
Genomic Characterization of Preclinical Prostate Cancer Cell Line Models
 and
and
Abstract
:1. Introduction
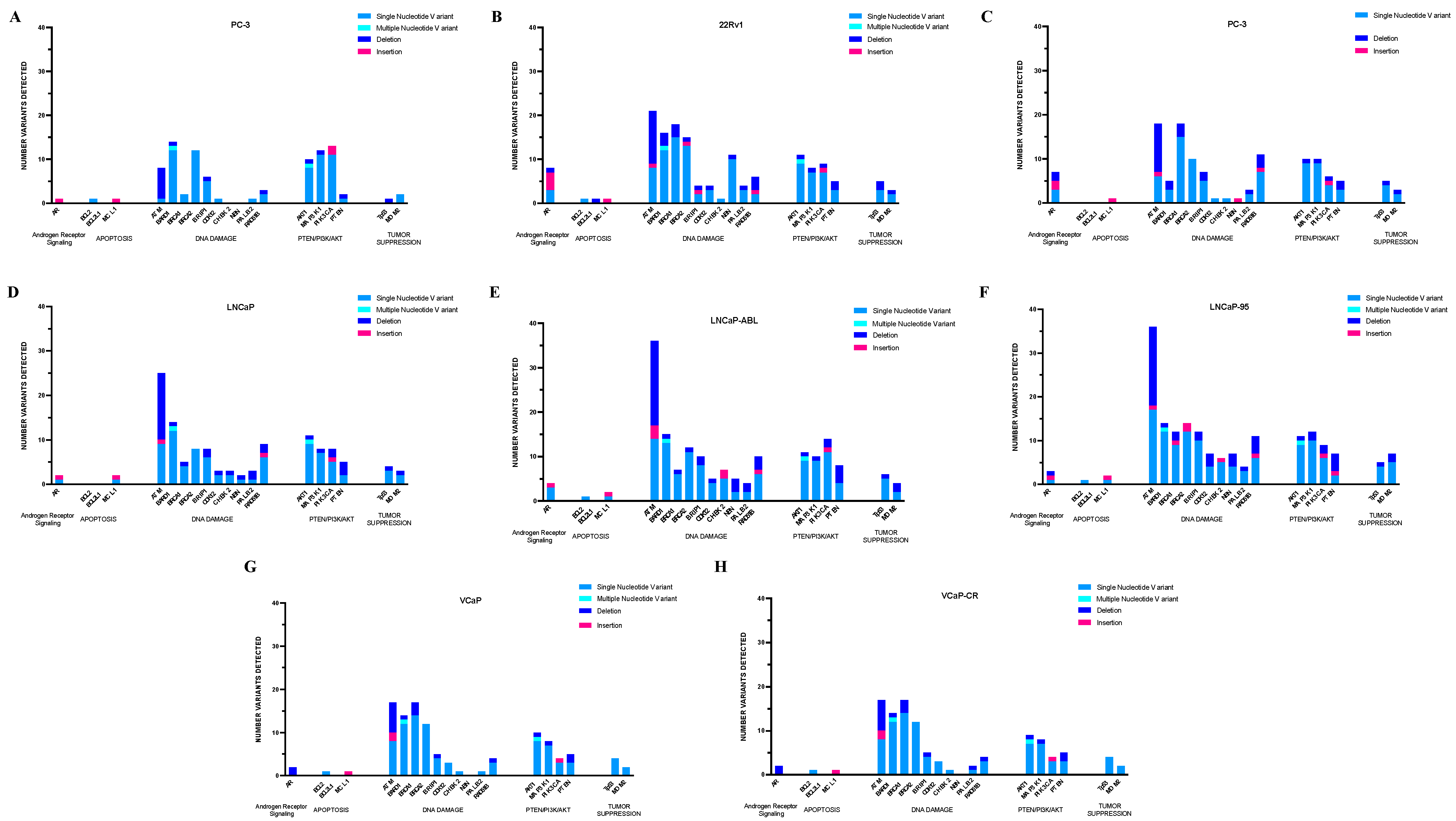
2. Results
2.1. Database Confirmation of Detected Coding Region Alterations
2.1.1. PC-3 Cells
2.1.2. 22RV1 Cells
2.1.3. Du-145 Cells
2.1.4. LNCaP Parental and Derivative Cell Lines
2.1.5. VCaP Parental and Derivative Cell Line
2.2. Clinically Actionable Alterations in Selected Pathways
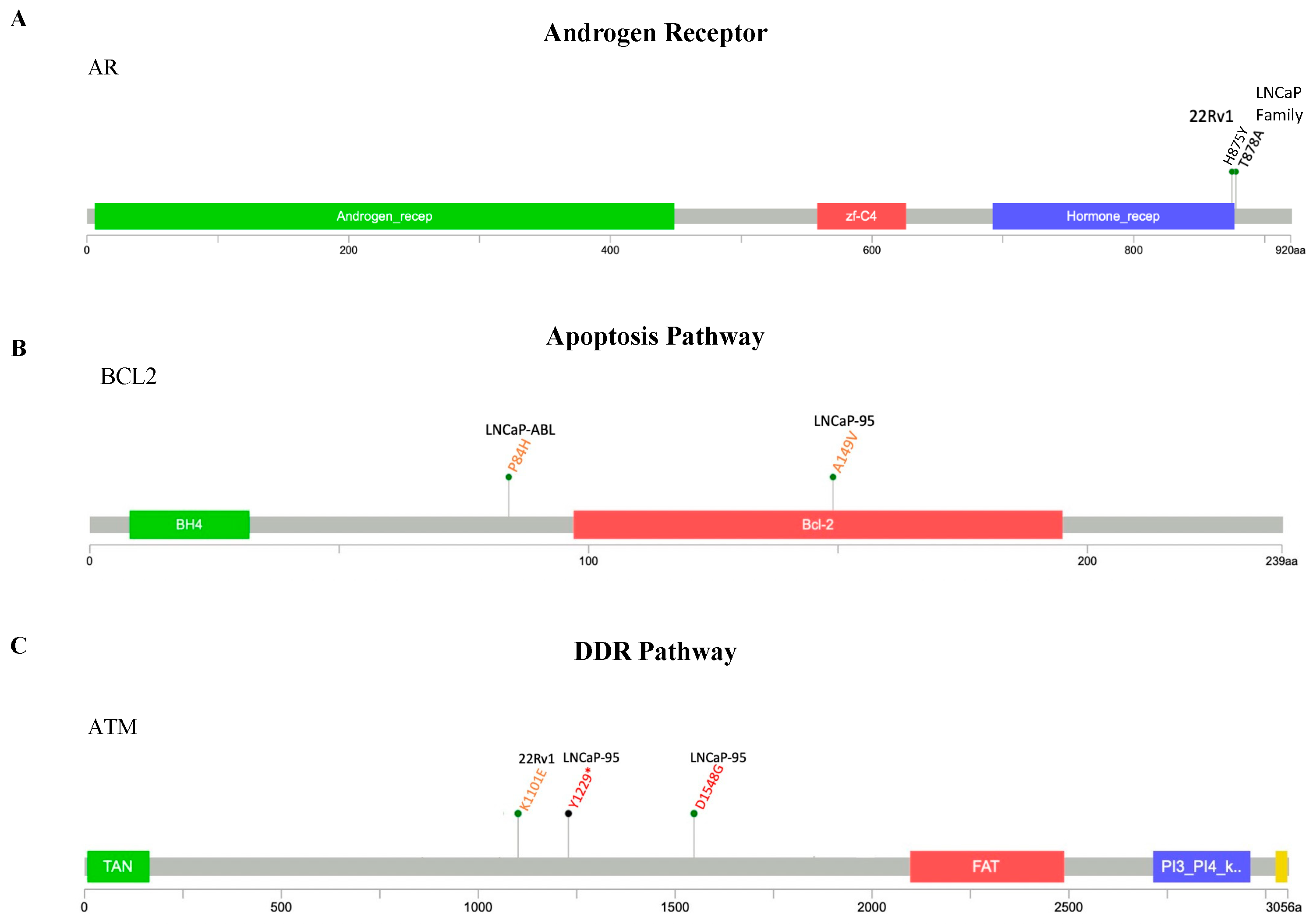
2.2.1. AR Pathway
2.2.2. Apoptosis Pathway
2.2.3. DDR Pathway
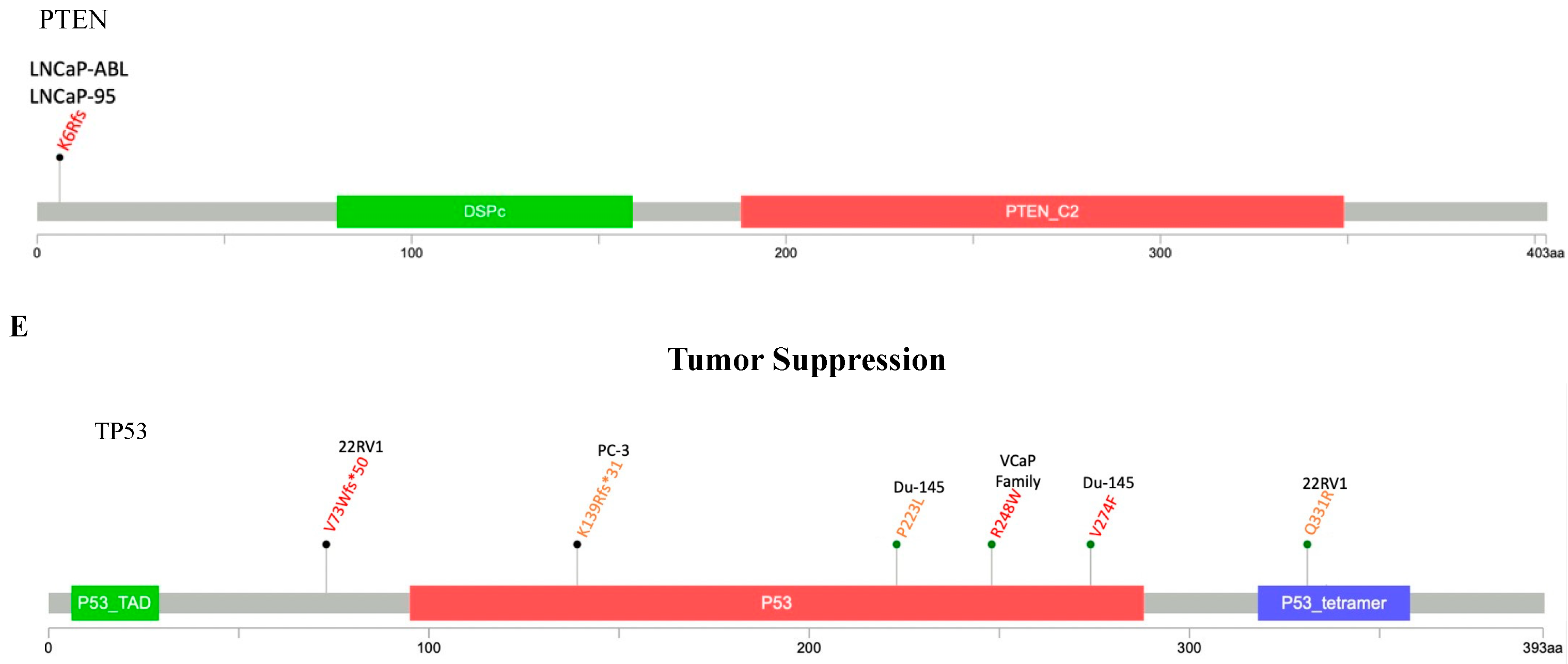
2.2.4. PTEN/PI3K/AKT Pathway
2.2.5. Tumor Suppression Pathway
2.3. Therapeutic Targeting of Cell Line-Specific Markers
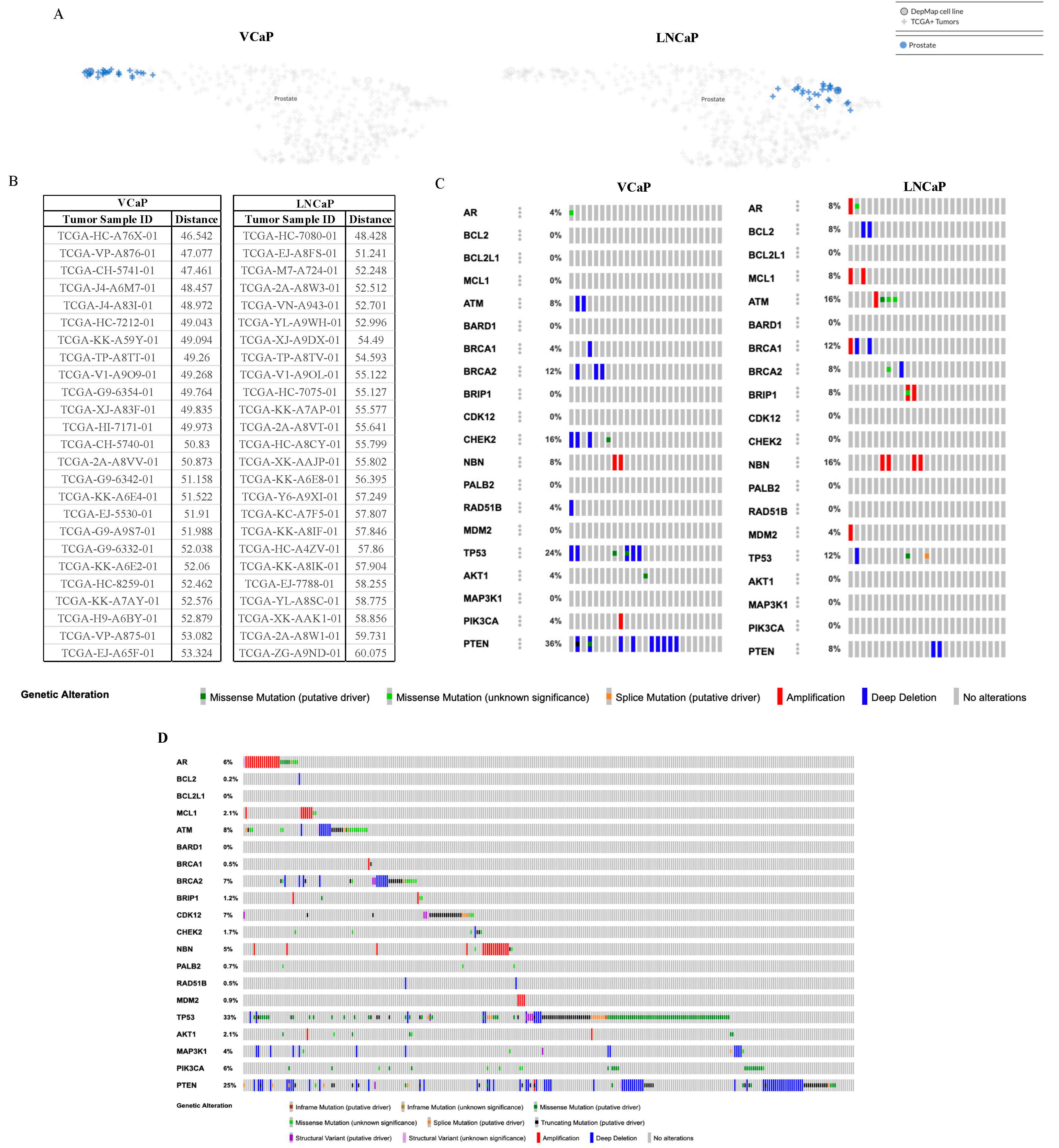
2.4. The Cancer Dependency Map Celligner Tool
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. DNA Extractions
4.3. Custom Illumina AmpliSeq Panel Construction
4.4. Library Preparation and Targeted Next-Generation Sequencing
4.5. Variant Identification and Analysis
4.6. Compounds
4.7. CellTiter-Glo Luminescent Cell Viability Assay
4.8. Population Data
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schmidt, K.T.; Huitema, A.D.R.; Chau, C.H.; Figg, W.D. Resistance to second-generation androgen receptor antagonists in prostate cancer. Nat. Rev. Urol. 2021, 18, 209–226. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Ben-David, R.; Mehrazin, R.; Yang, W.; Tewari, A.K.; Kyprianou, N. Novel signatures of prostate cancer progression and therapeutic resistance. Expert. Opin. Ther. Targets 2023, 27, 1195–1206. [Google Scholar] [CrossRef] [PubMed]
- Nakazawa, M.; Paller, C.; Kyprianou, N. Mechanisms of Therapeutic Resistance in Prostate Cancer. Curr. Oncol. Rep. 2017, 19, 13. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekar, T.; Yang, J.C.; Gao, A.C.; Evans, C.P. Targeting molecular resistance in castration-resistant prostate cancer. BMC Med. 2015, 13, 206. [Google Scholar] [CrossRef] [PubMed]
- Scher, H.I.; Halabi, S.; Tannock, I.; Morris, M.; Sternberg, C.N.; Carducci, M.A.; Eisenberger, M.A.; Higano, C.; Bubley, G.J.; Dreicer, R.; et al. Design and end points of clinical trials for patients with progressive prostate cancer and castrate levels of testosterone: Recommendations of the Prostate Cancer Clinical Trials Working Group. J. Clin. Oncol. 2008, 26, 1148–1159. [Google Scholar] [CrossRef] [PubMed]
- Sobel, R.E.; Sadar, M.D. Cell lines used in prostate cancer research: A compendium of old and new lines—Part 1. J. Urol. 2005, 173, 342–359. [Google Scholar] [CrossRef] [PubMed]
- van Bokhoven, A.; Varella-Garcia, M.; Korch, C.; Johannes, W.U.; Smith, E.E.; Miller, H.L.; Nordeen, S.K.; Miller, G.J.; Lucia, M.S. Molecular characterization of human prostate carcinoma cell lines. Prostate 2003, 57, 205–225. [Google Scholar] [CrossRef] [PubMed]
