol. 2019, 21, 1287–1301. [Google Scholar] [CrossRef] [PubMed]
- Kasikara, C.; Davra, V.; Calianese, D.; Geng, K.; Spires, T.E.; Quigley, M.; Wichroski, M.; Sriram, G.; Suarez-Lopez, L.; Yaffe, M.B.; et al. Pan-TAM Tyrosine Kinase Inhibitor BMS-777607 Enhances Anti-PD-1 mAb Efficacy in a Murine Model of Triple-Negative Breast Cancer. Cancer Res. 2019, 79, 2669–2683. [Google Scholar] [CrossRef]
- Rho, J.K.; Choi, Y.J.; Kim, S.Y.; Kim, T.W.; Choi, E.K.; Yoon, S.J.; Park, B.M.; Park, E.; Bae, J.H.; Choi, C.M.; et al. MET and AXL inhibitor NPS-1034 exerts efficacy against lung cancer cells resistant to EGFR kinase inhibitors because of MET or AXL activation. Cancer Res. 2014, 74, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Torka, R.; Penzes, K.; Gusenbauer, S.; Baumann, C.; Szabadkai, I.; Orfi, L.; Keri, G.; Ullrich, A. Activation of HER3 interferes with antitumor effects of Axl receptor tyrosine kinase inhibitors: Suggestion of combination therapy. Neoplasia 2014, 16, 301–318. [Google Scholar] [CrossRef] [PubMed]
- Iida, M.; McDaniel, N.K.; Kostecki, K.L.; Welke, N.B.; Kranjac, C.A.; Liu, P.; Longhurst, C.; Bruce, J.Y.; Hong, S.; Salgia, R.; et al. AXL regulates neuregulin1 expression leading to cetuximab resistance in head and neck cancer. BMC Cancer 2022, 22, 447. [Google Scholar] [CrossRef] [PubMed]
- Toulany, M.; Iida, M.; Lettau, K.; Coan, J.P.; Rebholz, S.; Khozooei, S.; Harari, P.M.; Wheeler, D.L. Targeting HER3-dependent activation of nuclear AKT improves radiotherapy of non-small cell lung cancer. Radiother. Oncol. 2022, 174, 92–100. [Google Scholar] [CrossRef]
- Citri, A.; Skaria, K.B.; Yarden, Y. The deaf and the dumb: The biology of ErbB-2 and ErbB-3. Exp. Cell Res. 2003, 284, 54–65. [Google Scholar] [CrossRef] [PubMed]
- Gaborit, N.; Lindzen, M.; Yarden, Y. Emerging anti-cancer antibodies and combination therapies targeting HER3/ERBB3. Hum. Vaccin. Immunother. 2016, 12, 576–592. [Google Scholar] [CrossRef] [PubMed]
- Majumder, A.; Sandhu, M.; Banerji, D.; Steri, V.; Olshen, A.; Moasser, M.M. The role of HER2 and HER3 in HER2-amplified cancers beyond breast cancers. Sci. Rep. 2021, 11, 9091. [Google Scholar] [CrossRef] [PubMed]
- Gan, H.K.; Millward, M.; Jalving, M.; Garrido-Laguna, I.; Lickliter, J.D.; Schellens, J.H.M.; Lolkema, M.P.; Van Herpen, C.L.M.; Hug, B.; Tang, L.; et al. A Phase I, First-in-Human Study of GSK2849330, an Anti-HER3 Monoclonal Antibody, in HER3-Expressing Solid Tumors. Oncologist 2021, 26, e1844–e1853. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.W.; Schram, A.M.; Hollebecque, A.; Nishino, K.; Macarulla, T.; Rha, S.Y.; Duruisseaux, M.; Liu, S.V.; Al Hallak, M.N.; Umemoto, K.; et al. The phase I/II eNRGy trial: Zenocutuzumab in patients with cancers harboring NRG1 gene fusions. Future Oncol. 2024. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, S.; Yonesaka, K.; Tanizaki, J.; Nonagase, Y.; Takegawa, N.; Haratani, K.; Kawakami, H.; Hayashi, H.; Takeda, M.; Tsurutani, J.; et al. Targeting of the HER2/HER3 signaling axis overcomes ligand-mediated resistance to trastuzumab in HER2-positive breast cancer. Cancer Med. 2019, 8, 1258–1268. [Google Scholar] [CrossRef] [PubMed]
- Krop, I.E.; Masuda, N.; Mukohara, T.; Takahashi, S.; Nakayama, T.; Inoue, K.; Iwata, H.; Yamamoto, Y.; Alvarez, R.H.; Toyama, T.; et al. Patritumab Deruxtecan (HER3-DXd), a Human Epidermal Growth Factor Receptor 3-Directed Antibody-Drug Conjugate, in Patients With Previously Treated Human Epidermal Growth Factor Receptor 3-Expressing Metastatic Breast Cancer: A Multicenter, Phase I/II Trial. J. Clin. Oncol. 2023, 41, 5550–5560. [Google Scholar] [CrossRef] [PubMed]
- Toth, G.; Szollosi, J.; Vereb, G. Quantitating ADCC against adherent cells: Impedance-based detection is superior to release, membrane permeability, or caspase activation assays in resolving antibody dose response. Cytom. A 2017, 91, 1021–1029. [Google Scholar] [CrossRef] [PubMed]
- Kute, T.; Stehle, J.R., Jr.; Ornelles, D.; Walker, N.; Delbono, O.; Vaughn, J.P. Understanding key assay parameters that affect measurements of trastuzumab-mediated ADCC against Her2 positive breast cancer cells. Oncoimmunology 2012, 1, 810–821. [Google Scholar] [CrossRef] [PubMed]
- Rebenku, I.; Lloyd, C.B.; Szollosi, J.; Vereb, G. Pixel-by-pixel autofluorescence corrected FRET in fluorescence microscopy improves accuracy for samples with spatially varied autofluorescence to signal ratio. Sci. Rep. 2023, 13, 2934. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ujlaky-Nagy, L.; Szöllősi, J.; Vereb, G. Disrupting EGFR–HER2 Transactivation by Pertuzumab in HER2-Positive Cancer: Quantitative Analysis Reveals EGFR Signal Input as Potential Predictor of Therapeutic Outcome. Int. J. Mol. Sci. 2024, 25, 5978. https://doi.org/10.3390/ijms25115978
Ujlaky-Nagy L, Szöllősi J, Vereb G. Disrupting EGFR–HER2 Transactivation by Pertuzumab in HER2-Positive Cancer: Quantitative Analysis Reveals EGFR Signal Input as Potential Predictor of Therapeutic Outcome. International Journal of Molecular Sciences. 2024; 25(11):5978. https://doi.org/10.3390/ijms25115978
Chicago/Turabian StyleUjlaky-Nagy, László, János Szöllősi, and György Vereb. 2024. "Disrupting EGFR–HER2 Transactivation by Pertuzumab in HER2-Positive Cancer: Quantitative Analysis Reveals EGFR Signal Input as Potential Predictor of Therapeutic Outcome" International Journal of Molecular Sciences 25, no. 11: 5978. https://doi.org/10.3390/ijms25115978
APA StyleUjlaky-Nagy, L., Szöllősi, J., & Vereb, G. (2024). Disrupting EGFR–HER2 Transactivation by Pertuzumab in HER2-Positive Cancer: Quantitative Analysis Reveals EGFR Signal Input as Potential Predictor of Therapeutic Outcome. International Journal of Molecular Sciences, 25(11), 5978. https://doi.org/10.3390/ijms25115978