

Human C15orf39 Inhibits Inflammatory Response via PRMT2 in Human Microglial HMC3 Cell Line

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. C15orf39 Was Down-Regulated in LPS/IFN-γ-Stimulated Human Microglia HMC3 Cells
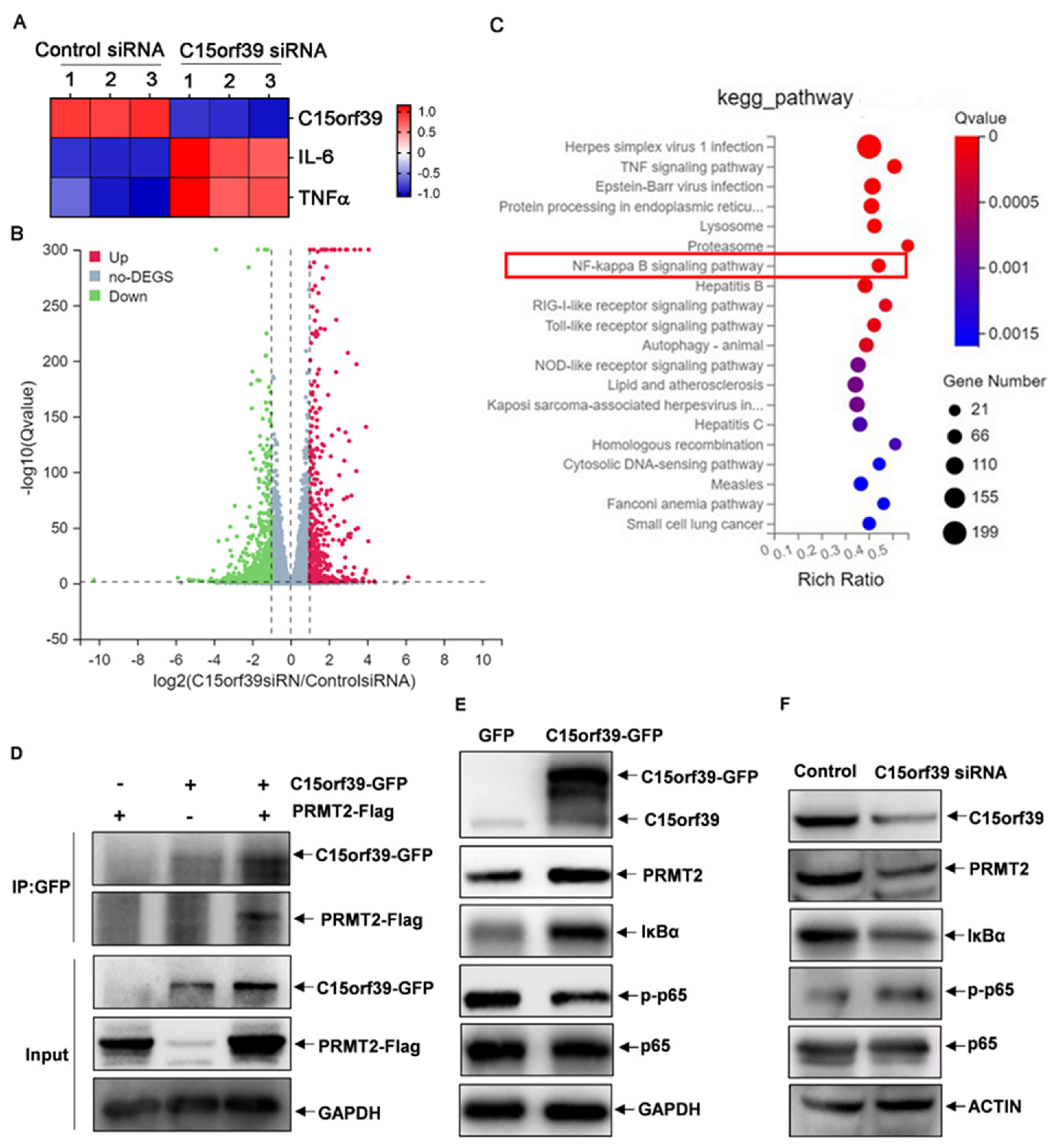
2.2. C15orf39 Overexpression Repressed IL-6 and TNFα Expression in LPS/IFN-γ-Stimulated HMC3 Cells
2.3. C15orf39 Knockdown Promoted IL-6 and TNFα Expression in LPS/IFN-γ-Stimulated HMC3 Cells
2.4. C15orf39 Inhibited NF-κB Activation by Interacting with PRMT2
2.5. C15orf39 Inhibited Microglial Inflammatory Response via PRMT2
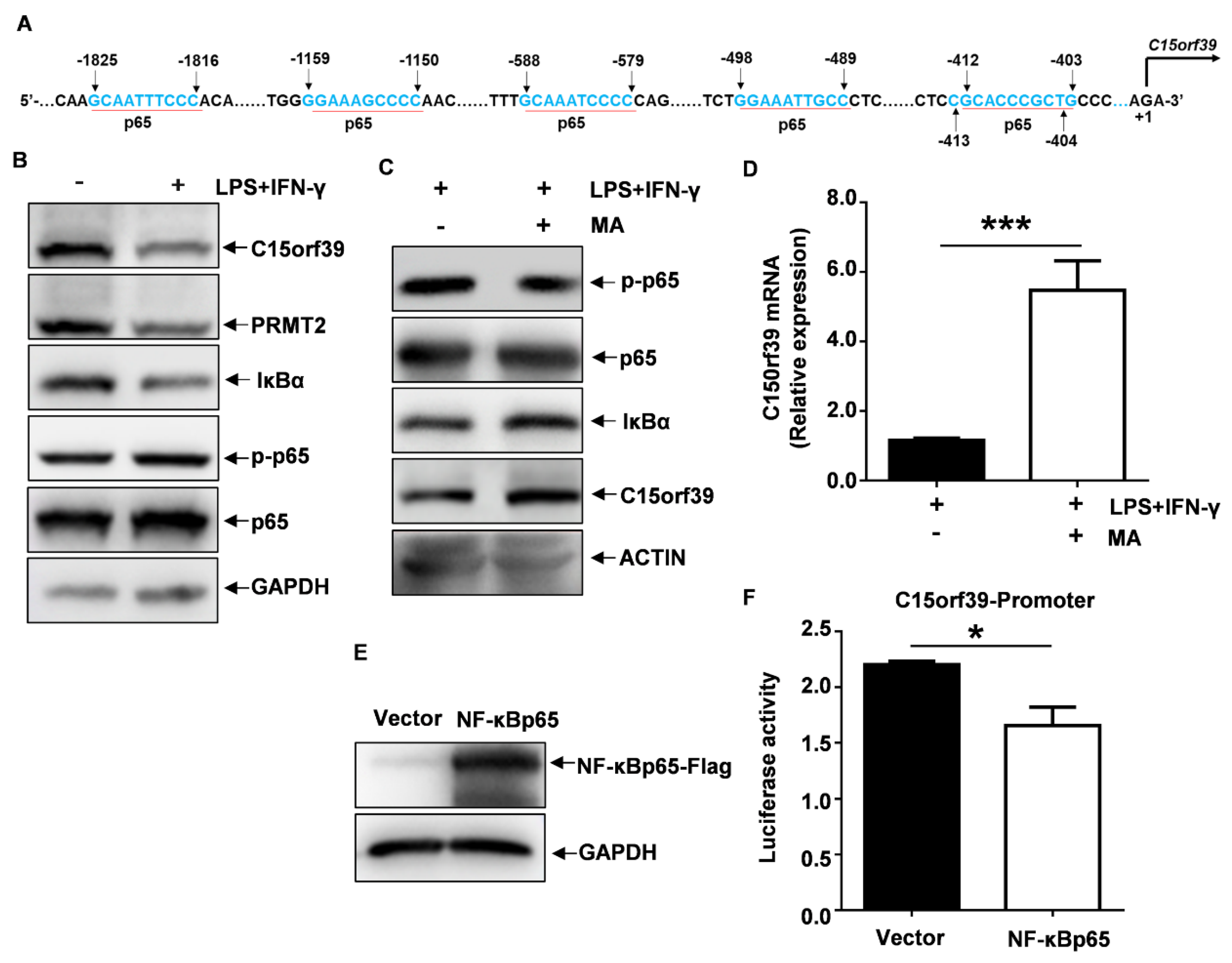
2.6. C15orf39 Inhibited NF-κB Signaling through Regulating Cytoplasmic PRMT2
2.7. NF-κBp65 Inhibited C15orf39 Transcription
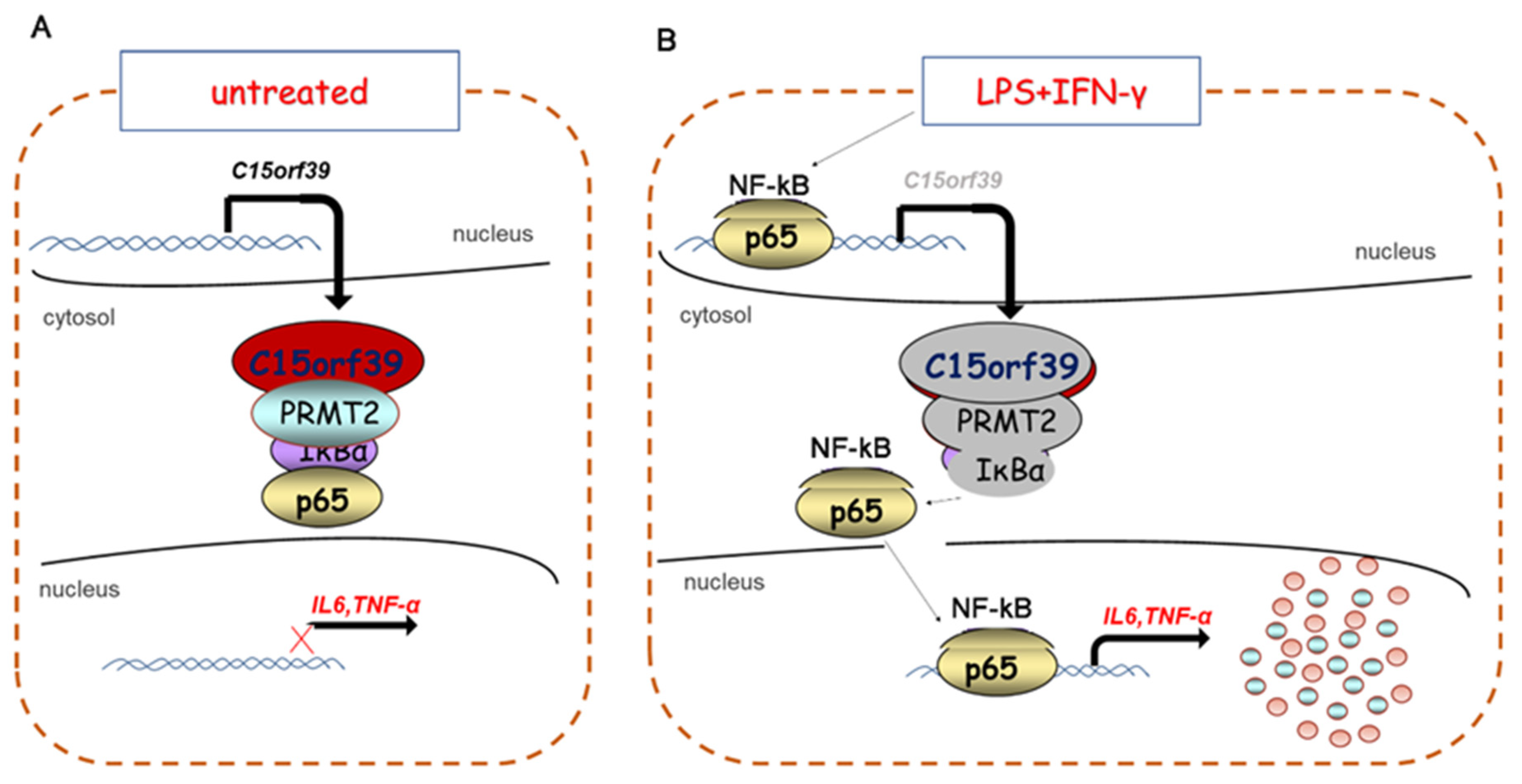
3. Discussion
4. Materials and Methods
4.1. Reagents and Antibodies
4.2. Cell Culture and Transfection
4.3. Immunofluorescence (IF)
4.4. Quantitative Real-Time PCR (qPCR)
4.5. Immunoprecipitation (IP)
4.6. Immunoblotting (IB)
4.7. Enzyme-Linked Immunosorbent Assay (ELISA)
4.8. Extraction of Nuclear and Cytoplasmic Protein
4.9. Dual-Luciferase Reporter Assay System
4.10. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| IL-6 | interleukin-6 |
| TNFα | tumor necrosis factor-α |
| AD | Alzheimer’s disease |
| LPS | lipopolysaccharide |
| IFN-γ | interferon-gamma |
| PRMT2 | protein arginine methyltransferase 2 |
| PRMT2 IP | PRMT2 interaction protein |
| MAPK1 | mitogen-Activated Protein Kinase 1 |
| MA | maslinic acid |
| IF | immunofluorescence |
| BSA | bovine serum albumin |
| RT | room temperature |
| qPCR | quantitative real-time PCR |
| IP | immunoprecipitation |
| IB | immunoblotting |
| WB | western blots |
| ELISA | enzyme-linked immunosorbent assay |
References
- Ginhoux, F.; Prinz, M. Origin of microglia: Current concepts and past controversies. Cold Spring Harb. Perspect. Biol. 2015, 7, a020537. [Google Scholar] [CrossRef] [PubMed]