- Robinson, D.; Van Allen, E.M.; Wu, Y.M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.M.; Montgomery, B.; Taplin, M.E.; Pritchard, C.C.; Attard, G.; et al. Integrative clinical genomics of advanced prostate cancer. Cell 2015, 161, 1215–1228. [Google Scholar] [CrossRef] [PubMed]
- Pham, M.T.; Gupta, A.; Gupta, H.; Vaghasia, A.; Skaist, A.; Garrison, M.A.; Coulter, J.B.; Haffner, M.C.; Zheng, S.L.; Xu, J.; et al. Identifying Phased Mutations and Complex Rearrangements in Human Prostate Cancer Cell Lines through Linked-Read Whole-Genome Sequencing. Mol. Cancer Res. 2022, 20, 1013–1020. [Google Scholar] [CrossRef]
- Franko, A.; Berti, L.; Guirguis, A.; Hennenlotter, J.; Wagner, R.; Scharpf, M.O.; de Angelis, M.H.; Wissmiller, K.; Lickert, H.; Stenzl, A.; et al. Characterization of Hormone-Dependent Pathways in Six Human Prostate-Cancer Cell Lines: A Gene-Expression Study. Genes 2020, 11, 1174. [Google Scholar] [CrossRef]
- Jividen, K.; Kedzierska, K.Z.; Yang, C.S.; Szlachta, K.; Ratan, A.; Paschal, B.M. Genomic analysis of DNA repair genes and androgen signaling in prostate cancer. BMC Cancer 2018, 18, 960. [Google Scholar] [CrossRef] [PubMed]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res. 2018, 47, D941–D947. [Google Scholar] [CrossRef] [PubMed]
- Baum, C.E.; Ockers, S.B.; English, B.C.; Price, D.K.; Sartor, O.; Figg, W.D. Androgen receptor sequence and variations in several common prostate cancer cell lines. Cancer Biol. Ther. 2010, 9, 383–388. [Google Scholar] [CrossRef] [PubMed]
- Ng, P.C.; Henikoff, S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003, 31, 3812–3814. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting functional effect of human missense mutations using PolyPhen-2. Curr. Protoc. Hum. Genet. 2013, 76, 7–20. [Google Scholar] [CrossRef] [PubMed]
- Reva, B.; Antipin, Y.; Sander, C. Determinants of protein function revealed by combinatorial entropy optimization. Genome Biol. 2007, 8, R232. [Google Scholar] [CrossRef] [PubMed]
- Reva, B.; Antipin, Y.; Sander, C. Predicting the functional impact of protein mutations: Application to cancer genomics. Nucleic Acids Res. 2011, 39, e118. [Google Scholar] [CrossRef]
- Shihab, H.A.; Rogers, M.F.; Gough, J.; Mort, M.; Cooper, D.N.; Day, I.N.; Gaunt, T.R.; Campbell, C. An integrative approach to predicting the functional effects of non-coding and coding sequence variation. Bioinformatics 2015, 31, 1536–1543. [Google Scholar] [CrossRef]
- Shihab, H.A.; Gough, J.; Cooper, D.N.; Stenson, P.D.; Barker, G.L.; Edwards, K.J.; Day, I.N.; Gaunt, T.R. Predicting the functional, molecular, and phenotypic consequences of amino acid substitutions using hidden Markov models. Hum. Mutat. 2013, 34, 57–65. [Google Scholar] [CrossRef]
- Nelson, P.S. Molecular states underlying androgen receptor activation: A framework for therapeutics targeting androgen signaling in prostate cancer. J. Clin. Oncol. 2012, 30, 644–646. [Google Scholar] [CrossRef] [PubMed]
- Wadosky, K.M.; Koochekpour, S. Molecular mechanisms underlying resistance to androgen deprivation therapy in prostate cancer. Oncotarget 2016, 7, 64447–64470. [Google Scholar] [CrossRef] [PubMed]
- Jernberg, E.; Bergh, A.; Wikstrom, P. Clinical relevance of androgen receptor alterations in prostate cancer. Endocr. Connect. 2017, 6, R146–R161. [Google Scholar] [CrossRef] [PubMed]
- Azad, A.A.; Volik, S.V.; Wyatt, A.W.; Haegert, A.; Le Bihan, S.; Bell, R.H.; Anderson, S.A.; McConeghy, B.; Shukin, R.; Bazov, J.; et al. Androgen Receptor Gene Aberrations in Circulating Cell-Free DNA: Biomarkers of Therapeutic Resistance in Castration-Resistant Prostate Cancer. Clin. Cancer Res. 2015, 21, 2315–2324. [Google Scholar] [CrossRef]
- Yoshida, T.; Kinoshita, H.; Segawa, T.; Nakamura, E.; Inoue, T.; Shimizu, Y.; Kamoto, T.; Ogawa, O. Antiandrogen bicalutamide promotes tumor growth in a novel androgen-dependent prostate cancer xenograft model derived from a bicalutamide-treated patient. Cancer Res. 2005, 65, 9611–9616. [Google Scholar] [CrossRef]
- Lallous, N.; Volik, S.V.; Awrey, S.; Leblanc, E.; Tse, R.; Murillo, J.; Singh, K.; Azad, A.A.; Wyatt, A.W.; LeBihan, S.; et al. Functional analysis of androgen receptor mutations that confer anti-androgen resistance identified in circulating cell-free DNA from prostate cancer patients. Genome Biol. 2016, 17, 10. [Google Scholar] [CrossRef]
- Steketee, K.; Timmerman, L.; Ziel-van der Made, A.C.; Doesburg, P.; Brinkmann, A.O.; Trapman, J. Broadened ligand responsiveness of androgen receptor mutants obtained by random amino acid substitution of H874 and mutation hot spot T877 in prostate cancer. Int. J. Cancer 2002, 100, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.; Sharief, Y.; Hamil, K.G.; Gregory, C.W.; Zang, D.Y.; Sar, M.; Gumerlock, P.H.; deVere White, R.W.; Pretlow, T.G.; Harris, S.E.; et al. Dehydroepiandrosterone activates mutant androgen receptors expressed in the androgen-dependent human prostate cancer xenograft CWR22 and LNCaP cells. Mol. Endocrinol. 1997, 11, 450–459. [Google Scholar] [CrossRef] [PubMed]
- Watson, P.A.; Arora, V.K.; Sawyers, C.L. Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat. Rev. Cancer 2015, 15, 701–711. [Google Scholar] [CrossRef]
- Steck, P.A.; Pershouse, M.A.; Jasser, S.A.; Yung, W.K.; Lin, H.; Ligon, A.H.; Langford, L.A.; Baumgard, M.L.; Hattier, T.; Davis, T.; et al. Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nat. Genet. 1997, 15, 356–362. [Google Scholar] [CrossRef]
- Juric, D.; Rodon, J.; Tabernero, J.; Janku, F.; Burris, H.A.; Schellens, J.H.M.; Middleton, M.R.; Berlin, J.; Schuler, M.; Gil-Martin, M.; et al. Phosphatidylinositol 3-Kinase alpha-Selective Inhibition With Alpelisib (BYL719) in PIK3CA-Altered Solid Tumors: Results From the First-in-Human Study. J. Clin. Oncol. 2018, 36, 1291–1299. [Google Scholar] [CrossRef]
- Turner, N.; Jhaveri, K.; Kalinsky, K.; Loibl, S.; Loi, S.; Im, S.-A.; Saura, C.; Schmid, P.; Schutzman, J.L.; Stout, T.J.; et al. Abstract OT-36-01: Phase III study of GDC-0077 or placebo (pbo) with palbociclib (P) + fulvestrant (F) in patients with PIK3CA-mutant/hormone receptor-positive/HER2-negative locally advanced or metastatic breast cancer (HR+/HER2- LA/MBC). Cancer Res. 2021, 81, OT-36-01. [Google Scholar] [CrossRef]
- Warren, A.; Chen, Y.; Jones, A.; Shibue, T.; Hahn, W.C.; Boehm, J.S.; Vazquez, F.; Tsherniak, A.; McFarland, J.M. Global computational alignment of tumor and cell line transcriptional profiles. Nat. Commun. 2021, 12, 22. [Google Scholar] [CrossRef] [PubMed]
- Stopsack, K.H.; Nandakumar, S.; Wibmer, A.G.; Haywood, S.; Weg, E.S.; Barnett, E.S.; Kim, C.J.; Carbone, E.A.; Vasselman, S.E.; Nguyen, B.; et al. Oncogenic Genomic Alterations, Clinical Phenotypes, and Outcomes in Metastatic Castration-Sensitive Prostate Cancer. Clin. Cancer Res. 2020, 26, 3230–3238. [Google Scholar] [CrossRef]
- Chatterjee, N.; Walker, G.C. Mechanisms of DNA damage, repair, and mutagenesis. Environ. Mol. Mutagen. 2017, 58, 235–263. [Google Scholar] [CrossRef] [PubMed]
- Alhmoud, J.F.; Woolley, J.F.; Al Moustafa, A.E.; Malki, M.I. DNA Damage/Repair Management in Cancers. Cancers 2020, 12, 1050. [Google Scholar] [CrossRef]
- Fong, P.C.; Boss, D.S.; Yap, T.A.; Tutt, A.; Wu, P.; Mergui-Roelvink, M.; Mortimer, P.; Swaisland, H.; Lau, A.; O’Connor, M.J.; et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N. Engl. J. Med. 2009, 361, 123–134. [Google Scholar] [CrossRef]
- Risdon, E.N.; Chau, C.H.; Price, D.K.; Sartor, O.; Figg, W.D. PARP Inhibitors and Prostate Cancer: To Infinity and Beyond BRCA. Oncologist 2021, 26, e115–e129. [Google Scholar] [CrossRef] [PubMed]
- Beatson, E.L.; Chau, C.H.; Price, D.K.; Figg, W.D. PARP inhibitors on the move in prostate cancer: Spotlight on Niraparib & update on PARP inhibitor combination trials. Am. J. Clin. Exp. Urol. 2022, 10, 252–257. [Google Scholar]
- Kwong, A.; Ng, E.K.; Wong, C.L.; Law, F.B.; Au, T.; Wong, H.N.; Kurian, A.W.; West, D.W.; Ford, J.M.; Ma, E.S. Identification of BRCA1/2 founder mutations in Southern Chinese breast cancer patients using gene sequencing and high resolution DNA melting analysis. PLoS ONE 2012, 7, e43994. [Google Scholar] [CrossRef]
- Serova-Sinilnikova, O.M.; Boutrand, L.; Stoppa-Lyonnet, D.; Bressac-de-Paillerets, B.; Dubois, V.; Lasset, C.; Janin, N.; Bignon, Y.J.; Longy, M.; Maugard, C.; et al. BRCA2 mutations in hereditary breast and ovarian cancer in France. Am. J. Hum. Genet. 1997, 60, 1236–1239. [Google Scholar] [PubMed]
- Holter, S.; Borgida, A.; Dodd, A.; Grant, R.; Semotiuk, K.; Hedley, D.; Dhani, N.; Narod, S.; Akbari, M.; Moore, M.; et al. Germline BRCA Mutations in a Large Clinic-Based Cohort of Patients With Pancreatic Adenocarcinoma. J. Clin. Oncol. 2015, 33, 3124–3129. [Google Scholar] [CrossRef] [PubMed]
- Kopic, S.; Eirich, K.; Schuster, B.; Hanenberg, H.; Varon-Mateeva, R.; Rittinger, O.; Schimpl, G.; Schindler, D.; Jones, N. Hepatoblastoma in a 4-year-old girl with Fanconi anaemia. Acta Paediatr. 2011, 100, 780–783. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.C.; Wang, Z.W.; Tsan, J.T.; Spillman, M.A.; Phung, A.; Xu, X.L.; Yang, M.C.; Hwang, L.Y.; Bowcock, A.M.; Baer, R. Identification of a RING protein that can interact in vivo with the BRCA1 gene product. Nat. Genet. 1996, 14, 430–440. [Google Scholar] [CrossRef] [PubMed]
- Huen, M.S.; Sy, S.M.; Chen, J. BRCA1 and its toolbox for the maintenance of genome integrity. Nat. Rev. Mol. Cell Biol. 2010, 11, 138–148. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, J.; Tauchi, H.; Sakamoto, S.; Nakamura, A.; Morishima, K.; Matsuura, S.; Kobayashi, T.; Tamai, K.; Tanimoto, K.; Komatsu, K. NBS1 localizes to gamma-H2AX foci through interaction with the FHA/BRCT domain. Curr. Biol. 2002, 12, 1846–1851. [Google Scholar] [CrossRef]
- Vivanco, I.; Sawyers, C.L. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat. Rev. Cancer 2002, 2, 489–501. [Google Scholar] [CrossRef] [PubMed]
- Taylor, B.S.; Schultz, N.; Hieronymus, H.; Gopalan, A.; Xiao, Y.; Carver, B.S.; Arora, V.K.; Kaushik, P.; Cerami, E.; Reva, B.; et al. Integrative genomic profiling of human prostate cancer. Cancer Cell 2010, 18, 11–22. [Google Scholar] [CrossRef]
- He, Y.; Sun, M.M.; Zhang, G.G.; Yang, J.; Chen, K.S.; Xu, W.W.; Li, B. Targeting PI3K/Akt signal transduction for cancer therapy. Signal Transduct. Target. Ther. 2021, 6, 425. [Google Scholar] [CrossRef]
- Glaviano, A.; Foo, A.S.C.; Lam, H.Y.; Yap, K.C.H.; Jacot, W.; Jones, R.H.; Eng, H.; Nair, M.G.; Makvandi, P.; Geoerger, B.; et al. PI3K/AKT/mTOR signaling transduction pathway and targeted therapies in cancer. Mol. Cancer 2023, 22, 138. [Google Scholar] [CrossRef]
- Jin, N.; Keam, B.; Cho, J.; Lee, M.J.; Kim, H.R.; Torosyan, H.; Jura, N.; Ng, P.K.; Mills, G.B.; Li, H.; et al. Therapeutic implications of activating noncanonical PIK3CA mutations in head and neck squamous cell carcinoma. J. Clin. Investig. 2021, 131, e150335. [Google Scholar] [CrossRef] [PubMed]
- Rönnstrand, L. Signal transduction via the stem cell factor receptor/c-Kit. Cell. Mol. Life Sci. 2004, 61, 2535–2548. [Google Scholar] [CrossRef]
- Nicholson, K.M.; Anderson, N.G. The protein kinase B/Akt signalling pathway in human malignancy. Cell Signal 2002, 14, 381–395. [Google Scholar] [CrossRef]
- Hers, I.; Vincent, E.E.; Tavaré, J.M. Akt signalling in health and disease. Cell Signal 2011, 23, 1515–1527. [Google Scholar] [CrossRef] [PubMed]
- Calleja, V.; Alcor, D.; Laguerre, M.; Park, J.; Vojnovic, B.; Hemmings, B.A.; Downward, J.; Parker, P.J.; Larijani, B. Intramolecular and intermolecular interactions of protein kinase B define its activation in vivo. PLoS Biol. 2007, 5, e95. [Google Scholar] [CrossRef]
- Calleja, V.; Laguerre, M.; Parker, P.J.; Larijani, B. Role of a novel PH-kinase domain interface in PKB/Akt regulation: Structural mechanism for allosteric inhibition. PLoS Biol. 2009, 7, e17. [Google Scholar] [CrossRef]
- Wu, W.I.; Voegtli, W.C.; Sturgis, H.L.; Dizon, F.P.; Vigers, G.P.; Brandhuber, B.J. Crystal structure of human AKT1 with an allosteric inhibitor reveals a new mode of kinase inhibition. PLoS ONE 2010, 5, e12913. [Google Scholar] [CrossRef] [PubMed]
- Parikh, C.; Janakiraman, V.; Wu, W.I.; Foo, C.K.; Kljavin, N.M.; Chaudhuri, S.; Stawiski, E.; Lee, B.; Lin, J.; Li, H.; et al. Disruption of PH-kinase domain interactions leads to oncogenic activation of AKT in human cancers. Proc. Natl. Acad. Sci. USA 2012, 109, 19368–19373. [Google Scholar] [CrossRef]
- Maehama, T.; Dixon, J.E. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J. Biol. Chem. 1998, 273, 13375–13378. [Google Scholar] [CrossRef]
- Suzuki, H.; Freije, D.; Nusskern, D.R.; Okami, K.; Cairns, P.; Sidransky, D.; Isaacs, W.B.; Bova, G.S. Interfocal heterogeneity of PTEN/MMAC1 gene alterations in multiple metastatic prostate cancer tissues. Cancer Res. 1998, 58, 204–209. [Google Scholar]
- Han, B.; Mehra, R.; Lonigro, R.J.; Wang, L.; Suleman, K.; Menon, A.; Palanisamy, N.; Tomlins, S.A.; Chinnaiyan, A.M.; Shah, R.B. Fluorescence in situ hybridization study shows association of PTEN deletion with ERG rearrangement during prostate cancer progression. Mod. Pathol. 2009, 22, 1083–1093. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Laitinen, S.; Khan, S.; Vihinen, M.; Kowalski, J.; Yu, G.; Chen, L.; Ewing, C.M.; Eisenberger, M.A.; Carducci, M.A.; et al. Copy number analysis indicates monoclonal origin of lethal metastatic prostate cancer. Nat. Med. 2009, 15, 559–565. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Lu, H. Signaling to p53: Ribosomal proteins find their way. Cancer Cell 2009, 16, 369–377. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Guo, M.; Wei, H.; Chen, Y. Targeting p53 pathways: Mechanisms, structures, and advances in therapy. Signal Transduct. Target. Ther. 2023, 8, 92. [Google Scholar] [CrossRef]
- Schlomm, T.; Iwers, L.; Kirstein, P.; Jessen, B.; Kollermann, J.; Minner, S.; Passow-Drolet, A.; Mirlacher, M.; Milde-Langosch, K.; Graefen, M.; et al. Clinical significance of p53 alterations in surgically treated prostate cancers. Mod. Pathol. 2008, 21, 1371–1378. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, A.M.; Attardi, L.D. Deconstructing networks of p53-mediated tumor suppression in vivo. Cell Death Differ. 2018, 25, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Sigal, A.; Rotter, V. Oncogenic mutations of the p53 tumor suppressor: The demons of the guardian of the genome. Cancer Res. 2000, 60, 6788–6793. [Google Scholar] [PubMed]
- Song, H.; Hollstein, M.; Xu, Y. p53 gain-of-function cancer mutants induce genetic instability by inactivating ATM. Nat. Cell Biol. 2007, 9, 573–580. [Google Scholar] [CrossRef]
- Landrum, M.J.; Lee, J.M.; Riley, G.R.; Jang, W.; Rubinstein, W.S.; Church, D.M.; Maglott, D.R. ClinVar: Public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2014, 42, D980–D985. [Google Scholar] [CrossRef]
- Chakravarty, D.; Gao, J.; Phillips, S.M.; Kundra, R.; Zhang, H.; Wang, J.; Rudolph, J.E.; Yaeger, R.; Soumerai, T.; Nissan, M.H.; et al. OncoKB: A Precision Oncology Knowledge Base. JCO Precis. Oncol. 2017, 1, 1–16. [Google Scholar] [CrossRef]
- Suehnholz, S.P.; Nissan, M.H.; Zhang, H.; Kundra, R.; Nandakumar, S.; Lu, C.; Carrero, S.; Dhaneshwar, A.; Fernandez, N.; Xu, B.W.; et al. Quantifying the Expanding Landscape of Clinical Actionability for Patients with Cancer. Cancer Discov. 2024, 14, 49–65. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed]
- de Bruijn, I.; Kundra, R.; Mastrogiacomo, B.; Tran, T.N.; Sikina, L.; Mazor, T.; Li, X.; Ochoa, A.; Zhao, G.; Lai, B.; et al. Analysis and Visualization of Longitudinal Genomic and Clinical Data from the AACR Project GENIE Biopharma Collaborative in cBioPortal. Cancer Res. 2023, 83, 3861–3867. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Figg Lab Panel | BRCAnalysis CDX | FoundationONE CDX | ProstateNext | Invitae PCa Panel | Color Hereditary Prostate | Fulgent | ||
|---|---|---|---|---|---|---|---|---|
| Androgen Receptor Signaling | AR | x | ||||||
| Apoptosis | BCL2 | x | ||||||
| BCL2L1 | x | |||||||
| MCL1 | x | |||||||
| DNA Damage Repair | ATM | x | x | x | x | x | x | |
| BARD1 | x | x | x | |||||
| BRCA1 | x | x | x | x | x | x | x | |
| BRCA2 | x | x | x | x | x | x | x | |
| BRIP1 | x | x | x | x | ||||
| CDK12 | x | x | ||||||
| CHEK2 | x | x | x | x | x | |||
| NBN | x | x | x | x | ||||
| PALB2 | x | x | x | x | x | |||
| RAD51B | x | x | ||||||
| PTEN/PI3K/AKT pathway | AKT1 | x | x | |||||
| MAP3K1 | x | |||||||
| PIK3CA | x | x | ||||||
| PTEN | x | x | ||||||
| Tumor Suppression | TP53 | x | x | x | x | x | ||
| MDM2 | x | |||||||
| AR | ATM | AKT1 | BARD1 | BCL2 | BCL2L1 | BRCA1 | BRCA2 | BRIP1 | CDK12 | CHEK2 | MAP3K1 | NBN | MCL1 | MDM2 | PALB2 | PIK3CA | PTEN | RAD51B | TP53 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PC-3 | INS-1 | DEL-7 | SNV-8 | SNV-12 | SNV-1 | SNV-2 | SNV-12 | SNV-5 | SNV-1 | SNV-11 | INS-1 | SNV-2 | SNV-1 | SNV-11 | DEL-1 | SNV-2 | DEL-1 | |||
| SNV-1 | MNV-1 | MNV-1 | DEL-1 | DEL-1 | INS-2 | SNV-1 | DEL-1 | |||||||||||||
| DEL-1 | DEL-1 | |||||||||||||||||||
| 22RV1 | INS-4 | DEL-12 | SNV-9 | SNV-12 | SNV-1 | DEL-1 | SNV-15 | SNV-13 | SNV-2 | SNV-3 | SNV-1 | SNV-7 | SNV-10 | INS-1 | SNV-2 | SNV-3 | SNV-7 | SNV-3 | SNV-2 | SNV-3 |
| SNV-3 | SNV-8 | MNV-1 | MNV-1 | DEL-3 | DEL-1 | DEL-1 | DEL-1 | DEL-1 | DEL-1 | DEL-1 | DEL-1 | DEL-1 | DEL-2 | DEL-3 | DEL-2 | |||||
| DEL-1 | INS-1 | DEL-1 | DEL-3 | INS-1 | INS-1 | INS-1 | INS-1 | |||||||||||||
| Du-145 | SNV-3 | DEL-11 | SNV-9 | SNV-3 | SNV-15 | SNV-10 | SNV-5 | SNV-1 | SNV-1 | SNV-9 | INS-1 | INS-1 | SNV-2 | SNV-2 | SNV-4 | SNV-3 | SNV-7 | SNV-4 | ||
| DEL-2 | SNV-6 | DEL-1 | DEL-2 | DEL-3 | DEL-2 | DEL-1 | DEL-1 | DEL-1 | DEL-1 | DEL-2 | DEL-3 | DEL-1 | ||||||||
| INS-2 | INS-1 | INS-1 | INS-1 | |||||||||||||||||
| LNCaP | SNV-1 | DEL-15 | SNV-9 | SNV-12 | SNV-4 | SNV-8 | SNV-6 | SNV-2 | SNV-2 | SNV-7 | SNV-1 | INS- 1 | SNV-2 | SNV-1 | SNV-5 | SNV-2 | SNV-6 | SNV-3 | ||
| INS-1 | SNV-9 | MNV-1 | MNV- 1 | DEL-1 | DEL-2 | DEL-1 | INS-1 | DEL-1 | DEL-1 | SNV-1 | DEL-1 | DEL-2 | DEL-2 | DEL-3 | INS-1 | DEL-1 | ||||
| INS-1 | DEL-1 | DEL-1 | INS-1 | DEL-2 | ||||||||||||||||
| LNCaP-ABL | SNV-3 | DEL-19 | SNV-9 | SNV-13 | SNV-1 | SNV-6 | SNV-11 | SNV-8 | SNV-4 | SNV-5 | SNV-9 | DEL-3 | INS-1 | SNV-2 | SNV-2 | SNV-11 | SNV-4 | SNV-6 | SNV-5 | |
| INS-1 | SNV-14 | MNV-1 | MNV-1 | DEL-1 | DEL-1 | DEL-2 | DEL-1 | INS-2 | DEL-1 | SNV-2 | SNV-1 | DEL-2 | DEL-2 | DEL-2 | DEL-4 | INS-1 | DEL-1 | |||
| INS-3 | DEL-1 | DEL-1 | INS-1 | DEL-3 | ||||||||||||||||
| LNCaP-95 | SNV-1 | DEL-18 | SNV-9 | SNV-12 | SNV-1 | SNV-9 | SNV-12 | SNV-10 | SNV-4 | SNV-5 | SNV-10 | SNV-4 | INS-1 | SNV-5 | SNV-3 | SNV-6 | SNV-2 | SNV-6 | SNV-4 | |
| INS-1 | SNV-17 | MNV-1 | MNV-1 | DEL-2 | DEL-2 | DEL-2 | DEL-3 | INS-1 | DEL-2 | DEL-3 | SNV-1 | DEL-2 | DEL-1 | DEL-2 | DEL-4 | DEL-4 | DEL-1 | |||
| DEL-1 | INS-1 | DEL-1 | DEL-1 | INS-1 | INS-1 | INS-1 | INS-1 | |||||||||||||
| VCaP | DEL-2 | DEL-7 | SNV-8 | SNV-12 | SNV-1 | SNV-14 | SNV-12 | SNV-4 | SNV-3 | SNV-1 | SNV-7 | INS-1 | SNV-2 | SNV-1 | SNV-3 | SNV-3 | SNV-3 | SNV-4 | ||
| SNV-8 | MNV-1 | MNV-1 | DEL-3 | DEL-1 | DEL-1 | INS-1 | DEL-2 | DEL-1 | ||||||||||||
| INS-2 | DEL-1 | DEL-1 | ||||||||||||||||||
| VCaP-CR | DEL-2 | DEL-7 | SNV-7 | SNV-12 | SNV-1 | SNV-14 | SNV-12 | SNV-4 | SNV-3 | SNV-1 | SNV-7 | INS-1 | SNV-2 | SNV-1 | SNV-3 | SNV-3 | SNV-3 | SNV-4 | ||
| SNV-8 | MNV-1 | MNV-1 | DEL-3 | DEL-1 | DEL-1 | INS-1 | DEL-2 | DEL-1 | ||||||||||||
| INS-2 | DEL-1 | DEL-1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beatson, E.L.; Risdon, E.N.; Napoli, G.C.; Price, D.K.; Chau, C.H.; Figg, W.D. