Abstract
Nitric oxide (NO) and reactive nitrogen species (RNS) exert profound biological impacts dictated by their chemistry. Understanding their spatial distribution is essential for deciphering their roles in diverse biological processes. This review establishes a framework for the chemical biology of NO and RNS, exploring their dynamic reactions within the context of cancer. Concentration-dependent signaling reveals distinctive processes in cancer, with three levels of NO influencing oncogenic properties. In this context, NO plays a crucial role in cancer cell proliferation, metastasis, chemotherapy resistance, and immune suppression. Increased NOS2 expression correlates with poor survival across different tumors, including breast cancer. Additionally, NOS2 can crosstalk with the proinflammatory enzyme cyclooxygenase-2 (COX-2) to promote cancer progression. NOS2 and COX-2 co-expression establishes a positive feed-forward loop, driving immunosuppression and metastasis in estrogen receptor-negative (ER-) breast cancer. Spatial evaluation of NOS2 and COX-2 reveals orthogonal expression, suggesting the unique roles of these niches in the tumor microenvironment (TME). NOS2 and COX2 niche formation requires IFN-γ and cytokine-releasing cells. These niches contribute to poor clinical outcomes, emphasizing their role in cancer progression. Strategies to target these markers include direct inhibition, involving pan-inhibitors and selective inhibitors, as well as indirect approaches targeting their induction or downstream effectors. Compounds from cruciferous vegetables are potential candidates for NOS2 and COX-2 inhibition offering therapeutic applications. Thus, understanding the chemical biology of NO and RNS, their spatial distribution, and their implications in cancer progression provides valuable insights for developing targeted therapies and preventive strategies.
1. Introduction
The study of the extensive physiologic effects of nitric oxide (NO) has given rise to a substantial biomedical field over the last decades. Its influence extends across numerous physiological and pathological processes: including chronic inflammatory diseases, cancer, cardiovascular diseases, and neurodegeneration. This diatomic radical has been identified as a mediator of both positive and negative effects [1] and its dualistic behavior, often referred to as the “Janus face of NO”, is best comprehended within a chemical context. NO and its downstream reactive nitrogen species (RNS) constitute diffusible gaseous molecules that exert their effects in time-, spatial-, and concentration-dependent manners [2]. Although many recognized pathways involve the interaction with biological macromolecules, the fundamental chemistry of NO/RNS and the modification of atomic targets emerge as the primary influencers of NO-mediated signaling pathways [3]. To provide a comprehensive framework for understanding the target interactions and biological responses associated with these reactive species in various diseases [4,5] we introduce the concept of ‘The Chemical Biology of NO and RNS’. This chemical roadmap offers insights into the concentration-, temporal-, and spatial-dependent mechanisms that impact diverse physiological and pathophysiological states.
The intriguing aspect of NO biology lies in its precise compartmentalization. A notable example is the controlled diffusion of NO from the endothelial layer to the vascular smooth muscle, leading to tissue bed relaxation. This containment is facilitated by the rapid reactivity of NO with erythrocytes and oxyhemoglobin (HbO2) [6]. While discussions on the concentration and temporal aspects of NO’s chemical biology have been prevalent, recent attention has turned towards appreciating the subtleties of its spatial considerations [7,8,9,10,11]. While NO donors may generate a uniform NO flux for study in a petri dish, NOS2 expression acts as a localized point source in situ. Consequently, an accurate mechanistic understanding of NO biology necessitates a thorough exploration of its spatial component.
In this review, we examine the principles of chemical biology to elucidate the spatial effects observed in cancer and the tumor microenvironment (TME). We delve into how the reactions of NO and tumor NOS2 and COX-2 localization are intricately intertwined, influencing the progression of numerous cancers toward unfavorable outcomes.
2. Principles of NO and RNS Chemical Biology
It is crucial to understand the fundamental chemistry of NO and RNS to elucidate their physical properties and spatial distribution [12]. NO, a diatomic radical molecule, engages in a wide spectrum of chemical reactions that vary based on concentration and location [1,11]. Two primary types of reactions are observed: direct effects, wherein NO binds directly to targets such as hemes in soluble guanylate cyclase (sGC) or the cytochrome P450 (CYP450) family, and other instances involve reactions with HbO2 to form nitrate—an essential scavenging mechanism for compartmentalizing NO in specific tissue beds [13,14]. Direct reactions also occur with oxy radicals and Fenton/peroxidase intermediates. Indirect effects encompass those resulting from RNS formed through various NO and nitrite reactions, prominently including S-nitrosation and nitration reactions. Direct mechanisms are associated with low NO fluxes (<100 nM), while indirect mechanisms involve higher NO levels or acidic nitrite-rich areas, such as in phagosomes or lysosomes [11,15] (Figure 1; Figure 2A).
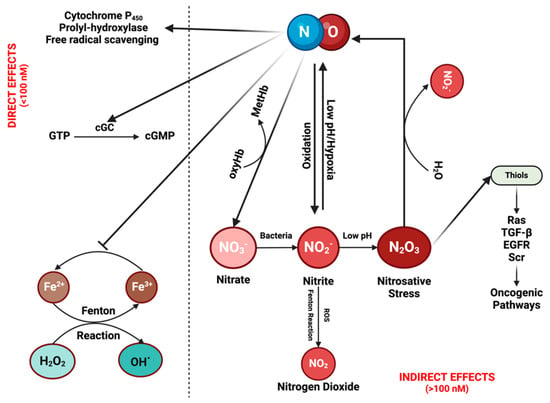
Figure 1.
The chemical biology of nitric oxide (NO) and reactive nitrogen species (RNS): NO can exert its effects either directly or indirectly. Direct effects are associated with low NO levels (<100 nM). At these concentrations, NO can activate the enzyme cyclic guanylyl cyclase (cGC), initiating the conversion of guanosine triphosphate (GTP) into cGMP. cGMP then serves as a signaling molecule in various physiological processes, such as vasodilation and neurotransmission. Moreover, it contributes to drug detoxification by interacting with proteins from the cytochrome P450 family, which can also eliminate NO itself. In addition, NO can induce the hypoxia-inducible factor (HIF) pathway through the inhibition of prolyl-hydroxylase, while also functioning as a free radical scavenger by inhibiting the Fenton reaction and reacting with reactive oxygen species (ROS). Indirect effects of NO are driven by its reactive nitrogen species (RNSs). These reactive species can undergo various reactions, leading to their recycling back into NO while simultaneously activating diverse oncogenic pathways. Figure Made in BioRender.com (accessed on 21 February 2024).
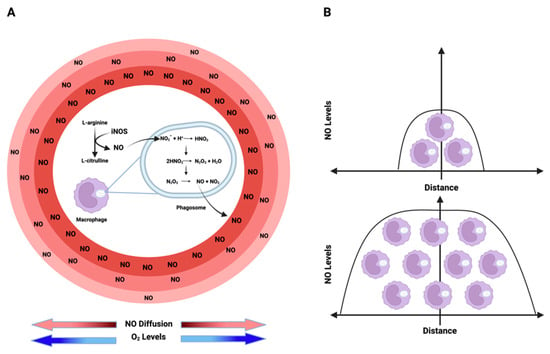
Figure 2.
Nitric oxide (NO) kinetics: (A) NO is enzymatically synthesized through the oxidation of L-arginine by iNOS. Conversely, nitrite (NO2−), the product of NO, acts as an alternative NO reservoir in acidic environments, such as phagosome/lysosome lumens. In these regions, nitrite undergoes a series of reactions, ultimately generating NO and NO2, which then diffuse from the source to the surroundings in an O2-dependent manner due to O2’s impact on NO half-life creating an NO flux gradient with higher levels around the source and lower levels as the distance from the source increases. (B) The extent to which NO can diffuse from its origin and the levels of this free radical in a specific environment depend on the cell density of iNOS-expressing cells. Higher densities of NO-producing cells result in elevated NO levels and an increased diffusion distance. Figure Made in BioRender.com (accessed on 21 February 2024).
There are additional direct reactions worth considering, such as oxidative stress mechanisms through reactive oxygen species (ROS) [2]. Metal oxo complexes derived from heme proteins, such as peroxidases, or non-heme iron complexes (as seen in the Fenton reaction), can oxidize nitrite to nitrogen dioxide (NO2). While hydrogen peroxide (H2O2) does not directly react with NO, the chemistry associated with Fenton and Haber-Weiss reactions exhibits a robust interaction with this oxygen radical. H2O2 reacts notably with many heme proteins, especially peroxidases, forming high-valent iron species that subsequently react with NO to produce nitrite (distinct from nitrate, as observed with HbO2/Myoglobin). The Fenton/Haber-Weiss cycle is central to the ROS cycle, where peroxide generates high-valent iron oxo species, such as ferric iron (Fe3+), from ferrous iron (Fe2+). To perpetuate the cycle, Fe3+ is then reduced back to Fe2+ by the oxide ion (O2−). NO serves as a potent antagonist to ROS chemistry at multiple stages, scavenging ferrous iron, high-valent iron (iron oxo), and H2O2 [16] (Figure 1).
Various sources of RNS play pivotal roles in cell signaling and contribute to inducing cellular and physiological stress [17]. The metabolism of NO by cells follows first order, while the autoxidation of NO is second order. The autoxidation of NO is significant not only in vitro but also under specific in vivo conditions and serves as a crucial source of RNS. The second-order dependence of NO indicates that the reaction’s half-life changes in proportion to NO concentration. Consequently, at low NO concentrations, the half-life concerning this autoxidation reaction can extend from minutes to hours, allowing for the occurrence of alternate reactions. However, with increasing NO levels, the reaction rate escalates exponentially, particularly in concentrated areas with high iNOS activity [11]. The gasses, NO and O2, exhibit 8–10 times higher concentrations in membranes, rendering the NO/O2 reaction 100 times faster than in hydrophilic regions [18]. Thiols, such as those associated with RAS, EGFR, and Src, can undergo nitrosation in these conditions, activating numerous oncogenic pathways. Generally, the rate of reaction with di-nitrogen trioxide N2O3 is significantly higher, nearly reaching diffusion-controlled levels, especially in low pKa thiols like GAPDH [11].
An additional source of NO and RNS comes from nitrite, either under acidic conditions or through oxidation by ROS interactions. Within acidic organelles like lysosomes and phagosomes, nitrite can undergo conversion, initially forming N2O3, which can further dissociate into NO and NO2. This equilibrium establishes a source from which NO can diffuse in an O2-dependent manner, interacting with different molecules [19] (Figure 2A; Figure 3). Furthermore, within lysosomes, this equilibrium facilitates thiol nitrosation and tyrosine nitration [20]. This RNS reservoir plays a crucial role in controlling viral replication, particularly within lysosomes. Nitrosation of proteases and other targets inhibits viral activity [21]. The density of iNOS-expressing cells determines the NO level and its diffusion from a source. As shown in Figure 2B, higher densities of iNOS-expressing cells coincide with higher levels of NO and increased diffusion distance from the source.
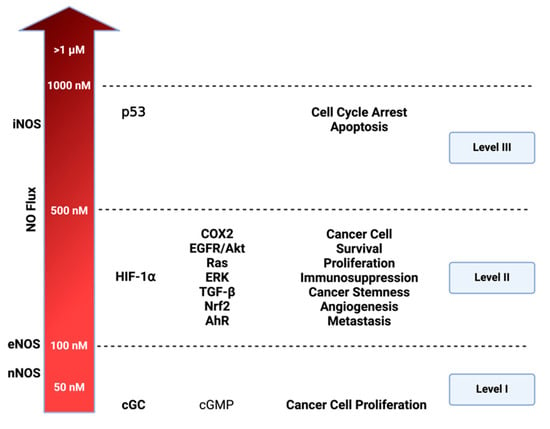
Figure 3.
Cellular signaling and nitric oxide (NO) levels: NO effects vary based on its concentration. Three distinct nitrosative levels are identified: Level 1 (<100 nM) is linked to cGMP-related cell signaling, contributing to cancer cell proliferation; Level 2 (100–500 nM) involves HIF-1⍺ activation and subsequent pathways promoting cancer progression; Level 3 (>500 nM) is associated with toxic NO effects, primarily triggered by p53 activation, leading to cell cycle arrest and apoptosis. Figure Made in BioRender.com (accessed on 21 February 2024).
A variety of reactions involving the interaction of NO and nitrite with metal peroxides play a significant role in biological processes. In hypoxic conditions, reduced metal complexes facilitate nitrite reduction to NO, inducing vasodilation. Conversely, the oxidation of nitrite, catalyzed by ROS-generating reactions, leads to the formation of NO2. Nitrite undergoes oxidation to NO2 via the Fenton/peroxidase mechanism, leading to nitration [22,23] (Figure 1). In summary, NO and nitrite engage in specific reactions with distinct targets, aligning with temporal and spatial orientations.
3. Understanding the Dynamics of NO Diffusion and Consumption for the Generation of High and Low NO Concentrations
Similarly to oxygen, NO is a gas with diffusible properties that exhibits higher solubility in hydrophobic compartments than in aqueous environments [24]. Constitutive endothelial nitric oxide synthase (eNOS) generates NO at lower fluxes, where NO diffuses from the site and reacts with HbO2 in nearby vessels, representing a predominant consumptive mechanism [25]. Conversely, inducible nitric oxide synthase (iNOS), encoded by the NOS2 gene, produces NO at higher concentrations, allowing sustained NO levels. At these concentrations, NO diffuses and is also consumed by cells in an O2-dependent manner (Figure 2A; Figure 3).
Interestingly, the rate of production/consumption of NO is similar in normoxic and hypoxic regions. This ultimately results in comparable NO fluxes in both areas, as both NO synthesis and consumption depend on oxygen concentration. Thus, at high O2 concentrations, NO synthesis and metabolism are maximized. On the other hand, at low O2 concentrations, both NO production and metabolism are reduced. Besides regulating NO production/consumption, O2 also impacts NO half-life and thus its diffusion rate. As mentioned, high levels of O2 increase NO metabolism reducing its half-life and consequently decreasing the distance of diffusion from the source. The opposite is true under low levels of O2 (Figure 2A). This is important because as detailed in the next topic of this review, the biological effects of NO are temporal-, spatial- and concentration-dependent. Consequently, the combination of production, consumption, and diffusion rates plays a crucial role in establishing a particular NO flux. Thus, the diffusion of NO from its source can be envisioned as establishing concentration topography, influencing cells based on their distance to the source [2].
4. Cellular Signaling and NO Levels
Levels of NO impact the physiology and pathology of biological processes in a concentration-dependent manner [11,12]. Constitutive isoforms like eNOS and neuronal nitric oxide synthase (nNOS) generate lower NO levels to maintain circulatory and neuronal systems homeostasis, respectively [26]. On the other hand, the inducible isoform, iNOS, can produce higher levels, leading to noxious effects with deleterious consequences (Figure 3). Dysregulated cytokine production, including tumor necrosis factor-alpha (TNF-α), interferon-gamma (INF-y), interleukin-1 alpha (IL-1α), and interleukin-6 (IL-6), can induce high levels of iNOS via nuclear factor-kappa B (NF-kB). This results in increased NO synthesis [27,28] and this dysregulation is observed in various chronic diseases like inflammatory bowel diseases and cancer [8,29].
The concentration-dependent signaling of NO and its RNS elucidates numerous distinctive processes in cancer and other diseases. Prior studies utilizing NO donors, such as diazeniumdiolates or NONOates, have proved to be an essential tool for mapping NO concentration and its effects on precise cellular signaling within physiologic and pathologic systems [30,31]. In vitro models employing various concentration temporal profiles have identified three distinct cellular signaling levels associated with cancer-related mechanisms. The first involves cGMP-related levels (<100 nM), the second encompasses oncogenic signaling levels (100–500 nM), and the third signifies nitrosative stress signaling (>500 nM) [3].
As illustrated in Figure 3, NO levels influence diverse mechanisms driving various oncogenic properties. In liver, prostate, pancreatic, and breast cancer, NO mechanisms have been identified at cGMP-related levels [32] These mechanisms involve a cGMP pathway that triggers ERK phosphorylation, subsequently activating TACE (ADAM17) to elevate NOTCH and EGF. cGMP pathway also influences ADAM10, which plays an important role in extracellular matrix (ECM) remodeling [33]. VEGF is also activated in these levels (<100 nM) promoting angiogenesis. At the higher level of nitrosative signaling (100–500 nM), NO interaction with membrane-bound thiols of low pKa and non-heme iron, such as prolyl hydroxylase, leads to an increase in major oncogenic signaling pathways: such as HIF-1, EGFR/Akt, RAS/ERK, TGF-β, Nrf2, and Aryl hydrocarbon receptor (AhR). At level 3 (>500 nM), the primary focus is the induction of p53 leading to cell cycle arrest and ultimately cell death. Under normal circumstances, p53 inhibits NOS2 by preventing its expression, leading to apoptosis of NOS2-expressing cells [2,34]. However, many cancers have mutated p53, losing the ability to inhibit NOS2 expression and other protumorigenic pathways [35], and chronic exposure to NO increases p53 mutations [36].
5. NO in Different Cancer Types
In the context of cancer, NO plays a crucial role in different aspects of cancer progression and response to treatment. This free radical is implicated in driving three key factors associated with unfavorable clinical outcomes in cancer: metastasis, resistance to chemo and radiotherapy, and immune suppression [8,37,38]. While some studies suggest positive effects of NO [39,40,41], the majority link high NOS2 expression to poor survival across a spectrum of hematological and solid tumors, including breast [8], pancreatic [42], nasopharyngeal carcinoma [43], colorectal [44,45], gastric [46], esophageal [47], prostate [48], melanoma [49], Hodgkin’s lymphoma [50], ovarian [51], squamous cell carcinoma [52], renal cell carcinoma [53], glioma [54], chondrosarcomas [55], Barrett’s esophageal adenocarcinoma [56], and in HCV-infected liver cancer patients [57].
Data extracted from the TCGA indicates a significant upregulation of NOS2 in ER- breast cancer patients compared to normal tissue, with a fold change of 13.9. Conversely, COX2 expression was decreased in the same patient cohort, although with a fold change of only 0.7 (Figure 4). These findings corroborate our earlier study, demonstrating the dynamic variation in NOS2 expression across estrogen receptor-negative ER- breast tumor tissues, contrasting with the relatively stable expression of COX2 [8]. Furthermore, Prueitt and colleagues analyzed NOS2 expression in a cohort of 248 breast cancer patients and reported that 29% and 41% of the patients exhibited moderate and high NOS2 expression, respectively [58].
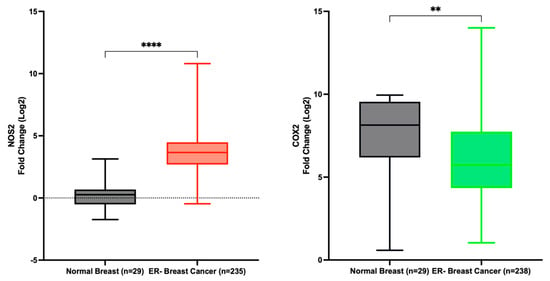
Figure 4.
NOS2 and PTGS2 (COX2) expression in normal breast and ER- breast cancer. **** p < 0.0001; ** p = 0.0032.
For instance, Stage II colorectal cancer patients with NOS2-high tumors experienced significantly reduced disease-free survival [59,60]. Similarly, elevated NOS2 tumor expression predicted poor survival in Stage III melanoma patients [61]. These findings underscore the association of NOS2 with higher tumor grade, increased lymph node positive status, and enhanced tumor vascularity, suggesting NOS2’s role as a promoter of cancer progression [59,60,61]. Mechanistically, NO is implicated in increased proliferation, adhesion, and migration of tumor cells [62]. NO has also been shown to promote angiogenesis and regulate the activity of a variety of matrix proteins associated with cancer progression and drug resistance [63].
While NOS2 can be considered as a univariate predictor of outcomes, its expression is not isolated from interactions with other significant inflammatory mechanisms. COX-2 is a pro-inflammatory enzyme that is often linked with NOS2 expression in inflammatory conditions and has also been correlated with adverse outcomes in cancer [7]. This mutual relationship between NOS2 and COX-2 has been identified as a mediator of cancer progression in different cancer types such as head and neck squamous cell carcinoma [64], breast [8], colon [44,65], and non-small cell lung cancers [66]. In addition, recent studies from our research group have revealed connections between elevated tumor NOS2 and COX-2 co-expression, chronic inflammation, and the development of more aggressive tumor phenotypes in ER- breast cancer [8]. Similarly, sustained co-expression of these pro-inflammatory markers promotes tumor progression in breast [7] and gastric cancers [67], as well as glioblastoma [68]. In summary, inflammation in the tumor microenvironment (TME) and NOS2 and COX-2 co-expression play pivotal roles in the mechanisms driving unfavorable clinical outcomes.
The interaction between NOS2 and COX-2 within the TME establishes a positive feed-forward loop, each activating pathways that enhance the expression of the other. The resulting products, NO and PGE2, create a multidimensional feedforward loop involving Th1 cytokines such as IL-6, IL-1, TNF-⍺, and IL-8, driving different mechanisms that contribute to metastasis, chemoresistance, and the development of cancer stemness.
In the TME, NO and PGE2 can induce immunosuppressive factors like IL-10 and TGF-β (Figure 5). NOS2 and COX2 expression have prognostic values in ER- breast cancer with hazard ratios (HR) of 6 [7] and 2.72 [69], respectively. Additionally, the expression of both markers is associated with a remarkable HR of 21 in five years. The synergistic action of these two inflammatory enzymes serves as a mechanism influencing almost every oncogenic pathway and immunosuppressive mechanism, ultimately leading to unfavorable clinical outcomes [7].
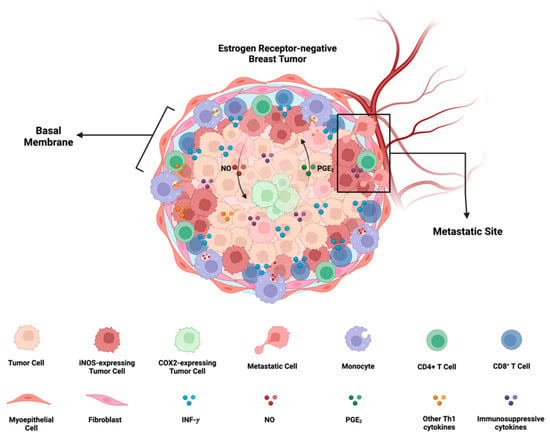
Figure 5.
iNOS/COX2 co-expression and its effects on ER- breast cancer progression: In ER- breast cancer, iNOS and COX2 exhibit orthogonal expression patterns. iNOS niches are located at the stroma-tumor interface, preventing the penetration of CD8+ T cells into the tumor, and are associated with a metastatic phenotype. On the other hand, COX2 niches are deeper within the tumor core, near immune-desert regions. There exists a positive feedforward loop between iNOS and COX2, where iNOS-derived nitric oxide (NO) induces COX2 activity, and COX2-derived PGE2 induces iNOS. The formation of these iNOS and COX2 niches depends on the presence of INF-γ-secreting cells, such as CD8+ T cells, and may be potentiated by other Th1 cytokines like TNF-⍺ and IL-1β. Furthermore, NO and PGE2 have the potential to induce the production of immunosuppressive cytokines such as IL-10 and TGF-β. Figure Made in BioRender.com (accessed on 24 February 2024).
In silico analysis corroborates the prognostic value of NOS2 and COX2 in TNBC patients [70,71]. Both markers are highly expressed in TNBC, and their expression is correlated with poor survival outcomes by promoting cancer stemness. Additionally, the co-expression of both markers was correlated with immunosuppression and metastasis in ER- breast cancer patients [8,70].
Under normal conditions, NOS2 and COX2 contribute to establishing a Th1 (IFN-γ) environment. During wound healing and tissue restoration, Th1 response plays a crucial role in restoring tissue homeostasis. In macrophages, an increase in NO leads to elevated p53 activation, inducing apoptosis and subsequently reducing the M1 macrophage population [72,73]. However, in cancer, NOS2 and COX2 can persist for extended periods, activating ATR/ATM, and leading to p53 stabilization [36]. P53, a major negative regulator of both NOS2 and COX2, binds to the TATA box, inhibiting the expression of these enzymes [74]. In p53-competent immune cells and other host cells, a high NO flux would cause a decrease in NOS2 and COX2. Interestingly, high NOS2 expression is associated with increased p53 mutation in breast cancer patients [30]. Additionally, the examination of NOS2 and p53 competency (null or mutant) is linked to elevated NOS2, supporting the negative relationship between NOS2 and p53 [75]. Because p53 is a negative regulator of NOS2 and COX2 expression, these results suggest that increased p53 mutation in tumor cells promotes persistent tumor NOS2 and COX2 expression.
6. Induction of NOS2 and COX2 in Cancer Cells
Over the past five decades, the activation of NOS2 and COX2 in murine macrophage models has been demonstrated to occur through the combination of IFN-γ with either cytokines (IL-1β and TNF-⍺) or lipopolysaccharide (LPS). While the most significant induction in murine myeloid cells was observed with a combination treatment of IFN-γ and LPS, the combination of INF-γ with TNF-⍺ and IL-1β yielded considerably lower effects [76]. On the other hand, the optimal induction of NOS2 and COX2 in murine and human tumor cells requires IFNγ, and its combination with TNF-⍺ and IL-1β was more effective than with LPS. These observations suggest variations in the pathways leading to NOS2 induction in different cell types. Comparing murine macrophages and tumor cells, a temporal distinction emerges, where the maximum NOS2 in macrophages and murine tumor cells occurs within 8 h to 24 h [77]. In human tumor cells, however, expression is delayed, occurring between 24 h to 48 h [8] (Figure 6). This temporal disparity implies that NOS2 expression in different cells may exhibit varying levels and sustainability of NO based upon orientation and timing of NOS2 expression.
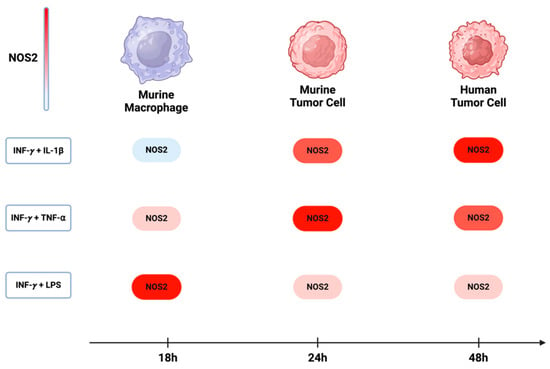
Figure 6.
NOS2 induction in murine and human cells: In murine macrophages, the maximum induction of NOS2 occurs with a combination of INF-y and LPS. However, in both murine and human tumor cells, the highest expression of NOS2 is achieved when INF-y is combined with either TNF-⍺ or IL-1β. The induction of NOS2 in murine macrophages occurs approximately 18 h after treatment, while in murine and human tumor cells, there is a delay, with induction occurring around 24 h to 48 h. Figure Made in BioRender.com (accessed on 24 February 2024).
A crucial aspect of NOS2 and COX2 is their orthogonal cellular expression in distinct cells. Traditionally, the Western blot concept assumed a homogeneous expression of both NOS2 and COX2 in every cell. However, microscopic examination and single cell-RNA-sequencing (sc-RNA-seq) have revealed orthogonal expression in both human and murine cells, with some cells expressing NOS2 and others expressing COX2 [8,77]. These results suggest that uniform stimulation of IFN-γ with other cytokines can create distinct phenotypes of cells expressing NOS2 or COX2. The orthogonal expression of NOS2 and COX2 implies unique roles for these cells in the TME, pointing to novel cellular neighborhoods. These cellular niches collaborate to sustain the expression of NOS2 and COX2 while also driving various oncogenic and immunosuppressive mechanisms [2,3] (Figure 5). Therefore, the spatial orientation of NOS2 and COX2 in the tissue provides crucial insights into the cellular neighborhoods that contribute to poor outcomes.
7. Human Breast Cancer and the Development of NOS2/COX2 Metastatic Niche Require CD8/IFNγ
The requirement of IFN-γ and TNF-⍺/IL-1β for the upregulation of NOS2 and COX2 in human tumor cells implies that the microenvironment inducing NOS2-rich regions requires cells present that are releasing IFN-γ and other proinflammatory cytokines. Lymphoid cells, including CD8+ T effector cells, CD4+ Th1, and NK cells, are potential sources, serving as major cytokine contributors under inflammatory conditions [78]. In our previous study [8,76], we demonstrated that stromal-restricted CD8+ T cells and limited lymphoid aggregates were closely positioned but orthogonal to tumor regions exhibiting high levels of NOS2 and COX2. Additionally, IFN-γ staining corresponded to regions with elevated NOS2 levels. On the other hand, in “immune-cold” areas of the tumor with low CD8+ T cells (immune deserts), there was notably less NOS2 and IFN-γ. Interestingly, NOS2 and COX2 were not co-expressed in the same cell, and areas with higher concentrations of NOS2 were associated with a specific metastatic cellular landscape. NOS2 was identified at the tumor-stromal interface, while COX2 was present in the vicinity of immune desert regions closer to the tumor core. This NOS2/COX2 spatial configuration in specified areas of the tumor indicated an increased metastatic potential. In summary, this spatial arrangement suggests a significant association between NOS2 and COX2 niches that drive poor clinical outcomes in ER- breast cancer (Figure 5).
Although NOS2/COX2 regions do not span the entire tumor and are not linked to a broader immune desert, these small focal points suggest a mechanism of cancer progression associated with poor outcomes. This association of NOS2 and COX2 induction and cancer progression was also observed in in vitro models using NO donors [7]. The crucial factor in the development of the NOS2 niche is the stroma restriction of CD8+ T cells [8]. Extrapolating from murine models of triple-negative breast (TNBC), it has been demonstrated that tumor NOS2 and COX2 play a vital role in CD8+ T cell stroma restriction, thus preventing their infiltration into the tumor core [76]. Therefore, IFN-γ originating from a proximal source, such as stroma-restricted CD8+ T effector cells, is pivotal in the formation of aggressive tumor NOS2 and COX2 niches that drive the NO and PGE2 feedforward loops, ultimately leading to increased metastasis and cancer stem cell (CSC) characteristics [7,8]. The identification of these niches presents novel therapeutic opportunities for the treatment of patients with these tumor signatures using clinically available NOS/COX inhibitors. Also, the discovery of inhibitory agents that limit the development of NOS2/COX2 niches could improve clinical outcomes by reducing metastasis, chemoresistance, and immunosuppression. This offers a potential playbook for screening novel and clinically available agents for the treatment and prevention of more aggressive cancers, such as TNBC.
8. Alternative Therapeutic Approaches for Preventing NOS2/COX2 Cellular Niche Development
A variety of direct and indirect strategies can be employed to diminish the formation of NOS2 and COX2 niches. There are different non-steroidal anti-inflammatory drugs (NSAIDs) that can target iNOS and/or COX2 and exhibit potential repurposing capacity for treating different types of cancers (Table 1). Targeting NOS2 and COX2 can involve: (1) reducing expression or targeting critical downstream effectors (indirect approach), and (2) inhibiting NO and PGE2 production from NOS2 and COX2 (direct approach) (Figure 7). These strategies are applicable in different contexts including chemoprevention and co-adjuvant treatment aiming to prevent cancer development, metastasis, and tumor recurrence. In preventive approaches, the focus is on well-tolerated long-term therapies with minimal side effects, while in contrast, more aggressive measures can be considered during active treatment phases [79].

Table 1.
List of non-cancer drugs that have shown some evidence of anticancer activity. Data extracted from https://www.anticancerfund.org/database-repurposing-drugs-oncology (accessed on 14 May 2024) and https://pubmed.ncbi.nlm.nih.gov/ (accessed on 14 May 2024).
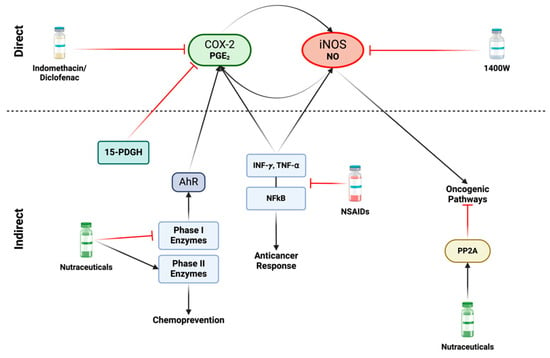
Figure 7.
Direct and indirect approaches for inhibiting iNOS and COX2: Various nonsteroidal anti-inflammatory drugs (NSAIDs), including indomethacin and diclofenac, as well as Celecoxib can directly inhibit COX2. Specific iNOS inhibitors, such as acetamidine compounds like 1400 W, demonstrate highly efficient and targeted effects. In terms of indirect approaches, the focus shifts to factors that induce iNOS and/or COX2 activity. A variety of NSAIDs play a role in inhibiting the production and release of proinflammatory cytokines, such as INF-γ and TNF-α, leading to subsequent inhibition of the NFκB pathway. However, inhibition of these proinflammatory mediators poses a challenge, as they are also crucial to anticancer responses, and their suppression may promote cancer progression. An alternative strategy to disrupt the iNOS/COX2 feedforward loop involves nutraceuticals, such as isothiocyanates and dithiolethione compounds. These compounds exhibit the capability to inhibit carcinogenic phase I enzymes while simultaneously inducing phase II detoxification enzymes, thereby promoting chemoprevention. Phase I enzymes can activate the Aryl hydrocarbon receptor (AhR), leading to COX2 activity induction. These natural compounds also serve as activators of the tumor suppressor protein phosphatase 2A (PP2A). This enzyme acts as an antagonist to various oncogenic pathways activated by nitric oxide. Figure Made in BioRender.com (accessed on 26 February 2024).
8.1. Direct Methods of NOS2 and COX2 Inhibition
Direct methods involve the use of compounds that decrease NO or PGE2 levels by inhibiting iNOS (NOS2) and COX2 enzyme activity, respectively (Figure 7). NOS and COX enzymes have both constitutive and induced isoforms: constitutive (nNOS, eNOS, and COX1) and induced (iNOS and COX2). Pan-inhibitors for both NOS and COX, which target both constitutive and induced isoforms, are available [154]. The advantage of pan-inhibitors lies in their ability to address various cancer-related mechanisms involving eNOS, such as angiogenesis and immune response regulation [155]. COX1 in certain situations inhibits apoptosis and processes other prostaglandins (PGs). Selective iNOS inhibitors, such as acetamidine compounds, were administered to patients for over two years with minimal side effects, offering an alternative to pan-inhibitors [156]. Alternatively, aminoguanidine (AG) is an iNOS-specific inhibitor utilized in humans for conditions like diabetes [157]. In murine models, AG significantly reduced metastasis [83]. However, AG is a weaker iNOS inhibitor compared to acetamidine compounds, such as 1400 W, and has off-target effects on other enzymes, including inhibiting diamine oxidase involved in polyamine synthesis [158].
There are FDA-approved compounds targeting COX2 that are clinically available. Celecoxib compounds and other COX2-specific agents provide targeted inhibition of PGE2 [159]. The use of these COX2-specific compounds presents a potential avenue for treatment. An alternative approach to PGE2 inhibition involves increasing 15-PDGH, an enzyme that metabolizes PGE2. NSAIDs, such as diclofenac and indomethacin, can inhibit COX while simultaneously boosting 15-PGDH. Some compounds have been developed to enhance 15-PDGH activity as an alternative to COX inhibition, however, these drugs can lead to gastrointestinal and cardiovascular effects [160]. Concerns about cardiovascular side effects with long-term use of pan NOS and COX inhibitors have been raised. However, in a recent phase 1/2 clinical trial, amlodipine and low-dose aspirin were given to prevent hypertension and thromboembolism, respectively in chemoresistant TNBC patients receiving the NOS inhibitor L-NMMA combined with docetaxel [161].
Moreover, in the same phase 1/2 clinical trial by Chung et al. [161], patients with chemorefractory breast cancer, locally advanced breast cancer (LABC), or metastatic TNBC were administered a regime composed of L-NMMA and docetaxel. It was reported a 45.8% overall response rate, 81.4% for patients with LABC, and 15.4% for metastatic TNBC patients. Additionally, another phase 1b/2 clinical trial with 15 metaplastic TNBC patients, a more aggressive form of TNBC, showed that the combination of L-NMMA and docetaxel results in an overall response of 20%, and progression-free survival (PFS) and overall survival (OS) of 4.5 months (range 3–7 m) and 12.8 months, respectively. Thus, inhibition of the nitric oxide pathway represents a promising and novel therapeutic option for TNBC. Further investigation with substantial cohorts is required to better understand the clinical benefits of NOS inhibitors in association with conventional chemotherapy [162].
8.2. Indirect Methods
An alternative strategy for direct inhibition involves targeting the induction of NOS2 and COX2, as well as the downstream products in their cellular signaling pathways. A variety of anti-inflammatory agents that target IFN-γ signaling are available [163,164]. However, a challenge arises due to the dichotomous nature of IFN-γ: while it can induce NOS2, it is also essential for a successful antitumor response [165]. Similar concerns arise when targeting NFκ-B and other components of the NOS2 and COX2 cellular signaling pathways, as this may interfere with critical anti-tumor pathways [166] (Figure 7). Therefore, the optimal strategy depends on the timing and context to ensure the most effective approach.
Different natural compounds aim to target the expression of NOS2 and COX2 [167]. These nutraceuticals encompass simple compounds like thiol-based agents and agonists of the antitumor PP2A [168]. Some dietary-based compounds like sulforaphane, have been transformed into pharmacological drugs that may have a role in this context [169]. Some of these nutraceutical compounds function as phase 1 enzyme inhibitors and phase 2 activators (Figure 7). Phase 1 enzymes are associated with procarcinogens, while phase 2 enzymes are part of anticarcinogenic systems [170].
Phase 1 enzymes exert various effects beyond inducing pro-carcinogenic P450 enzymes. They also play a role in regulating DNA and histone methylation in enzymes like BRCA1 [171]. In addition, these enzymes can induce AhR activation promoting COX2 expression [172] (Figure 7). The products of indoleamine 2,3-dioxygenase (IDO), induced by IFN-γ, can enhance AhR, leading to the production of kynurenines that exert diverse impacts on immunosuppression [173]. AhR activation is further linked to the induction of epithelial-mesenchymal transition (EMT), metastasis, and cancer stemness [174]. Hence, conditions that activate AhR, leading to increased COX2 and NOS2 via S-nitrosation enhance cellular resistance to treatment and promote EMT, metastasis, and immunosuppression.
In contrast, phase II enzymes are activated through NrF2/keap1, initiating a cascade that primarily protects against xenobiotics [175]. NrF2 increases antioxidant compounds, such as HO-1, playing a role in chemoprevention [176]. However, when these antioxidant defenses are activated in the context of cancer it leads to chemoresistance and immune suppression [177]. NO can activate NrF2 through S-nitrosation of keap1, releasing NrF2 and rendering cells more resistant to xenobiotics and electrophilic stress [178].
As mentioned earlier, several compounds classified as phase 1 inhibitors exhibit the ability to activate phase 2 enzymes. A subset of sulfur-based compounds, mainly found in cruciferous vegetables, exemplifies this dual action, including dithiolethiones [179] and isothiocyanates [180]. These thiol-based compounds extend their impact beyond phase transitions, demonstrating potent effects on additional targets, including the inhibition of NFκB [181,182]. Among dithiolethione compounds, anethole dithiolethione (ADT) stands out for its ability to activate PP2A by targeting SET, a pro-oncogenic protein that inhibits various anti-tumor proteins such as NMEM23 (known for its anti-metastatic properties) and PP2A [183,184,185]. PP2A is a crucial serine-threonine phosphatase that serves to inhibit numerous oncogenic pathways activated by NO (Figure 7). From a cancer cell-centric perspective, the activation of PP2A and the inhibition of pathways like NFκB, Akt, and ERK could be indicative of tumor regression [186,187,188]. These nutraceuticals present an opportunity to explore insights gained from spatial analysis of NOS2/COX2 and the chemical biology of NO and PGs for potential therapeutic applications in cancer.
9. Conclusions
Understanding the localization of NOS2/COX2 in tumors offers valuable insights and an opportunity to devise new approaches for targeting and preventing the progression of advanced cancers. The discussion above emphasizes that small, concentrated regions of NOS2-expressing tumor cells, compared to the entire tumor, can significantly impact survival. These small NOS2-rich areas generate elevated levels of NO, primarily formed through IFN-γ/cytokine stimulation. Identifying these NOS2 niches within tumors highlights a specific cellular mechanism associated with adverse outcomes. In addition, the pro-inflammatory enzyme, COX2, can potentiate NOS2 expression through a positive feed-forward loop. Therefore, spatial implications of NOS2/COX2 and the chemical biology of NO and RNS serve as a crucial roadmap for the design of new clinical trials supporting the development of innovative therapies that disrupt the nitric oxide signaling pathway and ultimately reduce immunosuppression, cancer stemness, and metastasis.
Author Contributions
All authors contributed equally to the writing, editing, and manuscript preparation. All authors have read and agreed to the published version of the manuscript.
Funding
This project was funded in whole or in part with federal funds from the Intramural Research Program of the NIH, National Cancer Institute, CCR, CIL (L.L.C., E.L.F., A.L.G., R.Y.S.C., L.A.R., and DAW). This project has been funded in part with federal funds from the Frederick National Laboratory for Cancer Research, National Institutes of Health, under contract 75N91019D00024 (R.L.M., W.F.H., and S.J.L.) and the Basic Science Program, Frederick National Laboratory for Cancer Research, Frederick, MD 21702 (SKA). This project was partly funded by the São Paulo Research Foundation (FAPESP), grants 2018/08107-2 and 2021/14642-0 (MCR).
Data Availability Statement
Not applicable.
Acknowledgments
The results shown here are in whole or part based upon data generated by the TCGA Research Network: https://www.cancer.gov/tcga. We acknowledge the Intramural Research Program of the NIH, National Cancer Institute, CCR, CIL; the Frederick National Laboratory for Cancer Research, NIH; and the São Paulo Research Foundation, FAPESP for funding this project.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Lundberg, J.O.; Weitzberg, E. Nitric Oxide Signaling in Health and Disease. Cell 2022, 185, 2853–2878. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.D. Breathing New Life into Nitric Oxide Signaling: A Brief Overview of the Interplay between Oxygen and Nitric Oxide. Redox Biol. 2015, 5, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Somasundaram, V.; Basudhar, D.; Bharadwaj, G.; No, J.H.; Ridnour, L.A.; Cheng, R.Y.S.; Fujita, M.; Thomas, D.D.; Anderson, S.K.; McVicar, D.W.; et al. Molecular Mechanisms of Nitric Oxide in Cancer Progression, Signal Transduction, and Metabolism. Antioxid. Redox Signal. 2019, 30, 1124–1143. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.D.; Heinecke, J.L.; Ridnour, L.A.; Cheng, R.Y.; Kesarwala, A.H.; Switzer, C.H.; McVicar, D.W.; Roberts, D.D.; Glynn, S.; Fukuto, J.M.; et al. Signaling and Stress: The Redox Landscape in NOS2 Biology. Free Radic. Biol. Med. 2015, 87, 204–225. [Google Scholar] [CrossRef] [PubMed]
- Haselden, W.D.; Kedarasetti, R.T.; Drew, P.J. Spatial and Temporal Patterns of Nitric Oxide Diffusion and Degradation Drive Emergent Cerebrovascular Dynamics. PLoS Comput. Biol. 2020, 16, e1008069. [Google Scholar] [CrossRef] [PubMed]
- Doyle, M.P.; Hoekstra, J.W. Oxidation of Nitrogen Oxides by Bound Dioxygen in Hemoproteins. J. Inorg. Biochem. 1981, 14, 351–358. [Google Scholar] [CrossRef] [PubMed]
- Basudhar, D.; Glynn, S.A.; Greer, M.; Somasundaram, V.; No, J.H.; Scheiblin, D.A.; Garrido, P.; Heinz, W.F.; Ryan, A.E.; Weiss, J.M.; et al. Coexpression of NOS2 and COX2 Accelerates Tumor Growth and Reduces Survival in Estrogen Receptor-Negative Breast Cancer. Proc. Natl. Acad. Sci. USA 2017, 114, 13030–13035. [Google Scholar] [CrossRef] [PubMed]
- Cheng, R.Y.S.; Ridnour, L.A.; Wink, A.L.; Gonzalez, A.L.; Femino, E.L.; Rittscher, H.; Somasundaram, V.; Heinz, W.F.; Coutinho, L.; Rangel, M.C.; et al. Interferon-Gamma Is Quintessential for NOS2 and COX2 Expression in ER- Breast Tumors That Lead to Poor Outcome. Cell Death Dis. 2023, 14, 319. [Google Scholar] [CrossRef] [PubMed]
- Mintz, J.; Vedenko, A.; Rosete, O.; Shah, K.; Goldstein, G.; Hare, J.M.; Ramasamy, R.; Arora, H. Current Advances of Nitric Oxide in Cancer and Anticancer Therapeutics. Vaccines 2021, 9, 94. [Google Scholar] [CrossRef]
- Li, J.Y.; Guo, Y.C.; Zhou, H.F.; Yue, T.T.; Wang, F.X.; Sun, F.; Wang, W.Z. Arginine Metabolism Regulates the Pathogenesis of Inflammatory Bowel Disease. Nutr. Rev. 2023, 81, 578–586. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.D.; Ridnour, L.A.; Isenberg, J.S.; Flores-Santana, W.; Switzer, C.H.; Donzelli, S.; Hussain, P.; Vecoli, C.; Paolocci, N.; Ambs, S.; et al. The Chemical Biology of Nitric Oxide: Implications in Cellular Signaling. Free Radic. Biol. Med. 2008, 45, 18–31. [Google Scholar] [CrossRef] [PubMed]
- Ridnour, L.A.; Thomas, D.D.; Switzer, C.; Flores-Santana, W.; Isenberg, J.S.; Ambs, S.; Roberts, D.D.; Wink, D.A. Molecular Mechanisms for Discrete Nitric Oxide Levels in Cancer. Nitric Oxide—Biol. Chem. 2008, 19, 73–76. [Google Scholar] [CrossRef] [PubMed]
- Olson, J.S.; Foley, E.W.; Rogge, C.; Tsai, A.L.; Doyle, M.P.; Lemon, D.D. NO Scavenging and the Hypertensive Effect of Hemoglobin-Based Blood Substitutes. Free Radic. Biol. Med. 2004, 36, 685–697. [Google Scholar] [CrossRef] [PubMed]
- Stasch, J.P.; Pacher, P.; Evgenov, O.V. Soluble Guanylate Cyclase as an Emerging Therapeutic Target in Cardiopulmonary Disease. Circulation 2011, 123, 2263–2273. [Google Scholar] [CrossRef]
- Hou, J.T.; Yu, K.K.; Sunwoo, K.; Kim, W.Y.; Koo, S.; Wang, J.; Ren, W.X.; Wang, S.; Yu, X.Q.; Kim, J.S. Fluorescent Imaging of Reactive Oxygen and Nitrogen Species Associated with Pathophysiological Processes. Chem 2020, 6, 832–866. [Google Scholar] [CrossRef]
- Radi, R. Oxygen Radicals, Nitric Oxide, and Peroxynitrite: Redox Pathways in Molecular Medicine. Proc. Natl. Acad. Sci. USA 2018, 115, 5839–5848. [Google Scholar] [CrossRef] [PubMed]
- Di Meo, S.; Reed, T.T.; Venditti, P.; Victor, V.M. Role of ROS and RNS Sources in Physiological and Pathological Conditions. Oxidative Med. Cell. Longev. 2016, 2016, 1245049. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Miller, M.J.S.; Joshi, M.S.; Thomas, D.D.; Lancaster, J.R. Accelerated Reaction of Nitric Oxide with O2 within the Hydrophobic Interior of Biological Membranes. Proc. Natl. Acad. Sci. USA 1998, 95, 2175–2179. [Google Scholar] [CrossRef] [PubMed]
- Pereira, C.; Ferreira, N.R.; Rocha, B.S.; Barbosa, R.M.; Laranjinha, J. The Redox Interplay between Nitrite and Nitric Oxide: From the Gut to the Brain. Redox Biol. 2013, 1, 276–284. [Google Scholar] [CrossRef] [PubMed]
- Bosworth, C.A.; Toledo, J.C.; Zmijewski, J.W.; Li, Q.; Lancaster, J.R. Dinitrosyliron Complexes and the Mechanism(s) of Cellular Protein Nitrosothiol Formation from Nitric Oxide. Proc. Natl. Acad. Sci. USA 2009, 106, 4671–4676. [Google Scholar] [CrossRef] [PubMed]
- Xiao, S.; Yuan, Z.; Huang, Y. The Potential Role of Nitric Oxide as a Therapeutic Agent against SARS-CoV-2 Infection. Int. J. Mol. Sci. 2023, 24, 17162. [Google Scholar] [CrossRef] [PubMed]
- Totzeck, M.; Hendgen-Cotta, U.B.; Luedike, P.; Berenbrink, M.; Klare, J.P.; Steinhoff, H.J.; Semmler, D.; Shiva, S.; Williams, D.; Kipar, A.; et al. Nitrite Regulates Hypoxic Vasodilation via Myoglobin-Dependent Nitric Oxide Generation. Circulation 2012, 126, 325–334. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Vanhoutte, P.M.; Leung, S.W.S. Vascular Nitric Oxide: Beyond ENOS. J. Pharmacol. Sci. 2015, 129, 83–94. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, E.M.; McGinity, C.; Wink, D.A.; McVicar, D.W. Nitric Oxide in Macrophage Immunometabolism: Hiding in Plain Sight. Metabolites 2020, 10, 429. [Google Scholar] [CrossRef]
- Wink, D.A.; Ridnour, L.A.; Hussain, S.P.; Harris, C.C. The Reemergence of Nitric Oxide and Cancer. Nitric Oxide—Biol. Chem. 2008, 19, 65–67. [Google Scholar] [CrossRef] [PubMed]
- Mishra, D.; Patel, V.; Banerjee, D. Nitric Oxide and S-Nitrosylation in Cancers: Emphasis on Breast Cancer. Breast Cancer Basic Clin. Res. 2020, 14, 1178223419882688. [Google Scholar] [CrossRef]
- Farlik, M.; Reutterer, B.; Schindler, C.; Greten, F.; Vogl, C.; Müller, M.; Decker, T. Nonconventional Initiation Complex Assembly by STAT and NF-ΚB Transcription Factors Regulates Nitric Oxide Synthase Expression. Immunity 2010, 33, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Kelleher, Z.T.; Matsumoto, A.; Stamler, J.S.; Marshall, H.E. NOS2 Regulation of NF-ΚB by S-Nitrosylation of P65. J. Biol. Chem. 2007, 282, 30667–30672. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.C.B.; Chao, T.N.; Cohen, N.A.; Mirza, N. Persistent Inflammation and Nitric Oxide Dysregulation Are Transcriptomic Blueprints of Subglottic Stenosis. Front. Immunol. 2021, 12, 748533. [Google Scholar] [CrossRef] [PubMed]
- Glynn, S.A.; Boersma, B.J.; Dorsey, T.H.; Yi, M.; Yfantis, H.G.; Ridnour, L.A.; Martin, D.N.; Switzer, C.H.; Hudson, R.S.; Wink, D.A.; et al. Increased NOS2 Predicts Poor Survival in Estrogen Receptor-Negative Breast Cancer Patients. J. Clin. Investig. 2010, 120, 3843–3854. [Google Scholar] [CrossRef] [PubMed]
- Heinecke, J.L.; Ridnour, L.A.; Cheng, R.Y.S.; Switzer, C.H.; Lizardo, M.M.; Khanna, C.; Glynn, S.A.; Hussain, S.P.; Young, H.A.; Ambs, S.; et al. Tumor Microenvironment-Based Feed-Forward Regulation of NOS2 in Breast Cancer Progression. Proc. Natl. Acad. Sci. USA 2014, 111, 6323–6328. [Google Scholar] [CrossRef] [PubMed]
- Mujoo, K.; Sharin, V.G.; Martin, E.; Choi, B.K.; Sloan, C.; Nikonoff, L.E.; Kots, A.Y.; Murad, F. Role of Soluble Guanylyl Cyclase-Cyclic GMP Signaling in Tumor Cell Proliferation. Nitric Oxide 2010, 22, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Geller, D.A.; Wink, D.A.; Cheng, B.; Billiar, T.R. NO and Hepatocellular Cancer. Br. J. Pharmacol. 2019, 177, 5459–5466. [Google Scholar] [CrossRef] [PubMed]
- Switzer, C.H.; Ridnour, L.A.; Cheng, R.; Heinecke, J.; Burke, A.; Glynn, S.; Ambs, S.; Wink, D.A. S-Nitrosation Mediates Multiple Pathways, That Lead to Tumor Progression in Estrogen Receptor—Negative Breast Cancer. Forum Immunopathol. Dis. Ther. 2012, 3, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhang, T.; Su, W.; Dou, Z.; Zhao, D.; Jin, X.; Lei, H.; Wang, J.; Xie, X.; Cheng, B.; et al. Mutant P53 in Cancer: From Molecular Mechanism to Therapeutic Modulation. Cell Death Dis. 2022, 13, 974. [Google Scholar] [CrossRef] [PubMed]
- Cheng, R.Y.S.; Burkett, S.; Ambs, S.; Moody, T.; Wink, D.A.; Ridnour, L.A. Chronic Exposure to Nitric Oxide Induces P53 Mutations and Malignant-like Features in Human Breast Epithelial Cells. Biomolecules 2023, 13, 311. [Google Scholar] [CrossRef] [PubMed]
- Jayaraman, P.; Parikh, F.; Lopez-Rivera, E.; Hailemichael, Y.; Clark, A.; Ma, G.; Cannan, D.; Ramacher, M.; Kato, M.; Overwijk, W.W.; et al. Tumor-Expressed Inducible Nitric Oxide Synthase Controls Induction of Functional Myeloid-Derived Suppressor Cells through Modulation of Vascular Endothelial Growth Factor Release. J. Immunol. 2012, 188, 5365–5376. [Google Scholar] [CrossRef] [PubMed]
- Switzer, C.H.; Glynn, S.A.; Cheng, R.Y.S.; Ridnour, L.A.; Green, J.E.; Ambs, S.; Wink, D.A. S-Nitrosylation of EGFR and Src Activates an Oncogenic Signaling Network in Human Basal-like Breast Cancer. Mol. Cancer Res. 2012, 10, 1203–1215. [Google Scholar] [CrossRef] [PubMed]
- Riganti, C.; Miraglia, E.; Viarisio, D.; Costamagna, C.; Pescarmona, G.; Ghigo, D.; Bosia, A. Nitric Oxide Reverts the Resistance to Doxorubicin in Human Colon Cancer Cells by Inhibiting the Drug Efflux. Cancer Res. 2005, 65, 516–525. [Google Scholar] [CrossRef] [PubMed]
- Li, C.Y.; Anuraga, G.; Chang, C.P.; Weng, T.Y.; Hsu, H.P.; Ta, H.D.K.; Su, P.F.; Chiu, P.H.; Yang, S.J.; Chen, F.W.; et al. Repurposing Nitric Oxide Donating Drugs in Cancer Therapy through Immune Modulation. J. Exp. Clin. Cancer Res. 2023, 42, 22. [Google Scholar] [CrossRef] [PubMed]
- Beppu, T.; Nishi, K.; Imoto, S.; Araki, W.; Setoguchi, I.; Ueda, A.; Suetsugi, N.; Ishima, Y.; Ikeda, T.; Otagiri, M.; et al. Novel Nitric Oxide Donor, Nitrated Phenylbutyrate, Induces Cell Death of Human Pancreatic Cancer Cells and Suppresses Tumor Growth of Cancer Xenografts. Oncol. Rep. 2022, 48, 178. [Google Scholar] [CrossRef] [PubMed]
- Fujita, M.; Imadome, K.; Endo, S.; Shoji, Y.; Yamada, S.; Imai, T. Nitric Oxide Increases the Invasion of Pancreatic Cancer Cells via Activation of the PI3K-AKT and RhoA Pathways after Carbon Ion Irradiation. FEBS Lett. 2014, 588, 3240–3250. [Google Scholar] [CrossRef] [PubMed]
- Bourouba, M.; Boukercha, A.; Zergoun, A.A.; Zebboudj, A.; Elhadjan, M.; Djenaoui, D.; Asselah, F.; Touil-Boukoffa, C. Increased Production of Nitric Oxide Correlates with Tumor Growth in Algerian Patients with Nasopharyngeal Carcinoma. Biomarkers 2012, 17, 618–624. [Google Scholar] [CrossRef] [PubMed]
- Cianchi, F.; Cortesini, C.; Fantappiè, O.; Messerini, L.; Schiavone, N.; Vannacci, A.; Nistri, S.; Sardi, I.; Baroni, G.; Marzocca, C.; et al. Inducible Nitric Oxide Synthase Expression in Human Colorectal Cancer Correlation with Tumor Angiogenesis. Am. J. Pathol. 2003, 162, 793–801. [Google Scholar] [CrossRef] [PubMed]
- Alam, A.; Smith, S.C.; Gobalakrishnan, S.; McGinn, M.; Yakovlev, V.A.; Rabender, C.S. Uncoupled Nitric Oxide Synthase Activity Promotes Colorectal Cancer Progression. Front. Oncol. 2023, 13, 1165326. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-Z.; Cao, Y.-Q.; Wu, J.-N.; Chen, M.; Cha, X.-Y. Expression of Nitric Oxide Synthase in Human Gastric Carcinoma and Its Relation to P53, PCNA. World J. Gastroenterol. 2005, 11, 46–50. [Google Scholar] [CrossRef] [PubMed]
- Barani, R.; Motalleb, G.; Maghsoudi, H. Evaluation of INOS Expression in Esophageal Cancer Patients. Gastrointest. Tumors 2016, 3, 44–58. [Google Scholar] [CrossRef] [PubMed]
- Burke, A.J.; McAuliffe, J.D.; Natoni, A.; Ridge, S.; Sullivan, F.J.; Glynn, S.A. Chronic Nitric Oxide Exposure Induces Prostate Cell Carcinogenesis, Involving Genetic Instability and a pro-Tumorigenic Secretory Phenotype. Nitric Oxide 2022, 127, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Sikora, A.G.; Gelbard, A.; Davies, M.A.; Sano, D.; Ekmekcioglu, S.; Kwon, J.; Hailemichael, Y.; Jayaraman, P.; Myers, J.N.; Grimm, E.A.; et al. Targeted Inhibition of Inducible Nitric Oxide Synthase Inhibits Growth of Human Melanoma In Vivo and Synergizes with Chemotherapy. Clin. Cancer Res. 2010, 16, 1834–1844. [Google Scholar] [CrossRef] [PubMed]
- Guida, G.; Culla, B.; Scirelli, T.; Bellone, G.; Sciascia, S.; Brussino, L.; Novero, D.; Palestro, G.; Heffler, E.; Gavarotti, P.; et al. Exhaled Nitric Oxide and Nitric Oxide Synthase Expression in Hodgkin’s Disease. Int. J. Immunopathol. Pharmacol. 2009, 22, 1027–1034. [Google Scholar] [CrossRef] [PubMed]
- Caneba, C.A.; Yang, L.; Baddour, J.; Curtis, R.; Win, J.; Hartig, S.; Marini, J.; Nagrath, D. Nitric Oxide Is a Positive Regulator of the Warburg Effect in Ovarian Cancer Cells. Cell Death Dis. 2014, 5, e1302. [Google Scholar] [CrossRef]
- Sangle, V.A.; Chaware, S.J.; Kulkarni, M.A.; Ingle, Y.C.; Singh, P.; Pooja, V.K. Elevated Tissue Nitric Oxide in Oral Squamous Cell Carcinoma. J. Oral Maxillofac. Pathol. 2018, 22, 35–39. [Google Scholar] [CrossRef] [PubMed]
- Ene, C.D.; Tampa, M.; Georgescu, S.R.; Matei, C.; Leulescu, I.M.T.; Dogaru, C.I.; Penescu, M.N.; Nicolae, I. Disturbances in Nitric Oxide Cycle and Related Molecular Pathways in Clear Cell Renal Cell Carcinoma. Cancers 2023, 15, 5797. [Google Scholar] [CrossRef] [PubMed]
- Kruglyakov, D.; Ojha, S.K.; Kartawy, M.; Tripathi, M.K.; Hamoudi, W.; Bazbaz, W.; Khaliulin, I.; Amal, H. Nitric Oxide Synthase Inhibition Prevents Cell Proliferation in Glioblastoma. J. Mol. Neurosci. 2023, 73, 875–883. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, S.A.; Lopes, A.; De Carvalho, A.L.; Rossi, B.M.; Da Cunha, I.W.; Soares, F.A.; Chung, W.T.; Alves, L.A. Nitric Oxide Synthases, Cyclooxygenase-2, Nitrotyrosine, and Angiogenesis in Chondrosarcoma and Their Relation to Prognosis. J. Bone Jt. Surg. 2010, 92, 1738–1746. [Google Scholar] [CrossRef] [PubMed]
- Vaninetti, N.; Williams, L.; Geldenhuys, L.; Porter, G.A.; Guernsey, D.L.; Casson, A.G. Regulation of CDX2 Expression in Esophageal Adenocarcinoma. Mol. Carcinog. 2009, 48, 965–974. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.A.; Dhar, D.K.; Yamaguchi, E.; Maruyama, S.; Sato, T.; Hayashi, H.; Ono, T.; Yamanoi, A.; Kohno, H.; Nagasue, N. Coexpression of Inducible Nitric Oxide Synthase and COX-2 in Hepatocellular Carcinoma and Surrounding Liver: Possible Involvement of COX-2 in the Angiogenesis of Hepatitis C Virus-Positive Cases. Clin. Cancer Res. 2001, 7, 1325–1332. [Google Scholar] [PubMed]
- Prueitt, R.L.; Boersma, B.J.; Howe, T.M.; Goodman, J.E.; Thomas, D.D.; Ying, L.; Pfiester, C.M.; Yfantis, H.G.; Cottrell, J.R.; Lee, D.H.; et al. Inflammation and IGF-I Activate the Akt Pathway in Breast Cancer. Int. J. Cancer 2007, 120, 796–805. [Google Scholar] [CrossRef] [PubMed]
- Gochman, E.; Mahajna, J.; Shenzer, P.; Dahan, A.; Blatt, A.; Elyakim, R.; Reznick, A.Z. The Expression of INOS and Nitrotyrosine in Colitis and Colon Cancer in Humans. Acta Histochem. 2012, 114, 827–835. [Google Scholar] [CrossRef] [PubMed]
- Zafirellis, K.; Zachaki, A.; Agrogiannis, G.; Gravani, K. Inducible Nitric Oxide Synthase Expression and Its Prognostic Significance in Colorectal Cancer. APMIS 2010, 118, 115–124. [Google Scholar] [CrossRef]
- Ding, Z.; Ogata, D.; Roszik, J.; Qin, Y.; Kim, S.H.; Tetzlaff, M.T.; Lazar, A.J.; Davies, M.A.; Ekmekcioglu, S.; Grimm, E.A. INOS Associates with Poor Survival in Melanoma: A Role for Nitric Oxide in the PI3K-AKT Pathway Stimulation and PTEN S-Nitrosylation. Front. Oncol. 2021, 11, 631766. [Google Scholar] [CrossRef]
- Alsharabasy, A.M.; Aljaabary, A.; Bohara, R.; Farràs, P.; Glynn, S.A.; Pandit, A. Nitric Oxide-Scavenging, Anti-Migration Effects, and Glycosylation Changes after Hemin Treatment of Human Triple-Negative Breast Cancer Cells: A Mechanistic Study. ACS Pharmacol. Transl. Sci. 2023, 6, 1416–1432. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.H.; Han, X.C.; Jia, M.K.; Jiang, W.D.; Wang, M.; Zhang, H.; Han, G.; Jiang, Y. Expressions of Inducible Nitric Oxide Synthase and Matrix Metalloproteinase-9 and Their Effects on Angiogenesis and Progression of Hepatocellular Carcinoma. World J. Gastroenterol. 2005, 11, 5931–5937. [Google Scholar] [CrossRef]
- Celenk, F.; Bayramoglu, I.; Akin Yilmaz, Þ.; Menevse, A.; Bayazit, Y. Expression of Cyclooxygenase-2, 12-Lipoxygenase, and Inducible Nitric Oxide Synthase in Head and Neck Squamous Cell Carcinoma. J. Craniofacial Surg. 2013, 24, 1114–1117. [Google Scholar] [CrossRef]
- Cianchi, F.; Cortesini, C.; Fantappie, O.; Messerini, L.; Sardi, I.; Lasagna, N.; Perna, F.; Fabbroni, V.; Felice, A.D.; Perigli, G.; et al. Cyclooxygenase-2 Activation Mediates the Proangiogenic Effect of Nitric Oxide in Colorectal Cancer. Clin. Cancer Res. 2004, 10, 2694–2704. [Google Scholar] [CrossRef]
- Marrogi, A.J.; Travis, W.D.; Welsh, J.A.; Khan, M.A.; Rahim, H.; Tazelaar, H.; Pairolero, P.; Trastek, V.; Jett, J.; Caporaso, N.E.; et al. Nitric Oxide Synthase, Cyclooxygenase 2, and Vascular Endothelial Growth Factor in the Angiogenesis of Non-Small Cell Lung Carcinoma. Clin. Cancer Res. 2000, 6, 4739–4744. [Google Scholar] [PubMed]
- Zhang, H.; Ding, C.; Suo, Z.; Kang, Y. Effect of Helicobacter Pylori on Cyclooxygenase-2 and Inducible Nitric Oxide Synthase in Patients with Gastric Precancerous Lesions and Its Clinical Significance. Exp. Ther. Med. 2015, 9, 2364–2368. [Google Scholar] [CrossRef] [PubMed]
- Hara, A.; Okayasu, I. Cyclooxygenase-2 and Inducible Nitric Oxide Synthase Expression in Human Astrocytic Gliomas: Correlation with Angiogenesis and Prognostic Significance. Acta Neuropathol. 2004, 108, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Glynn, S.A.; Prueitt, R.L.; Ridnour, L.A.; Boersma, B.J.; Dorsey, T.M.; Wink, D.A.; Goodman, J.E.; Yfantis, H.G.; Lee, D.H.; Ambs, S. COX-2 Activation Is Associated with Akt Phosphorylation and Poor Survival in ER-Negative, HER2-Positive Breast Cancer. BMC Cancer 2010, 10, 626. [Google Scholar] [CrossRef] [PubMed]
- Granados-Principal, S.; Liu, Y.; Guevara, M.L.; Blanco, E.; Choi, D.S.; Qian, W.; Patel, T.; Rodriguez, A.A.; Cusimano, J.; Weiss, H.L.; et al. Inhibition of INOS as a Novel Effective Targeted Therapy against Triple-Negative Breast Cancer. Breast Cancer Res. 2015, 17, 25. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Hachim, M.Y.; Hachim, I.Y.; Dai, M.; Lo, C.; Al Raffa, F.; Ali, S.; Lebrun, J.J. Cyclooxygenase-2 Regulates TGFβ-Induced Cancer Stemness in Triple-Negative Breast Cancer. Sci. Rep. 2017, 7, 40258. [Google Scholar] [CrossRef] [PubMed]
- Marei, H.E.; Althani, A.; Afifi, N.; Hasan, A.; Caceci, T.; Pozzoli, G.; Morrione, A.; Giordano, A.; Cenciarelli, C. P53 Signaling in Cancer Progression and Therapy. Cancer Cell Int. 2021, 21, 703. [Google Scholar] [CrossRef] [PubMed]
- Brockhaus, F.; Bruè, B. P53 Accumulation in Apoptotic Macrophages Is an Energy Demanding Process That Precedes Cytochrome c Release in Response to Nitric Oxide. Oncogene 1999, 18, 6403–6410. [Google Scholar] [CrossRef] [PubMed]
- Bortolanza, M.; Padovan-Neto, F.E.; Cavalcanti-Kiwiatkoski, R.; Dos Santos-Pereira, M.; Mitkovski, M.; Raisman-Vozari, R.; Del-Bel, E. Are Cyclooxygenase-2 and Nitric Oxide Involved in the Dyskinesia of Parkinson’s Disease Induced by L-DOPA? Philos. Trans. R. Soc. B Biol. Sci. 2015, 370, 20140190. [Google Scholar] [CrossRef] [PubMed]
- Li, C.Q.; Pang, B.; Kiziltepe, T.; Trudel, L.J.; Engelward, B.P.; Dedon, P.C.; Wogan, G.N. Threshold Effects of Nitric Oxide-Induced Toxicity and Cellular Responses in Wild-Type and P53-Null Human Lymphoblastoid Cells. Chem. Res. Toxicol. 2006, 19, 399–406. [Google Scholar] [CrossRef] [PubMed]
- Somasundaram, V.; Ridnour, L.A.; Cheng, R.Y.; Walke, A.J.; Kedei, N.; Bhattacharyya, D.D.; Wink, A.L.; Edmondson, E.F.; Butcher, D.; Warner, A.C.; et al. Systemic Nos2 Depletion and Cox Inhibition Limits TNBC Disease Progression and Alters Lymphoid Cell Spatial Orientation and Density. Redox Biol. 2022, 58, 102529. [Google Scholar] [CrossRef] [PubMed]
- Somasundaram, V.; Gilmore, A.C.; Basudhar, D.; Palmieri, E.M.; Scheiblin, D.A.; Heinz, W.F.; Cheng, R.Y.S.; Ridnour, L.A.; Altan-Bonnet, G.; Lockett, S.J.; et al. Inducible Nitric Oxide Synthase-Derived Extracellular Nitric Oxide Flux Regulates Proinflammatory Responses at the Single Cell Level. Redox Biol. 2020, 28, 101354. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, L.S.; Barreto, A.S.; Bomfim, L.G.S.; Gomes, M.C.; Ferreira, N.L.C.; da Cruz, G.S.; Magalhães, L.S.; de Jesus, A.R.; Palatnik-de-Sousa, C.B.; Corrêa, C.B.; et al. Multifunctional, TNF-α and IFN-γ-Secreting CD4 and CD8 T Cells and CD8High T Cells Are Associated with the Cure of Human Visceral Leishmaniasis. Front. Immunol. 2021, 12, 773983. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, N.; Ansari, M.Y.; Haqqi, T.M. Role of INOS in Osteoarthritis: Pathological and Therapeutic Aspects. J. Cell. Physiol. 2020, 235, 6366–6376. [Google Scholar] [CrossRef] [PubMed]
- Ausina, P.; Branco, J.R.; Demaria, T.M.; Esteves, A.M.; Leandro, J.G.B.; Ochioni, A.C.; Mendonça, A.P.M.; Palhano, F.L.; Oliveira, M.F.; Abou-Kheir, W.; et al. Acetylsalicylic Acid and Salicylic Acid Present Anticancer Properties against Melanoma by Promoting Nitric Oxide-Dependent Endoplasmic Reticulum Stress and Apoptosis. Sci. Rep. 2020, 10, 19617. [Google Scholar] [CrossRef] [PubMed]
- Mädge, J.C.; Stallmach, A.; Kleebusch, L.; Schlattmann, P. Meta-Analysis of Aspirin-Guided Therapy of Colorectal Cancer. J. Cancer Res. Clin. Oncol. 2022, 148, 1407–1417. [Google Scholar] [CrossRef] [PubMed]
- Skriver, C.; Maltesen, T.; Dehlendorff, C.; Skovlund, C.W.; Schmidt, M.; Sorensen, H.T.; Friis, S. Long-Term Aspirin Use and Cancer Risk: A 20-Year Cohort Study. J. Natl. Cancer Inst. 2024, 116, 530–538. [Google Scholar] [CrossRef] [PubMed]
- Mohamad, N.A.; Cricco, G.P.; Sambuco, L.A.; Croci, M.; Medina, V.A.; Gutiérrez, A.S.; Bergoc, R.M.; Rivera, E.S.; Martín, G.A. Aminoguanidine Impedes Human Pancreatic Tumor Growth and Metastasis Development in Nude Mice. World J. Gastroenterol. 2009, 15, 1065–1071. [Google Scholar] [CrossRef] [PubMed]
- Meurling, L.; Marquez, M.; Holmberg, A.R. Polymer-Conjugated Guanidine Is a Potentially Useful Anti-Tumor Agent. Int. J. Oncol. 2009, 35, 281–285. [Google Scholar] [CrossRef] [PubMed]
- Najafi, A.; Behnam, B.; Jafari, E.; Anani, H.; Karami-Mohajeri, S. Aminoguanidine Induced Apoptosis in Human Hepatocarcinoma HepG2 Cells. Gene Rep. 2021, 25, 101329. [Google Scholar] [CrossRef]
- Mayorek, N.; Naftali-Shani, N.; Grunewald, M. Diclofenac Inhibits Tumor Growth in a Murine Model of Pancreatic Cancer by Modulation of VEGF Levels and Arginase Activity. PLoS ONE 2010, 5, e12715. [Google Scholar] [CrossRef] [PubMed]
- Hofer, M.; Hoferová, Z.; Fedoročko, P.; Macková, N.O. Hematopoiesis-Stimulating and Anti-Tumor Effects of Repeated Administration of Diclofenac in Mice with Transplanted Fibrosarcoma Cells. Physiol. Res. 2002, 51, 629–632. [Google Scholar] [CrossRef] [PubMed]
- Hamoya, T.; Fujii, G.; Miyamoto, S.; Takahashi, M.; Totsuka, Y.; Wakabayashi, K.; Toshima, J.; Mutoh, M. Effects of NSAIDs on the Risk Factors of Colorectal Cancer: A Mini Review. Genes Environ. 2016, 38, 6. [Google Scholar] [CrossRef] [PubMed]
- Krischak, G.D.; Augat, P.; Blakytny, R.; Claes, L.; Kinzl, L.; Beck, A. The Non-Steroidal Anti-Inflammatory Drug Diclofenac Reduces Appearance of Osteoblasts in Bone Defect Healing in Rats. Arch. Orthop. Trauma Surg. 2007, 127, 453–458. [Google Scholar] [CrossRef] [PubMed]
- Ruocco, M.R.; Cecere, F.; Iuliano, A.; Albano, F.; Zappelli, C.; Castellano, I.; Grimaldi, P.; Masullo, M.; De Vendittis, E. Diclofenac-Induced Apoptosis in the Neuroblastoma Cell Line SH-SY5Y: Possible Involvement of the Mitochondrial Superoxide Dismutase. J. Biomed. Biotechnol. 2010, 2010, 801726. [Google Scholar] [CrossRef] [PubMed]
- Valle, B.L.; D’Souza, T.; Becker, K.G.; Wood, W.H.; Zhang, Y.; Wersto, R.P.; Morin, P.J. Non-Steroidal Anti-Inflammatory Drugs Decrease E2F1 Expression and Inhibit Cell Growth in Ovarian Cancer Cells. PLoS ONE 2013, 8, e61836. [Google Scholar] [CrossRef] [PubMed]
- Prüser, J.L.; Ramer, R.; Wittig, F.; Ivanov, I.; Merkord, J.; Hinz, B. The Monoacylglycerol Lipase Inhibitor JZL184 Inhibits Lung Cancer Cell Invasion and Metastasis via the CB1 Cannabinoid Receptor. Mol. Cancer Ther. 2021, 20, 787–802. [Google Scholar] [CrossRef] [PubMed]
- Galve-Roperh, I.; Sánchez, C.; Cortés, M.L.; del Pulgar, T.G.; Izquierdo, M.; Guzmán, M. Anti-Tumoral Action of Cannabinoids: Involvement of Sustained Ceramide Accumulation and Extracellular Signal-Regulated Kinase Activation. Nat. Med. 2000, 6, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Myint, Z.; St. Clair, W.H.; Strup, S.; Wang, P.; James, A.C.; Yan, D.; Ellis, C.S.; Otto, D.E.; DiPaola, R.S.; Arnold, S.M.; et al. A Phase I Does-Escalation and Expansion Study of Epidiolex (Cannabidiol) in Patients with Biochemically Recurrent Prostate Cancer. J. Clin. Oncol. 2022, 40 (Suppl. S6), 257. [Google Scholar] [CrossRef]
- Weiss, M.C.; Giaddui, M.; Kjelstrom, S.; Erebor, E.; Meske, S.; Saeed, L.; Ruiz, K.A.; Ghaneie, A.; Hibbs, J.; Hong, J.; et al. Safety and Efficacy of Cannabidiol in the Management of Chemotherapy-Induced Peripheral Neuropathy. J. Clin. Oncol. 2023, 41, 12020. [Google Scholar] [CrossRef]
- Liu, B.; Yan, S.; Qu, L.; Zhu, J. Celecoxib Enhances Anticancer Effect of Cisplatin and Induces Anoikis in Osteosarcoma via PI3K/Akt Pathway. Cancer Cell Int. 2017, 17, 1. [Google Scholar] [CrossRef] [PubMed]
- Chiang, S.L.; Velmurugan, B.K.; Chung, C.M.; Lin, S.H.; Wang, Z.H.; Hua, C.H.; Tsai, M.H.; Kuo, T.M.; Yeh, K.T.; Chang, P.Y.; et al. Preventive Effect of Celecoxib Use against Cancer Progression and Occurrence of Oral Squamous Cell Carcinoma. Sci. Rep. 2017, 7, 6235. [Google Scholar] [CrossRef] [PubMed]
- Shirakawa, K.; Wang, L.; Man, N.; Maksimoska, J.; Sorum, A.W.; Lim, H.W.; Lee, I.S.; Shimazu, T.; Newman, J.C.; Schrö Der, S.; et al. Salicylate, Diflunisal and Their Metabolites Inhibit CBP/P300 and Exhibit Anticancer Activity. Elife 2016, 5, e11156. [Google Scholar] [CrossRef] [PubMed]
- Coşkun, G.P.; Djikic, T.; Hayal, T.B.; Türkel, N.; Yelekçi, K.; Şahin, F.; Küçükgüzel, G. Synthesis, Molecular Docking and Anticancer Activity of Diflunisal Derivatives as Cyclooxygenase Enzyme Inhibitors. Molecules 2018, 23, 1969. [Google Scholar] [CrossRef]
- Shigemura, K.; Shirakawa, T.; Wada, Y.; Kamidono, S.; Fujisawa, M.; Gotoh, A. Antitumor Effects of Etodolac, a Selective Cyclooxygenase-II Inhibitor, against Human Prostate Cancer Cell Lines In Vitro and In Vivo. Urology 2005, 66, 1239–1244. [Google Scholar] [CrossRef] [PubMed]
- Shibata, M.; Shimura, T.; Gonda, K.; Ohki, S.; Koyama, Y.; Sakurai, K.; Tomita, R.; Takenoshita, S. Etodolac Inhibits Interleukin-6 Production and Improves Survival Combined with Chemotherapy in a Colon 26 Cachexia Model. Ann. Cancer Res. Ther. 2010, 18, 1–5. [Google Scholar] [CrossRef]
- Okamoto, A.; Shirakawa, T.; Bito, T.; Shigemura, K.; Hamada, K.; Gotoh, A.; Fujisawa, M.; Kawabata, M. Etodolac, a Selective Cyclooxygenase-2 Inhibitor, Induces Upregulation of E-Cadherin and Has Antitumor Effect on Human Bladder Cancer Cells In Vitro and In Vivo. Urology 2008, 71, 156–160. [Google Scholar] [CrossRef] [PubMed]
- Orun, O.; Tiber, P.M.; Sevinç, S.K. Apoptotic Effects of Etodolac in Breast Cancer Cell Cultures. In Nonsteroidal Anti-Inflammatory Drugs; InTech: London, UK, 2017. [Google Scholar] [CrossRef]
- Murata, S.; Adachi, M.; Kioi, M.; Torigoe, S.; Ijichi, K.; Hasegawa, Y.; Ogawa, T.; Bhayani, M.K.; Lai, S.Y.; Mitsudo, K.; et al. Etodolac Improves 5-FU Sensitivity of Head and Neck Cancer Cells through Inhibition of Thymidylate Synthase. Anticancer Res. 2011, 31, 2893–2898. [Google Scholar] [PubMed]
- Dhar, S.S.; Shahi, U.P.; Kumar, D.; Mishra, R.; Kaser, P.; Dewangan, S.; Mandal, A.; Choudhary, S.; Aggarwal, L.M.; Asthana, A.K.; et al. Addition of Etoricoxib During Concurrent Chemo-Radiation of Cervical Cancer Patients Could Result in Faster Resolution of Gross Disease: A Prospective Single-Institution Study. Indian J. Gynecol. Oncol. 2020, 18, 1. [Google Scholar] [CrossRef]
- Saini, M.K.; Sharma, P.; Kaur, J.; Sanyal, S.N.; Saini, M.K. Inhibition of Colon Carcinogenesis with Etoricoxib the Cyclooxygenase-2 Inhibitor Etoricoxib Is a Potent Chemopreventive Agent of Colon Carcinogenesis in the Rat Model. J. Environ. Pathol. Toxicol. Oncol. 2009, 28, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Rosati, M.S.; Di Seri, M.; Baciarello, G.; LO Russo, V.; Grassi, P.; Marchetti, L.; Giovannoni, S.; Basile, M.L.; Frati, L. Etoricoxib and Anastrozole in Adjuvant Early Breast Cancer: ETAN Trial (Phase III). J. Clin. Oncol. 2011, 29 (Suppl. S15), 533. [Google Scholar] [CrossRef]
- Sikka, M. Abstract A28: Mechanistic Study Involving the Combined Antiproliferative Effect of Etoricoxib- Cyclooxygenase-2 Inhibitor and Cholecystokinin-2 Receptor Antagonist in Human Pancreatic Cancer Cells. Mol. Cancer Res. 2016, 14 (Suppl. S11), A28. [Google Scholar] [CrossRef]
- Wang, X.; Ye, X.; Zhang, Y.; Ji, F. Flurbiprofen Suppresses the Inflammation, Proliferation, Invasion and Migration of Colorectal Cancer Cells via COX2. Oncol. Lett. 2020, 20, 132. [Google Scholar] [CrossRef] [PubMed]
- Yang, N.; Liang, Y.; Yang, P.; Jiang, L. Flurbiprofen Inhibits Cell Proliferation in Thyroid Cancer through Interrupting HIP1R-Induced Endocytosis of PTEN. Eur. J. Med. Res. 2022, 27, 29. [Google Scholar] [CrossRef] [PubMed]
- Cui, B.; Cheng, X.; Zhang, X.; Chen, L.; Pang, W.; Liu, Y.; Yang, Z.; Li, H.; He, X.; Li, X.; et al. Anti-Cancer Activity and Mechanism of Flurbiprofen Organoselenium Compound RY-1-92 in Non-Small Cell Lung Cancer. RSC Med. Chem. 2024, 15, 1737–1745. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.E.; Beebe-Donk, J.; Doss, H.; Doss, D.B. Aspirin, Ibuprofen, and Other Non-Steroidal Anti-Inflammatory Drugs in Cancer Prevention: A Critical Review of Non-Selective COX-2 Blockade (Review). Oncol. Rep. 2005, 13, 559–583. [Google Scholar] [CrossRef] [PubMed]
- Bittoni, M.A.; Carbone, D.P.; Harris, R.E. Ibuprofen and Fatal Lung Cancer: A Brief Report of the Prospective Results from the Third National Health and Nutrition Examination Survey (NHANES III). Mol. Clin. Oncol. 2017, 6, 917–920. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.; Zhang, X.; Du, R.; Gao, W.; Wang, J.; Bao, Y.; Yang, W.; Luo, N.; Li, J. Ibuprofen Mediates Histone Modification to Diminish Cancer Cell Stemness Properties via a COX2-Dependent Manner. Br. J. Cancer 2020, 123, 730–741. [Google Scholar] [CrossRef] [PubMed]
- Okda, T.; Abd-Εlghaffar, S.; Katary, M.; Abd-αlhaseeb, M. Chemopreventive and Anticancer Activities of Indomethacin and Vitamin D Combination on Colorectal Cancer Induced by 1,2-dimethylhydrazine in Rats. Biomed. Rep. 2020, 14, 27. [Google Scholar] [CrossRef] [PubMed]
- Ackerstaff, E.; Gimi, B.; Artemov, D.; Bhujwalla, Z.M. Anti-Inflammatory Agent Indomethacin Reduces Invasion and Alters Metabolism in a Human Breast Cancer Cell Line. Neoplasia 2007, 9, 222–235. [Google Scholar] [CrossRef]
- Segovia-Mendoza, M.; Camacho-Camacho, C.; Rojas-Oviedo, I.; Prado-Garcia, H.; Barrera, D.; Martínez-Reza, I.; Larrea, F.; García-Becerra, R. An Organotin Indomethacin Derivative Inhibits Cancer Cell Proliferation and Synergizes the Antiproliferative Effects of Lapatinib in Breast Cancer Cells. Am. J. Cancer Res. 2020, 10, 3358–3369. [Google Scholar]
- Noori, S.; Rajabi, S.; Tavirani, M.R.; Shokri, B.; Zarghi, A. Anti-Breast Cancer Activities of Ketoprofen-RGD Conjugate by Targeting Breast Cancer Stem-Like Cells and Parental Cells. Anticancer Agents Med. Chem. 2021, 21, 1027–1036. [Google Scholar] [CrossRef] [PubMed]
- Patra, I.; Naser, R.H.; Hussam, F.; Hameed, N.M.; Kadhim, M.M.; Ahmad, I.; Awadh, S.A.; Hamad, D.A.; Parra, R.M.R.; Mustafa, Y.F. Ketoprofen Suppresses Triple Negative Breast Cancer Cell Growth by Inducing Apoptosis and Inhibiting Autophagy. Mol. Biol. Rep. 2023, 50, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Gor, R.; Gharib, A.; Dharshini Balaji, P.; Madhavan, T.; Ramalingam, S. Inducing Cytotoxicity in Colon Cancer Cells and Suppressing Cancer Stem Cells by Dolasetron and Ketoprofen through Inhibition of RNA Binding Protein PUM1. Toxics 2023, 11, 669. [Google Scholar] [CrossRef] [PubMed]
- Afshari, H.; Noori, S.; Shokri, B.; Zarghi, A. Co-Treatment of Naringenin and Ketoprofen-RGD Suppresses Cell Proliferation via Calmodulin/PDE/CAMP/PKA Axis Pathway in Leukemia and Ovarian Cancer Cells. Iran. J. Pharm. Res. 2023, 22, e136131. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Cheng, S.; Fu, G.; Ji, F.; Wang, C.; Cao, M. Postoperative Administration of Ketorolac Averts Morphine-Induced Angiogenesis and Metastasis in Triple-Negative Breast Cancer. Life Sci. 2020, 251, 117604. [Google Scholar] [CrossRef] [PubMed]
- Hudson, L.G.; Cook, L.S.; Grimes, M.M.; Muller, C.Y.; Adams, S.F.; Wandinger-Ness, A. Dual Actions of Ketorolac in Metastatic Ovarian Cancer. Cancers 2019, 11, 1049. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Wang, Q.; Li, L.; Chen, Y.; Cui, J.; Liu, M.; Zhang, N.; Liu, Z.; Han, J.; Wang, Z. Ketoprofen and Loxoprofen Platinum(IV) Complexes Displaying Antimetastatic Activities by Inducing DNA Damage, Inflammation Suppression, and Enhanced Immune Response. J. Med. Chem. 2021, 64, 17920–17935. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Jia, B.; Zhang, Q.; Zhang, Y. Meclofenamic Acid Restores Gefinitib Sensitivity by Downregulating Breast Cancer Resistance Protein and Multidrug Resistance Protein 7 via FTO/M6A-Demethylation/c-Myc in Non-Small Cell Lung Cancer. Front. Oncol. 2022, 12, 870636. [Google Scholar] [CrossRef] [PubMed]
- Saglam, B.S.; Kanli, A.; Yanar, S.; Kasap, M.; Akpinar, G. Investigation of the Effect of Meclofenamic Acid on the Proteome of LNCaP Cells Reveals Changes in Alternative Polyadenylation and Splicing Machinery. Med. Oncol. 2022, 39, 190. [Google Scholar] [CrossRef] [PubMed]
- Seyyedi, R.; Talebpour Amiri, F.; Farzipour, S.; Mihandoust, E.; Hosseinimehr, S.J. Mefenamic Acid as a Promising Therapeutic Medicine against Colon Cancer in Tumor-Bearing Mice. Med. Oncol. 2022, 39, 18. [Google Scholar] [CrossRef] [PubMed]
- Hosseinimehr, S.J.; Safavi, Z.; Kangarani Farahani, S.; Noaparst, Z.; Ghasemi, A.; Asgarian-Omran, H. The Synergistic Effect of Mefenamic Acid with Ionizing Radiation in Colon Cancer. J. Bioenerg. Biomembr. 2019, 51, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Ho Woo, D.; Han, I.-S.; Jung, G. Mefenamic Acid-Induced Apoptosis in Human Liver Cancer Cell-Lines through Caspase-3 Pathway. Life Sci. 2004, 75, 2439–2449. [Google Scholar] [CrossRef] [PubMed]
- Shiiba, M.; Yamagami, H.; Yamamoto, A.; Minakawa, Y.; Okamoto, A.; Kasamatsu, A.; Sakamoto, Y.; Uzawa, K.; Takiguchi, Y.; Tanzawa, H. Mefenamic Acid Enhances Anticancer Drug Sensitivity via Inhibition of Aldo-Keto Reductase 1C Enzyme Activity. Oncol. Rep. 2017, 37, 2025–2032. [Google Scholar] [CrossRef] [PubMed]
- Naruse, T.; Nishida, Y.; Hosono, K.; Ishiguro, N. Meloxicam Inhibits Osteosarcoma Growth, Invasiveness and Metastasis by COX-2-Dependent and Independent Routes. Carcinogenesis 2006, 27, 584–592. [Google Scholar] [CrossRef] [PubMed]
- Guangshun, S.; Guoqiang, S.; Xin, C.; Xiangyi, K.; Wubin, Z.; Zhitao, L.; Zhiying, Z.; Hongyong, C.; Chengyu, L.; Yongxiang, X.; et al. Meloxicam Inhibits Hepatocellular Carcinoma Progression and Enhances the Sensitivity of Immunotherapy via the MicroRNA-200/PD-L1 Pathway. J. Oncol. 2022, 2022, 4598573. [Google Scholar] [CrossRef] [PubMed]
- Goldman, A. Meloxicam Inhibits the Growth of Colorectal Cancer Cells. Carcinogenesis 1998, 19, 2195–2199. [Google Scholar] [CrossRef] [PubMed]
- Bersuder, E.; Terciolo, C.; Lechevrel, M.; Martin, E.; Quesnelle, C.; Freund, J.N.; Reimund, J.M.; Gross, I. Mesalazine Initiates an Anti-Oncogenic β-Catenin/MUCDHL Negative Feed-Back Loop in Colon Cancer Cells by Cell-Specific Mechanisms. Biomed. Pharmacother. 2022, 146, 112543. [Google Scholar] [CrossRef] [PubMed]
- Swami, D.; Siddiqui, S.; Kumar, U.; Devarajan, S.; Aich, J. One of the 5-Aminosalicylates Drug, Mesalamine as a Drug Repurposing Lead against Breast Cancer. Bull. Natl. Res. Cent. 2022, 46, 249. [Google Scholar] [CrossRef]
- Jung, H.-J.; Seo, I.; Jha, B.; Suh, S.-I.; Baek, W.-K. Miconazole Induces Autophagic Death in Glioblastoma Cells via Reactive Oxygen Species-mediated Endoplasmic Reticulum Stress. Oncol. Lett. 2021, 21, 335. [Google Scholar] [CrossRef] [PubMed]
- Ho, C.; Chang, A.; Hsu, C.; Tsai, T.; Lin, Y.; Chou, K.; Chen, H.; Lin, J.; Chen, P.; Hwang, T.I. Miconazole Induces Protective Autophagy in Bladder Cancer Cells. Environ. Toxicol. 2021, 36, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.-Y.; Shiau, M.-Y.; Ou, Y.-C.; Huang, Y.-C.; Chen, C.-C.; Cheng, C.-L.; Chiu, K.-Y.; Wang, S.-S.; Tsai, K.-J. Miconazole Induces Apoptosis via the Death Receptor 5-Dependent and Mitochondrial-Mediated Pathways in Human Bladder Cancer Cells. Oncol. Rep. 2017, 37, 3606–3616. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.-H.; Jeng, J.-H.; Wang, Y.-J.; Tseng, C.-J.; Liang, Y.-C.; Chen, C.-H.; Lee, H.-M.; Lin, J.-K.; Lin, C.-H.; Lin, S.-Y.; et al. Antitumor Effects of Miconazole on Human Colon Carcinoma Xenografts in Nude Mice through Induction of Apoptosis and G0/G1 Cell Cycle Arrest. Toxicol. Appl. Pharmacol. 2002, 180, 22–35. [Google Scholar] [CrossRef] [PubMed]
- Tone, M.; Iwahori, K.; Shiroyama, T.; Futami, S.; Naito, Y.; Fukushima, K.; Miyake, K.; Koyama, S.; Hirata, H.; Nagatomo, I.; et al. Impact of Minocycline on Outcomes of EGFR-Mutant Non-Small Cell Lung Cancer Patients Treated with EGFR-TKIs. Sci. Rep. 2023, 13, 8313. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Yang, J.; Liu, H.; Lan, J.; Xu, Y.; Wu, X.; Mao, Y.; Wu, D.; Pan, K.; Zhang, T. Minocycline Binds and Inhibits LYN Activity to Prevent STAT3-Meditated Metastasis of Colorectal Cancer. Int. J. Biol. Sci. 2022, 18, 2540–2552. [Google Scholar] [CrossRef]
- Chu, M.; Wang, T.; Sun, A.; Chen, Y. Nimesulide Inhibits Proliferation and Induces Apoptosis of Pancreatic Cancer Cells by Enhancing Expression of PTEN. Exp. Ther. Med. 2018, 16, 370–376. [Google Scholar] [CrossRef] [PubMed]
- Sawangrat, K.; Morishita, M.; Kusamori, K.; Katsumi, H.; Sakane, T.; Yamamoto, A. Effects of Various Pharmaceutical Excipients on the Intestinal Transport and Absorption of Sulfasalazine, a Typical Substrate of Breast Cancer Resistance Protein Transporter. J. Pharm. Sci. 2018, 107, 2946–2956. [Google Scholar] [CrossRef] [PubMed]
- Wada, F.; Koga, H.; Akiba, J.; Niizeki, T.; Iwamoto, H.; Ikezono, Y.; Nakamura, T.; Abe, M.; Masuda, A.; Sakaue, T.; et al. High Expression of CD44v9 and xCT in Chemoresistant Hepatocellular Carcinoma: Potential Targets by Sulfasalazine. Cancer Sci. 2018, 109, 2801–2810. [Google Scholar] [CrossRef] [PubMed]
- Garcia, C.G.; Kahn, S.A.; Geraldo, L.H.M.; Romano, I.; Domith, I.; e Silva, D.C.L.; dos Santos Assunção, F.; Ferreira, M.J.; Portugal, C.C.; de Souza, J.M.; et al. Combination Therapy with Sulfasalazine and Valproic Acid Promotes Human Glioblastoma Cell Death Through Imbalance of the Intracellular Oxidative Response. Mol. Neurobiol. 2018, 55, 6816–6833. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.H.; Shin, D.; Lee, J.; Jung, A.R.; Roh, J.-L. CISD2 Inhibition Overcomes Resistance to Sulfasalazine-Induced Ferroptotic Cell Death in Head and Neck Cancer. Cancer Lett. 2018, 432, 180–190. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Pathi, S.S.; Safe, S. Sulindac Sulfide Inhibits Colon Cancer Cell Growth and Downregulates Specificity Protein Transcription Factors. BMC Cancer 2015, 15, 974. [Google Scholar] [CrossRef] [PubMed]
- Yin, T.; Wang, G.; Ye, T.; Wang, Y. Sulindac, a Non-Steroidal Anti-Inflammatory Drug, Mediates Breast Cancer Inhibition as an Immune Modulator. Sci. Rep. 2016, 6, 19534. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Yang, A.L.; Chung, Y.T.; Zhang, W.; Liao, J.; Yang, G.-Y. Sulindac Inhibits Pancreatic Carcinogenesis in LSL-KrasG12D-LSL-Trp53R172H-Pdx-1-Cre Mice via Suppressing Aldo-Keto Reductase Family 1B10 (AKR1B10). Carcinogenesis 2013, 34, 2090–2098. [Google Scholar] [CrossRef] [PubMed]
- Abdelrahim, M.; Baker, C.H.; Abbruzzese, J.L.; Safe, S. Tolfenamic Acid and Pancreatic Cancer Growth, Angiogenesis, and Sp Protein Degradation. J. Natl. Cancer Inst. 2006, 98, 855–868. [Google Scholar] [CrossRef] [PubMed]
- Papineni, S.; Chintharlapalli, S.; Abdelrahim, M.; Lee, S.-O.; Burghardt, R.; Abudayyeh, A.; Baker, C.; Herrera, L.; Safe, S. Tolfenamic Acid Inhibits Esophageal Cancer through Repression of Specificity Proteins and C-Met. Carcinogenesis 2009, 30, 1193–1201. [Google Scholar] [CrossRef] [PubMed]
- Pathi, S.; Li, X.; Safe, S. Tolfenamic Acid Inhibits Colon Cancer Cell and Tumor Growth and Induces Degradation of Specificity Protein (Sp) Transcription Factors. Mol. Carcinog. 2014, 53, E53–E61. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.U.; Shin, Y.S.; Hwang, H.S.; Baek, S.J.; Lee, S.-H.; Kim, C.-H. Tolfenamic Acid Induces Apoptosis and Growth Inhibition in Head and Neck Cancer: Involvement of NAG-1 Expression. PLoS ONE 2012, 7, e34988. [Google Scholar] [CrossRef] [PubMed]
- Stuehr, D.J.; Haque, M.M. Nitric Oxide Synthase Enzymology in the 20 Years after the Nobel Prize. Br. J. Pharmacol. 2019, 176, 177–188. [Google Scholar] [CrossRef] [PubMed]
- Doman, A.J.; Tommasi, S.; Perkins, M.V.; McKinnon, R.A.; Mangoni, A.A.; Nair, P.C. Chemical Similarities and Differences among Inhibitors of Nitric Oxide Synthase, Arginase and Dimethylarginine Dimethylaminohydrolase-1: Implications for the Design of Novel Enzyme Inhibitors Modulating the Nitric Oxide Pathway. Bioorg. Med. Chem. 2022, 72, 116970. [Google Scholar] [CrossRef] [PubMed]
- Grottelli, S.; Amoroso, R.; Macchioni, L.; D’Onofrio, F.; Fettucciari, K.; Bellezza, I.; Maccallini, C. Acetamidine-Based INOS Inhibitors as Molecular Tools to Counteract Inflammation in BV2 Microglial Cells. Molecules 2020, 25, 2646. [Google Scholar] [CrossRef]
- Yamagishi, S.I. Advanced Glycation End-Products. In Brenner’s Encyclopedia of Genetics, 2nd ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2013; pp. 36–38. [Google Scholar] [CrossRef]
- Dover, A.R.; Chia, S.; Ferguson, J.W.; Cruden, N.L.; Megson, I.L.; Fox, K.A.A.; Newby, D.E. Inducible Nitric Oxide Synthase Activity Does Not Contribute to the Maintenance of Peripheral Vascular Tone in Patients with Heart Failure. Clin. Sci. 2006, 111, 275–280. [Google Scholar] [CrossRef]
- Katz, J.A. COX-2 Inhibition: What We Learned-A Controversial Update on Safety Data. Pain Med. 2013, 14, S29–S34. [Google Scholar] [CrossRef] [PubMed]
- Tai, H.H.; Chi, X.; Tong, M. Regulation of 15-Hydroxyprostaglandin Dehydrogenase (15-PGDH) by Non-Steroidal Anti-Inflammatory Drugs (NSAIDs). Prostaglandins Other Lipid Mediat. 2011, 96, 37–40. [Google Scholar] [CrossRef] [PubMed]
- Chung, A.W.; Anand, K.; Anselme, A.C.; Chan, A.A.; Gupta, N.; Venta, L.A.; Schwartz, M.R.; Qian, W.; Xu, Y.; Zhang, L.; et al. A Phase 1/2 Clinical Trial of the Nitric Oxide Synthase Inhibitor L-NMMA and Taxane for Treating Chemoresistant Triple-Negative Breast Cancer. Sci. Transl. Med. 2021; 13, eabj5070. [Google Scholar]
- Puri, A.; Ordonez, A.; Anselme, A.C.; Guzman, L.; Niravath, P.; Chang, J.C. Phase 1B/2 Clinical Trial Targeting Nitric Oxide in the Treatment of Chemo-Refractory Metaplastic Triple-Negative Breast Cancer Patients. Cancer Res. 2022; 82, (Suppl. S4), P5-17-07. [Google Scholar] [CrossRef]
- Park, J.S.; Woo, M.S.; Kim, S.Y.; Kim, W.K.; Kim, H.S. Repression of Interferon-γ-Induced Inducible Nitric Oxide Synthase (INOS) Gene Expression in Microglia by Sodium Butyrate Is Mediated through Specific Inhibition of ERK Signaling Pathways. J. Neuroimmunol. 2005, 168, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Kulesza, A.; Paczek, L.; Burdzinska, A. The Role of COX-2 and PGE2 in the Regulation of Immunomodulation and Other Functions of Mesenchymal Stromal Cells. Biomedicines 2023, 11, 445. [Google Scholar] [CrossRef] [PubMed]
- Zaidi, M.R. The Interferon-Gamma Paradox in Cancer. J. Interferon Cytokine Res. 2019, 39, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Ma, C.; Zhang, Z.; Zhang, H.; Hu, H. NF-ΚB Signaling in Inflammation and Cancer. MedComm 2021, 2, 618–653. [Google Scholar] [CrossRef] [PubMed]
- Moita, E.; Gil-Izquierdo, A.; Sousa, C.; Ferreres, F.; Silva, L.R.; Valentão, P.; Domínguez-Perles, R.; Baenas, N.; Andrade, P.B. Integrated Analysis of COX-2 and INOS Derived Inflammatory Mediators in LPS-Stimulated RAW Macrophages Pre-Exposed to Echium plantagineum L. Bee Pollen Extract. PLoS ONE 2013, 8, e59131. [Google Scholar] [CrossRef] [PubMed]
- Abdellatif, K.R.A.; Abdelall, E.K.A.; Elshemy, H.A.H.; Philoppes, J.N.; Hassanein, E.H.M.; Kahk, N.M. Optimization of Pyrazole-Based Compounds with 1,2,4-Triazole-3-Thiol Moiety as Selective COX-2 Inhibitors Cardioprotective Drug Candidates: Design, Synthesis, Cyclooxygenase Inhibition, Anti-Inflammatory, Ulcerogenicity, Cardiovascular Evaluation, and Molecular Modeling Studies. Bioorg. Chem. 2021, 114, 105122. [Google Scholar] [CrossRef] [PubMed]
- Shan, Y.; Zhao, R.; Geng, W.; Lin, N.; Wang, X.; Du, X.; Wang, S. Protective Effect of Sulforaphane on Human Vascular Endothelial Cells against Lipopolysaccharide-Induced Inflammatory Damage. Cardiovasc. Toxicol. 2010, 10, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Vrânceanu, M.; Galimberti, D.; Banc, R.; Dragoş, O.; Cozma-Petruţ, A.; Hegheş, S.C.; Voştinaru, O.; Cuciureanu, M.; Stroia, C.M.; Miere, D.; et al. The Anticancer Potential of Plant-Derived Nutraceuticals via the Modulation of Gene Expression. Plants 2022, 11, 2524. [Google Scholar] [CrossRef] [PubMed]
- Justenhoven, C. Polymorphisms of Phase I and Phase II Enzymes and Breast Cancer Risk. Front. Genet. 2012, 3, 258. [Google Scholar] [CrossRef]
- Köhle, C.; Bock, K.W. Coordinate Regulation of Phase I and II Xenobiotic Metabolisms by the Ah Receptor and Nrf2. Biochem. Pharmacol. 2007, 73, 1853–1862. [Google Scholar] [CrossRef] [PubMed]
- Salazar, F.; Awuah, D.; Negm, O.H.; Shakib, F.; Ghaemmaghami, A.M. The Role of Indoleamine 2,3-Dioxygenase-Aryl Hydrocarbon Receptor Pathway in the TLR4-Induced Tolerogenic Phenotype in Human DCs. Sci. Rep. 2017, 7, 43337. [Google Scholar] [CrossRef] [PubMed]
- Therachiyil, L.; Krishnankutty, R.; Ahmad, F.; Mateo, J.M.; Uddin, S.; Korashy, H.M. Aryl Hydrocarbon Receptor Promotes Cell Growth, Stemness Like Characteristics, and Metastasis in Human Ovarian Cancer via Activation of PI3K/Akt, β-Catenin, and Epithelial to Mesenchymal Transition Pathways. Int. J. Mol. Sci. 2022, 23, 6395. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.M.U.; Luo, L.; Namani, A.; Wang, X.J.; Tang, X. Nrf2 Signaling Pathway: Pivotal Roles in Inflammation. Biochim. Biophys. Acta—Mol. Basis Dis. 2017, 1863, 585–597. [Google Scholar] [CrossRef] [PubMed]
- Ghareghomi, S.; Moosavi-Movahedi, F.; Saso, L.; Habibi-Rezaei, M.; Khatibi, A.; Hong, J.; Moosavi-Movahedi, A.A. Modulation of Nrf2/HO-1 by Natural Compounds in Lung Cancer. Antioxidants 2023, 12, 735. [Google Scholar] [CrossRef]
- Jung, A.Y.; Cai, X.; Thoene, K.; Obi, N.; Jaskulski, S.; Behrens, S.; Flesch-Janys, D.; Chang-Claude, J. Antioxidant Supplementation and Breast Cancer Prognosis in Postmenopausal Women Undergoing Chemotherapy and Radiation Therapy. Am. J. Clin. Nutr. 2019, 109, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Um, H.C.; Jang, J.H.; Kim, D.H.; Lee, C.; Surh, Y.J. Nitric Oxide Activates Nrf2 through S-Nitrosylation of Keap1 in PC12 Cells. Nitric Oxide—Biol. Chem. 2011, 25, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Munday, R. Dithiolethiones for Cancer Chemoprevention: Where Do We Stand? Mol. Cancer Ther. 2008, 7, 3470–3479. [Google Scholar] [CrossRef] [PubMed]
- Esteve, M. Mechanisms Underlying Biological Effects of Cruciferous Glucosinolate-Derived Isothiocyanates/Indoles: A Focus on Metabolic Syndrome. Front. Nutr. 2020, 7, 111. [Google Scholar] [CrossRef] [PubMed]
- Sailaja, B.S.; Aita, R.; Maledatu, S.; Ribnicky, D.; Verzi, M.P.; Raskin, I. Moringa Isothiocyanate-1 Regulates Nrf2 and NF-ΚB Pathway in Response to LPS-Driven Sepsis and Inflammation. PLoS ONE 2021, 16, e0248691. [Google Scholar] [CrossRef] [PubMed]
- Switzer, C.H.; Cheng, R.Y.S.; Ridnour, L.A.; Murray, M.C.; Tazzari, V.; Sparatore, A.; Del Soldato, P.; Hines, H.B.; Glynn, S.A.; Ambs, S.; et al. Dithiolethiones Inhibit NF-ΚB Activity via Covalent Modification in Human Estrogen Receptor-Negative Breast Cancer. Cancer Res. 2012, 72, 2394–2404. [Google Scholar] [CrossRef]
- Switzer, C.H.; Ridnour, L.A.; Cheng, R.Y.S.; Sparatore, A.; Del Soldato, P.; Moody, T.W.; Vitek, M.P.; Roberts, D.D.; Wink, D.A. Dithiolethione Compounds Inhibit Akt Signaling in Human Breast and Lung Cancer Cells by Increasing PP2A Activity. Oncogene 2009, 28, 3837–3846. [Google Scholar] [CrossRef] [PubMed]
- Switzer, C.H.; Glynn, S.A.; Ridnour, L.A.; Cheng, R.Y.S.; Vitek, M.P.; Ambs, S.; Wink, D.A. Nitric Oxide and Protein Phosphatase 2A Provide Novel Therapeutic Opportunities in ER-Negative Breast Cancer. Trends Pharmacol. Sci. 2011, 32, 644–651. [Google Scholar] [CrossRef]
- Dacol, E.C.; Wang, S.; Chen, Y.; Lepique, A.P. The Interaction of SET and Protein Phosphatase 2A as Target for Cancer Therapy. Biochim. Biophys. Acta—Rev. Cancer 2021, 1876, 188578. [Google Scholar] [CrossRef] [PubMed]
- Pandey, P.; Seshacharyulu, P.; Das, S.; Rachagani, S.; Ponnusamy, M.P.; Yan, Y.; Johansson, S.L.; Datta, K.; Lin, M.F.; Batra, S.K. Impaired Expression of Protein Phosphatase 2A Subunits Enhances Metastatic Potential of Human Prostate Cancer Cells through Activation of AKT Pathway. Br. J. Cancer 2013, 108, 2590–2600. [Google Scholar] [CrossRef] [PubMed]
- Tohmé, R.; Izadmehr, S.; Gandhe, S.; Tabaro, G.; Vallabhaneni, S.; Thomas, A.; Vasireddi, N.; Dhawan, N.S.; Ma’ayan, A.; Sharma, N.; et al. Direct Activation of PP2A for the Treatment of Tyrosine Kinase Inhibitor-Resistant Lung Adenocarcinoma. JCI Insight 2019, 4, e125693. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, A.; Sugiyama, G.; Ohyama, Y.; Kumamaru, W.; Yamada, T.; Mori, Y. Regulation of NF-KB Signalling through the PR55β-RelA Interaction in Osteoblasts. In Vivo 2020, 34, 601–608. [Google Scholar] [CrossRef] [PubMed]
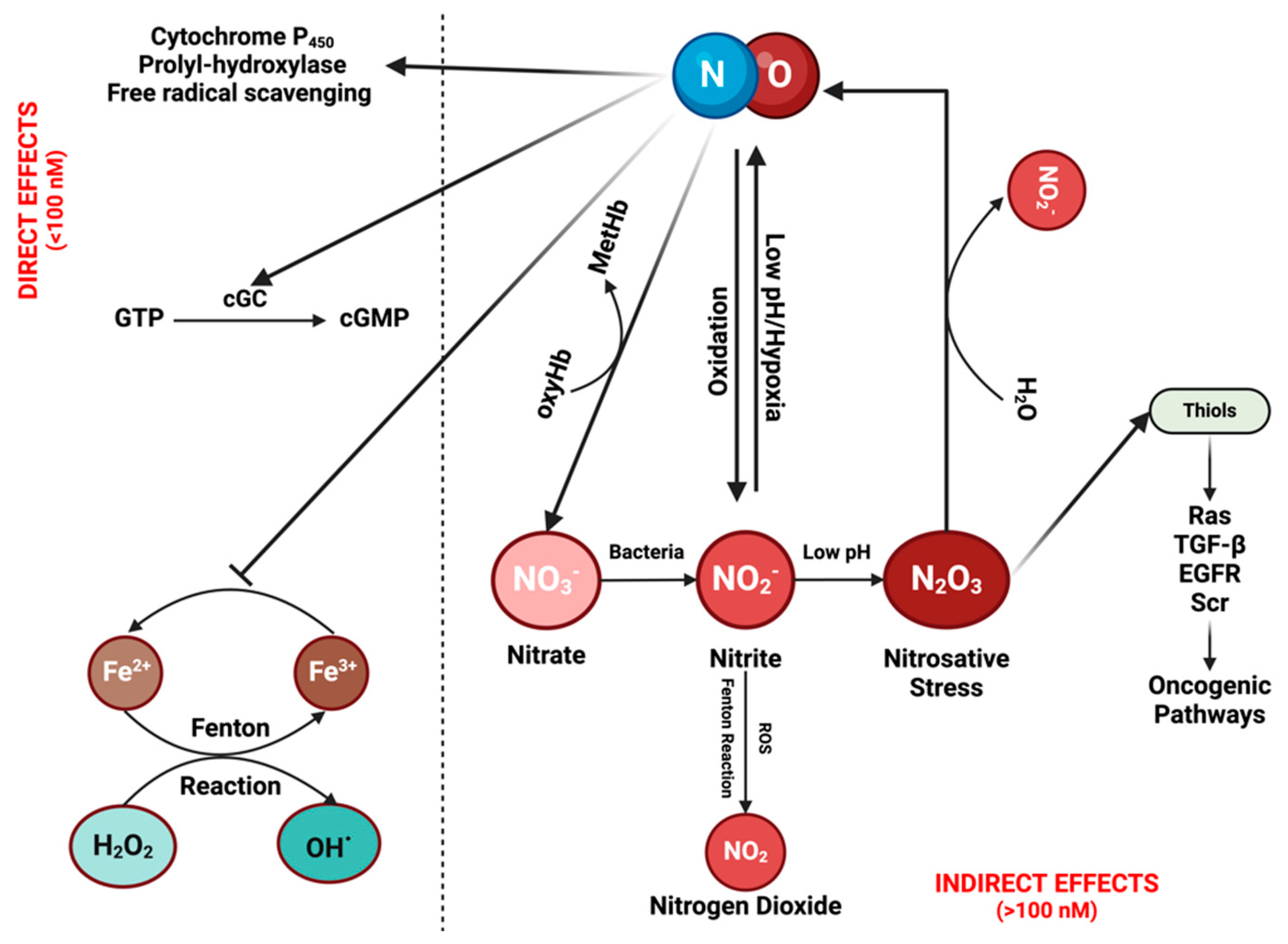
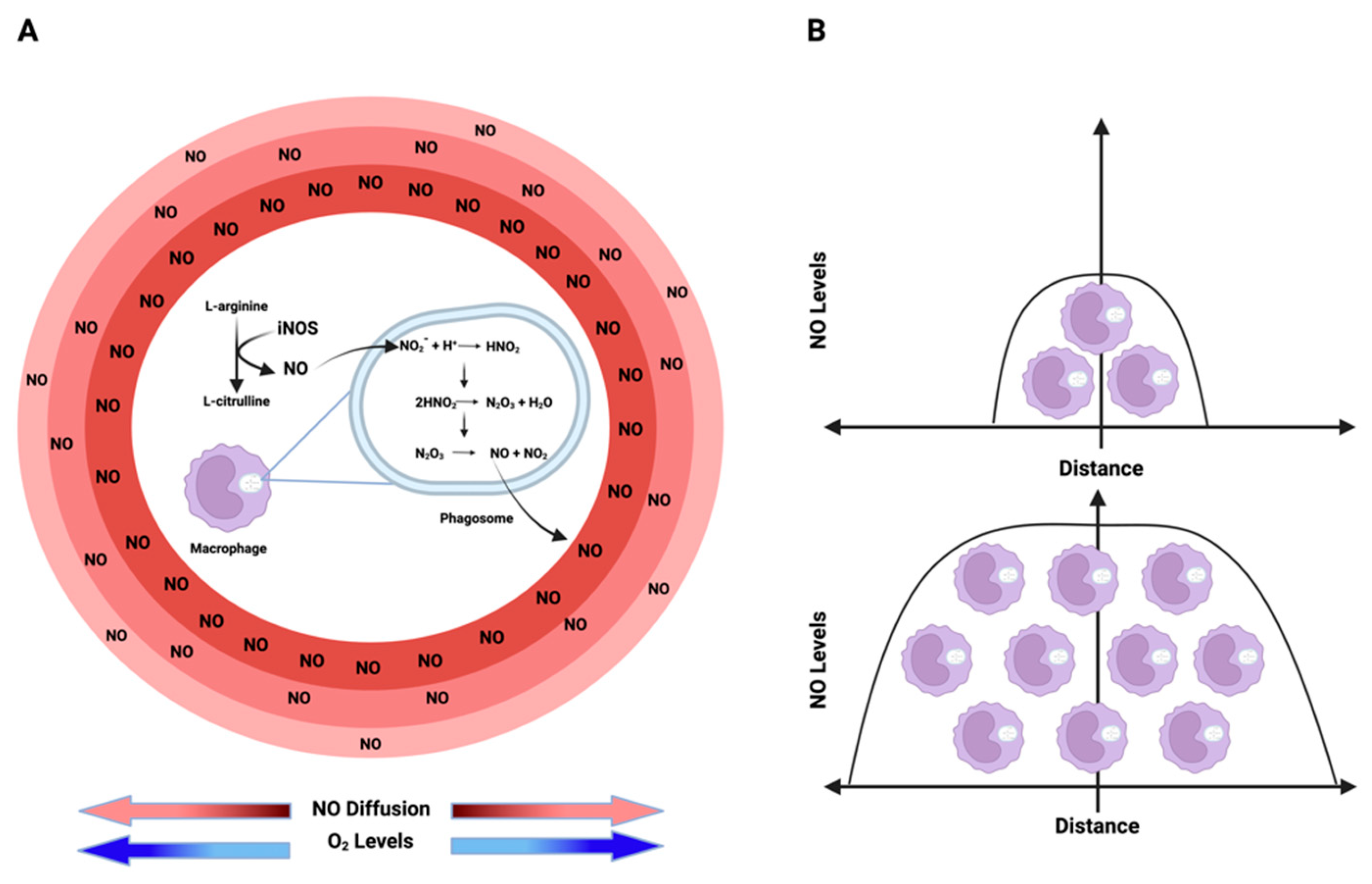
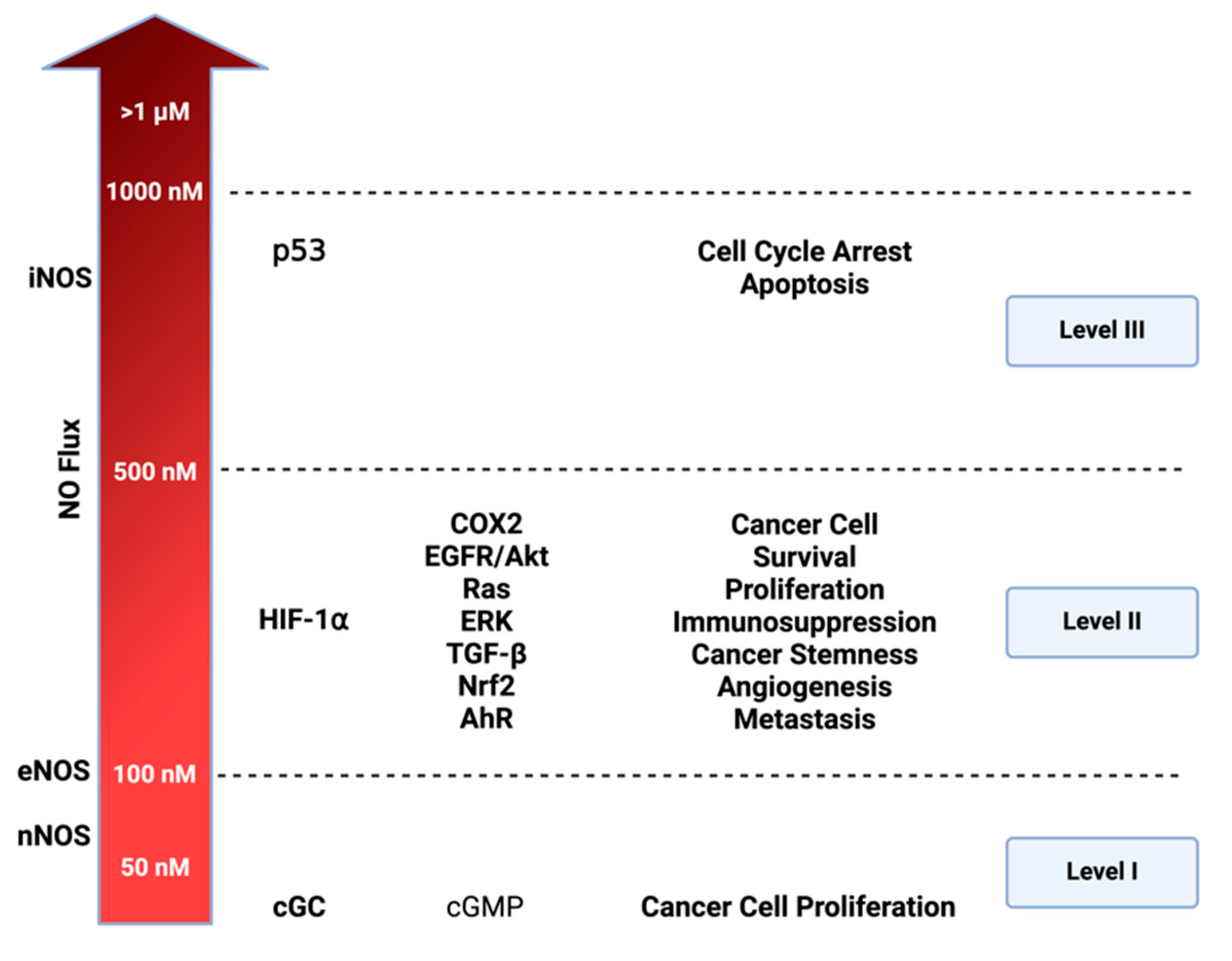
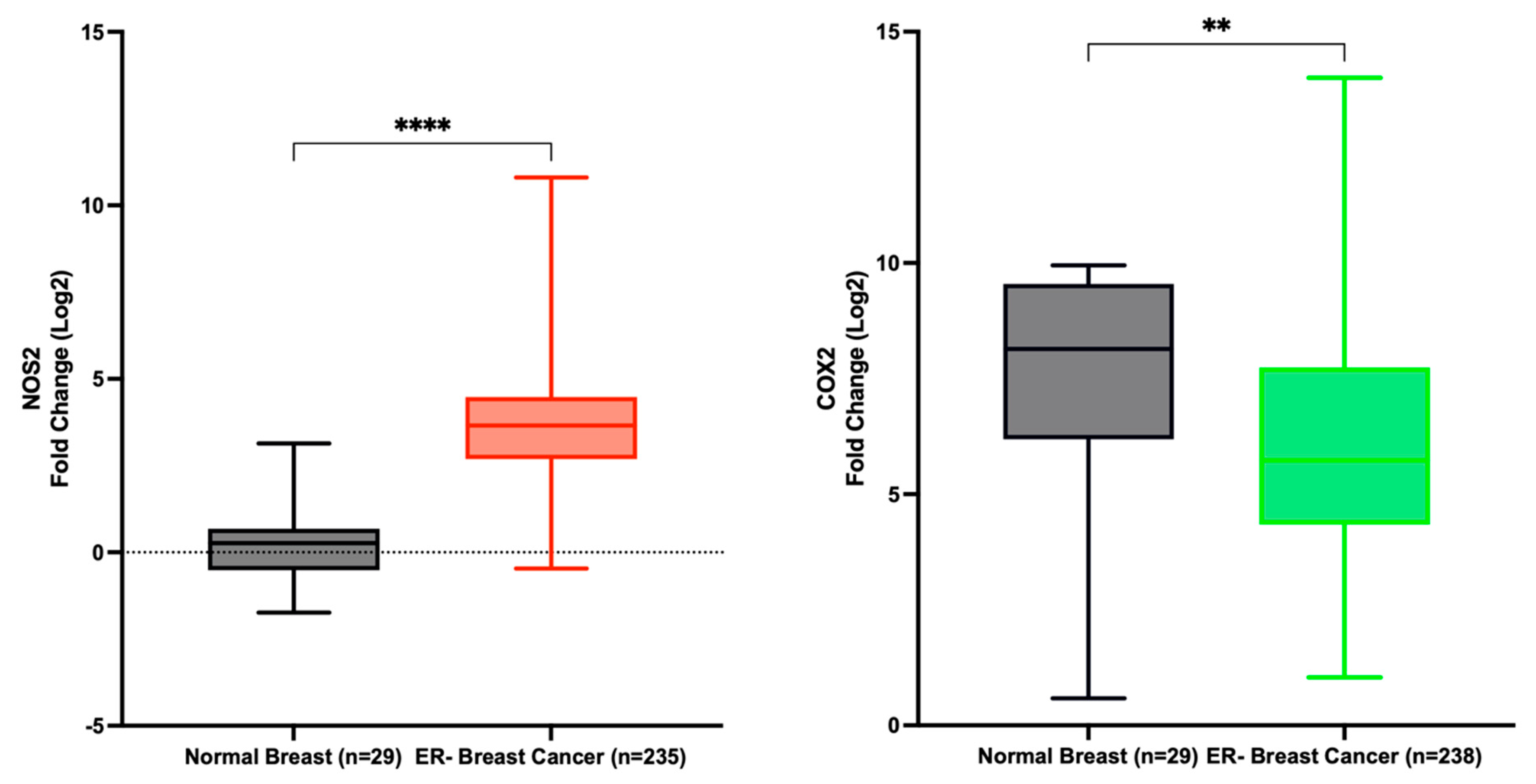
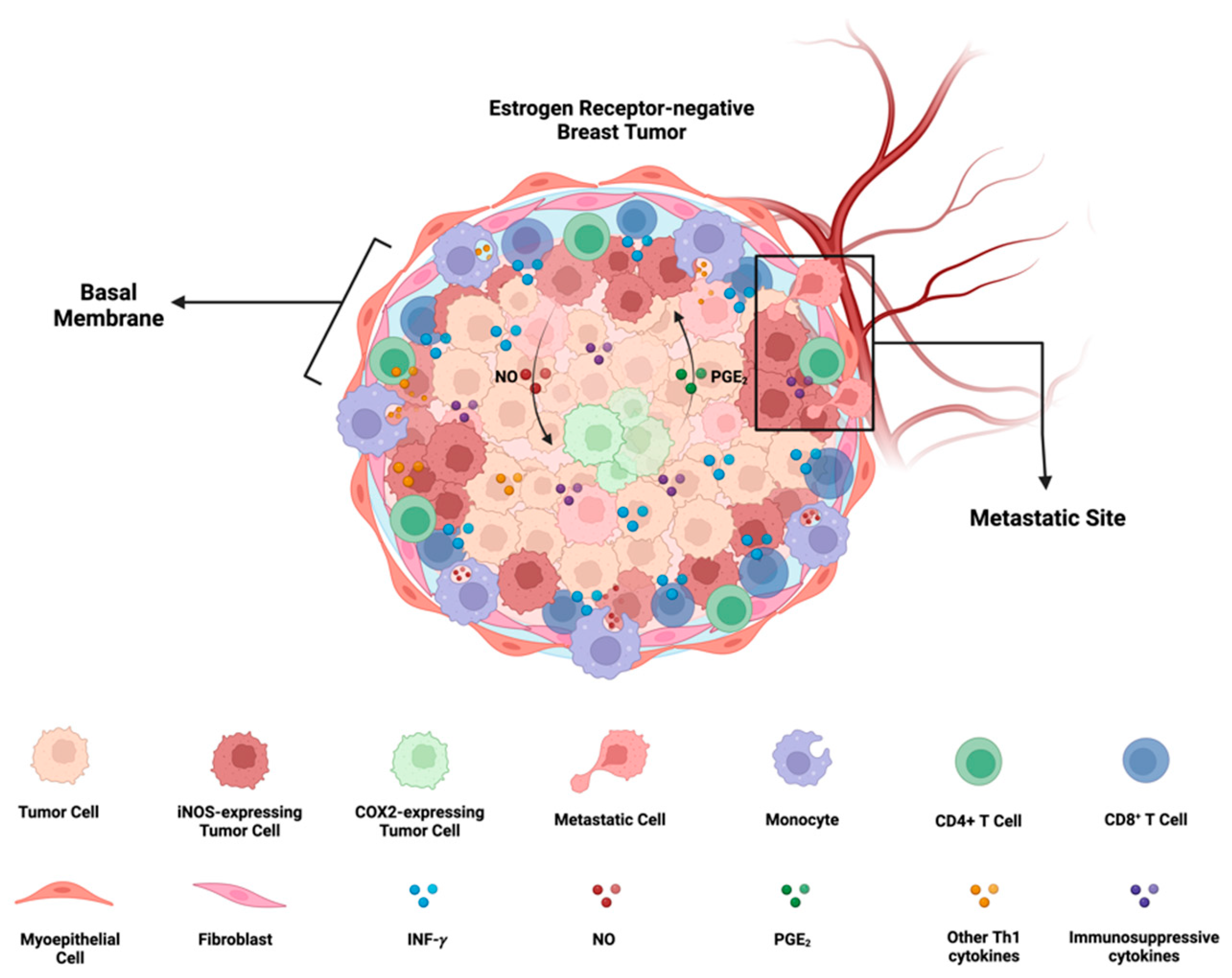
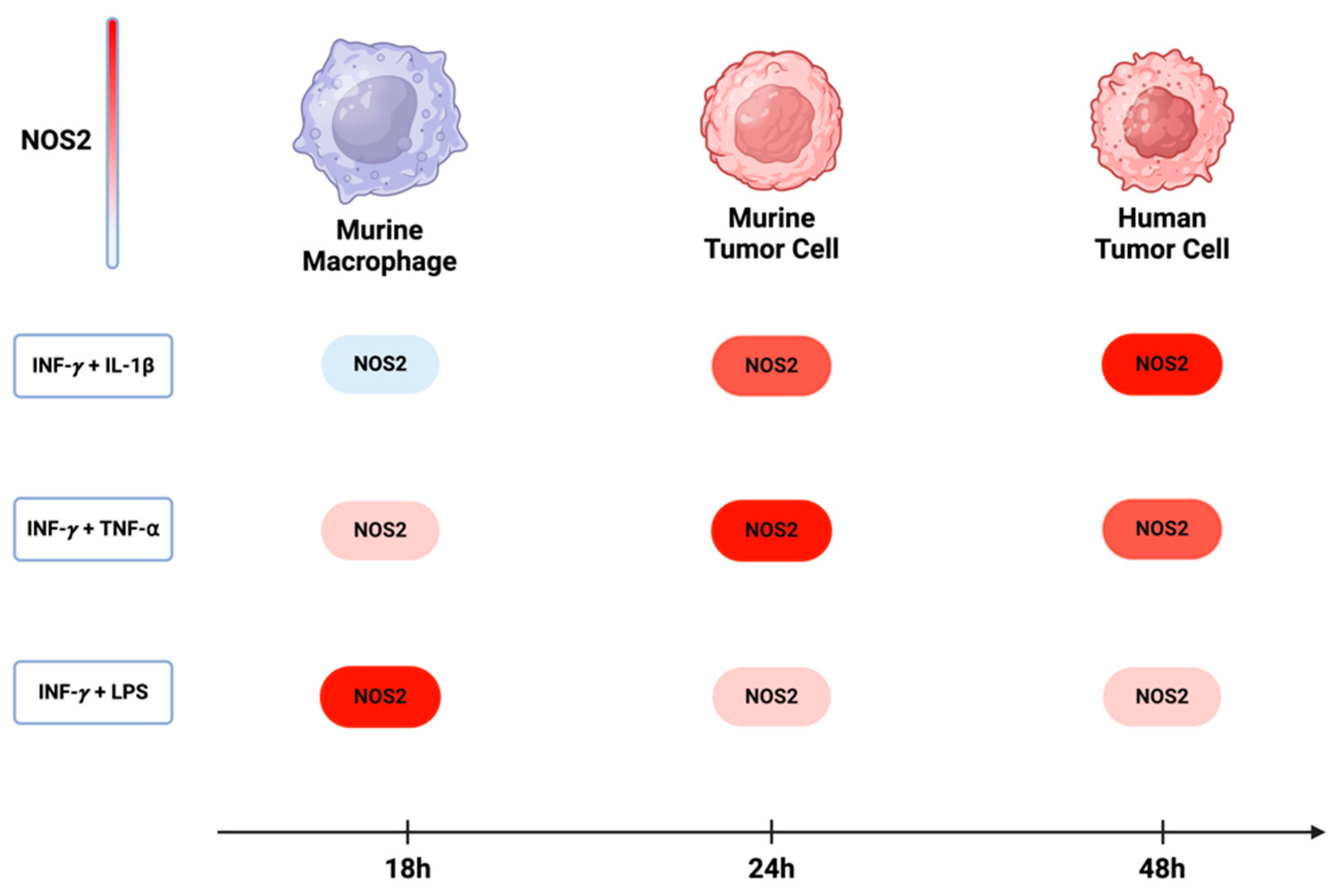
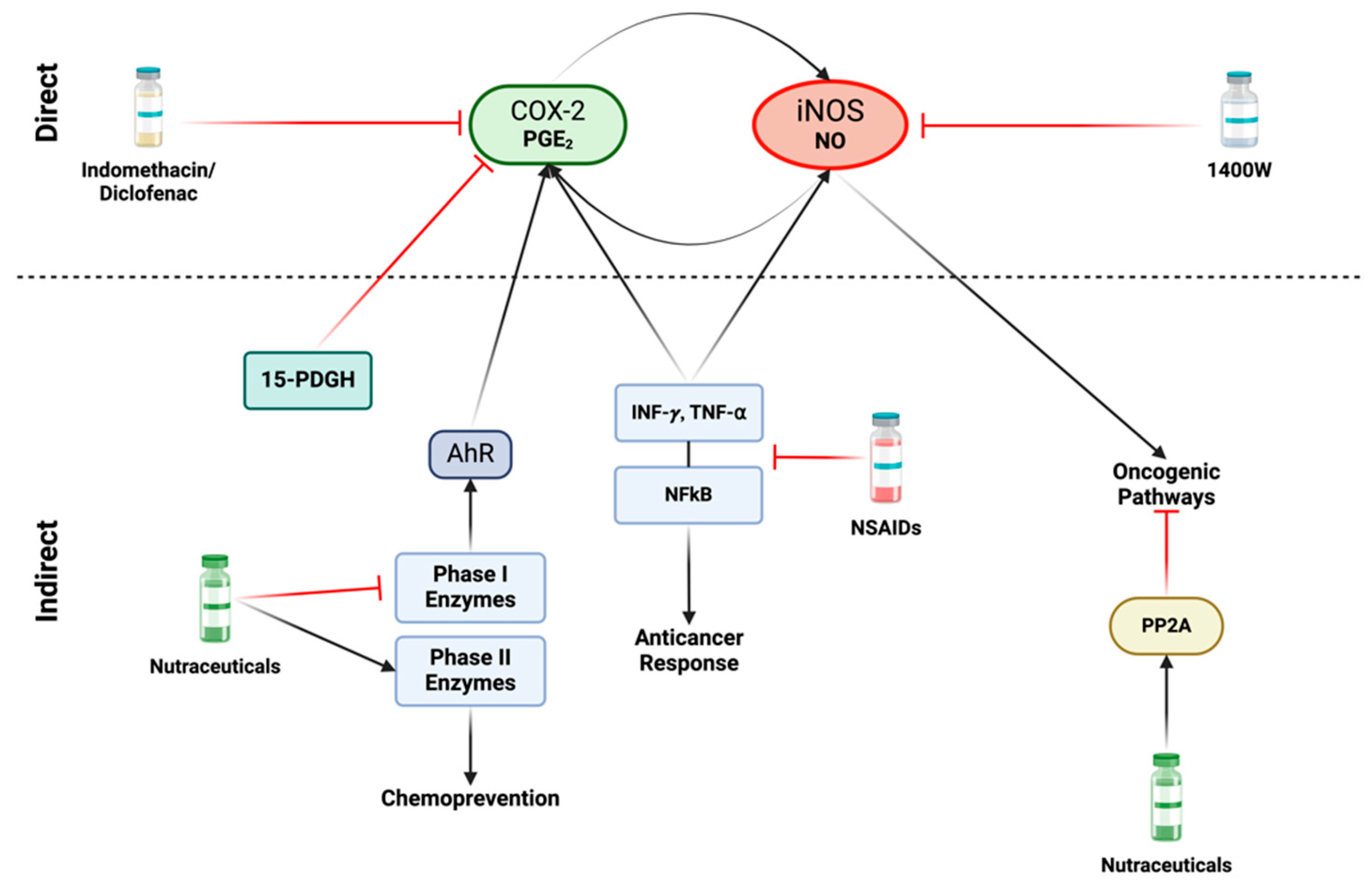
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).