- Ransohoff, R.M.; Perry, V.H. Microglial physiology: Unique stimuli, specialized responses. Annu. Rev. Immunol. 2009, 27, 119–145. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Jia, L.; Liu, C.-C.; Rong, Z.; Zhong, L.; Yang, L.; Chen, X.-F.; Fryer, J.D.; Wang, X.; Zhang, Y.-W.; et al. TREM2 Promotes Microglial Survival by Activating Wnt/beta-Catenin Pathway. J. Neurosci. 2017, 37, 1772–1784. [Google Scholar] [CrossRef] [PubMed]
- Glass, C.K.; Natoli, G. Molecular control of activation and priming in macrophages. Nat. Immunol. 2016, 17, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Baik, S.H.; Kang, S.; Lee, W.; Choi, H.; Chung, S.; Kim, J.-I.; Mook-Jung, I. A Breakdown in Metabolic Reprogramming Causes Microglia Dysfunction in Alzheimer’s Disease. Cell Metab. 2019, 30, 493–507e6. [Google Scholar] [CrossRef]
- Wang, G.; Li, X.; Li, N.; Wang, X.; He, S.; Li, W.; Fan, W.; Li, R.; Liu, J.; Hou, S. Icariin alleviates uveitis by targeting peroxiredoxin 3 to modulate retinal microglia M1/M2 phenotypic polarization. Redox Biol. 2022, 52, 102297. [Google Scholar] [CrossRef]
- Murray, P.J.; Allen, J.E.; Biswas, S.K.; Fisher, E.A.; Gilroy, D.W.; Goerdt, S.; Gordon, S.; Hamilton, J.A.; Ivashkiv, L.B.; Lawrence, T.; et al. Macrophage activation and polarization: Nomenclature and experimental guidelines. Immunity 2014, 41, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Toyoda, H.; Miyagawa, T.; Koike, A.; Kanbayashi, T.; Imanishi, A.; Sagawa, Y.; Kotorii, N.; Kotorii, T.; Hashizume, Y.; Ogi, K.; et al. A polymorphism in CCR1/CCR3 is associated with narcolepsy. Brain Behav. Immun. 2015, 49, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Tansey, K.E.; Cameron, D.; Hill, M.J. Genetic risk for Alzheimer’s disease is concentrated in specific macrophage and microglial transcriptional networks. Genome Med. 2018, 10, 14. [Google Scholar] [CrossRef]
- Porcellini, E.; Ianni, M.; Carbone, I.; Franceschi, M.; Licastro, F. Monocyte chemoattractant protein-1 promoter polymorphism and plasma levels in alzheimer’s disease. Immun. Ageing 2013, 10, 6. [Google Scholar] [CrossRef]
- Cai, Z.; Hussain, M.D.; Yan, L.-J. Microglia, neuroinflammation, and beta-amyloid protein in Alzheimer’s disease. Int. J. Neurosci. 2014, 124, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Tuppo, E.E.; Arias, H.R. The role of inflammation in Alzheimer’s disease. Int. J. Biochem. Cell Biol. 2005, 37, 289–305. [Google Scholar] [CrossRef] [PubMed]
- Sayers, E.W.; Beck, J.; Bolton, E.E.; Bourexis, D.; Brister, J.R.; Canese, K.; Comeau, D.C.; Funk, K.; Kim, S.; Klimke, W.; et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2021, 49, D10–D17. [Google Scholar] [CrossRef] [PubMed]
- Stelzer, G.; Rosen, N.; Plaschkes, I.; Zimmerman, S.; Twik, M.; Fishilevich, S.; Stein, T.I.; Nudel, R.; Lieder, I.; Mazor, Y.; et al. The GeneCards Suite: From Gene Data Mining to Disease Genome Sequence Analyses. Curr. Protoc. Bioinform. 2016, 54, 1.30.1–1.30.33. [Google Scholar] [CrossRef]
- Li, J.; Yu, C.; Ni, S.; Duan, Y. Identification of Core Genes and Screening of Potential Targets in Intervertebral Disc Degeneration Using Integrated Bioinformatics Analysis. Front. Genet. 2022, 13, 864100. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Fassl, A.; Vaites, L.P.; Fu, S.; Sicinski, P.; Paulo, J.A.; Gygi, S.P. Interrogating Kinase-Substrate Relationships with Proximity Labeling and Phosphorylation Enrichment. J. Proteome Res. 2022, 21, 494–506. [Google Scholar] [CrossRef]
- Gu, W.; Wang, Y.; Qiu, Z.; Dong, J.; Wang, Y.; Chen, J. Mitogen-activated protein kinase signaling is involved in nonylphenol-induced proinflammatory cytokines secretion by BV2 microglia. J. Appl. Toxicol. 2018, 38, 958–967. [Google Scholar] [CrossRef] [PubMed]
- Ganesh, L.; Yoshimoto, T.; Moorthy, N.C.; Akahata, W.; Boehm, M.; Nabel, E.G.; Nabel, G.J. Protein methyltransferase 2 inhibits NF-kappaB function and promotes apoptosis. Mol. Cell. Biol. 2006, 26, 3864–3874. [Google Scholar] [CrossRef] [PubMed]
- Dalloneau, E.; Pereira, P.L.; Brault, V.; Nabel, E.G.; Hérault, Y. Prmt2 regulates the lipopolysaccharide-induced responses in lungs and macrophages. J. Immunol. 2011, 187, 4826–4834. [Google Scholar] [CrossRef] [PubMed]
- Ijaq, J.; Chandrasekharan, M.; Poddar, R.; Bethi, N.; Sundararajan, V.S. Annotation and curation of uncharacterized proteins-challenges. Front. Genet. 2015, 6, 119. [Google Scholar] [CrossRef]
- Huang, S.; Li, J.; Wu, S.; Zheng, Z.; Wang, C.; Li, H.; Zhao, L.; Zhang, X.; Huang, H.; Huang, C.; et al. C4 orf19 inhibits colorectal cancer cell proliferation by competitively binding to Keap1 with TRIM25 via the USP17/Elk-1/CDK6 axis. Oncogene 2023, 42, 1333–1346. [Google Scholar] [CrossRef]
- Meyer, R.; Wolf, S.S.; Obendorf, M. PRMT2, a member of the protein arginine methyltransferase family, is a coactivator of the androgen receptor. J. Steroid Biochem. Mol. Biol. 2007, 107, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Min, Z.-Q.; Jiang, M.-J.; Liu, X.-L.; Yuan, S.-P.; Chen, P.-A.; Wang, C.-H.; Chen, Y.-J.; Dai, X.-P. Advances in Research on Protein Arginine Methyltransferase 2: Functions and Diseases. Protein Pept. Lett. 2024, 31, 25–42. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, S.; Vargas, J.; Hoffmann, A. Signaling via the NFkappaB system. Wiley Interdiscip. Rev. Syst. Biol. Med. 2016, 8, 227–241. [Google Scholar] [CrossRef] [PubMed]
- Wan, F.; Lenardo, M.J. Specification of DNA binding activity of NF-kappaB proteins. Cold Spring Harb. Perspect. Biol. 2009, 1, a000067. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.S.; Ghosh, S. Shared principles in NF-kappaB signaling. Cell 2008, 132, 344–362. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.L.; Somma, D.; Kerrigan, D.; McIntyre, Z.; Cole, J.J.; Liang, K.L.; A Kiely, P.; Keeshan, K.; Carmody, R.J. The regulation of sequence specific NF-kappaB DNA binding and transcription by IKKbeta phosphorylation of NF-kappaB p50 at serine 80. Nucleic Acids Res. 2019, 47, 11151–11163. [Google Scholar] [CrossRef] [PubMed]
- Alpha Scientists in Reproductive Medicine and ESHRE Special Interest Group of Embryology; Embryology ESIGo. The Istanbul Consensus Workshop on Embryo Assessment: Proceedings of an Expert Meeting. Hum. Reprod. 2011, 26, 1270–1283. [Google Scholar] [CrossRef] [PubMed]
- Leeman, J.R.; Gilmore, T.D. Alternative splicing in the NF-kappaB signaling pathway. Gene 2008, 423, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Todorov, V.T.; Völkl, S.; Müller, M.; Bohla, A.; Klar, J.; Kunz-Schughart, L.A.; Hehlgans, T.; Kurtz, A. Tumor necrosis factor-alpha activates NFkappaB to inhibit renin transcription by targeting cAMP-responsive element. J. Biol. Chem. 2004, 279, 1458–1467. [Google Scholar] [CrossRef]
- Han, Y.-M.; Kim, M.S.; Jo, J.; Shin, D.; Kwon, S.-H.; Seo, J.B.; Kang, D.; Lee, B.D.; Ryu, H.; Hwang, E.M.; et al. Decoding the temporal nature of brain GR activity in the NFkappaB signal transition leading to depressive-like behavior. Mol. Psychiatry 2021, 26, 5087–5096. [Google Scholar] [CrossRef]
- Lian, H.; Shim, D.J.; Gaddam, S.S.; Rodriguez-Rivera, J.; Bitner, B.R.; Pautler, R.G.; Robertson, C.S.; Zheng, H. IkappaBalpha deficiency in brain leads to elevated basal neuroinflammation and attenuated response following traumatic brain injury: Implications for functional recovery. Mol. Neurodegener. 2012, 7, 47. [Google Scholar] [CrossRef]
- Li, W.; Li, Q.L.; Xu, Q.Y.; Wang, X.; Yang, T. Tp47 promoted the phagocytosis of HMC3 cells though autophagy induced by endoplamic reticlum stress. J. Eur. Acad. Dermatol. Venereol. 2022, 36, 2224–2234. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Fang, Y.; Zhai, B.; Liu, X.; Zhu, G.; Zhou, S.; Xu, Y.; Wang, X.; Su, W.; Wang, R. Gm40600 promotes CD4+ T-cell responses by interacting with Ahnak. Immunology 2021, 164, 190–206. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Xu, R.; Zhai, B.; Zhou, S.; Wang, X.; Wang, R. Gm614 Protects Germinal Center B Cells from Death by Suppressing Caspase-1 Transcription in Lupus-Prone Mice. Front. Immunol. 2020, 11, 585726. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Xu, R.; Zhai, B.; Fang, Y.; Hou, C.; Xing, C.; Xiao, H.; Chen, G.; Wang, X.; Ma, N.; et al. Hspa13 Promotes Plasma Cell Production and Antibody Secretion. Front. Immunol. 2020, 11, 913. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhang, Y.; Wei, Y.; Wang, Z.; Zhu, G.; Fang, Y.; Zhai, B.; Xu, R.; Han, G.; Chen, G.; et al. The E3 ubiquitin ligase Itch is required for B-cell development. Sci. Rep. 2019, 9, 421. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wei, Y.; Xiao, H.; Liu, X.; Zhang, Y.; Han, G.; Chen, G.; Hou, C.; Ma, N.; Shen, B.; et al. A novel IL-23 p19/Ebi3 (IL-39) cytokine mediates inflammation in Lupus-like mice. Eur. J. Immunol. 2016, 46, 1343–1350. [Google Scholar] [CrossRef]
- Wang, R.-X.; Yu, C.-R.; Dambuza, I.M.; Mahdi, R.M.; Dolinska, M.B.; Sergeev, Y.V.; Wingfield, P.T.; Kim, S.-H.; E Egwuagu, C. Interleukin-35 induces regulatory B cells that suppress autoimmune disease. Nat. Med. 2014, 20, 633–641. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; He, Y.; Zhai, B.; Hou, C.; Xu, R.; Xing, C.; Wang, X.; Ma, N.; Han, G.; Wang, R. The E3 ubiquitin ligase Itch deficiency promotes antigen-driven B-cell responses in mice. Eur. J. Immunol. 2021, 51, 103–114. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, Z.; Xiao, H.; Liu, X.; Zhu, G.; Yu, D.; Han, G.; Chen, G.; Hou, C.; Ma, N.; et al. Foxd3 suppresses interleukin-10 expression in B cells. Immunology 2017, 150, 478–488. [Google Scholar] [CrossRef]
- Ma, N.; Fang, Y.; Xu, R.; Zhai, B.; Hou, C.; Wang, X.; Jiang, Z.; Wang, L.; Liu, Q.; Han, G.; et al. Ebi3 promotes T- and B-cell division and differentiation via STAT3. Mol. Immunol. 2019, 107, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.; Wang, X.; Xiao, H.; Liu, X.; Fang, Y.; Zhai, B.; Xu, R.; Han, G.; Chen, G.; Hou, C.; et al. Both Notch1 and its ligands in B cells promote antibody production. Mol. Immunol. 2017, 91, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhu, G.; Xiao, H.; Liu, X.; Han, G.; Chen, G.; Hou, C.; Shen, B.; Li, Y.; Ma, N.; et al. CD19 regulates ADAM28-mediated Notch2 cleavage to control the differentiation of marginal zone precursors to MZ B cells. J. Cell. Mol. Med. 2017, 21, 3658–3669. [Google Scholar] [CrossRef]
- Fang, Y.; Xu, R.; Zhai, B.; Hou, C.; Ma, N.; Wang, L.; Han, G.; Jiang, Z.; Wang, R. Gm40600 suppressed SP 2/0 isograft tumor by reducing Blimp1 and Xbp1 proteins. BMC Cancer 2019, 19, 700. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Fang, Y.; Hou, C.; Zhai, B.; Jiang, Z.; Ma, N.; Wang, L.; Han, G.; Wang, R. BC094916 suppressed SP 2/0 xenograft tumor by down-regulating Creb1 and Bcl2 transcription. Cancer Cell Int. 2018, 18, 138. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, M.; Xu, Y.; Zhu, G.; Zeng, Q.; Gao, R.; Qiu, J.; Su, W.; Wang, R. Human C15orf39 Inhibits Inflammatory Response via PRMT2 in Human Microglial HMC3 Cell Line. Int. J. Mol. Sci. 2024, 25, 6025. https://doi.org/10.3390/ijms25116025
Zhang M, Xu Y, Zhu G, Zeng Q, Gao R, Qiu J, Su W, Wang R. Human C15orf39 Inhibits Inflammatory Response via PRMT2 in Human Microglial HMC3 Cell Line. International Journal of Molecular Sciences. 2024; 25(11):6025. https://doi.org/10.3390/ijms25116025
Chicago/Turabian StyleZhang, Min, Yaqi Xu, Gaizhi Zhu, Qi Zeng, Ran Gao, Jinming Qiu, Wenting Su, and Renxi Wang. 2024. "Human C15orf39 Inhibits Inflammatory Response via PRMT2 in Human Microglial HMC3 Cell Line" International Journal of Molecular Sciences 25, no. 11: 6025. https://doi.org/10.3390/ijms25116025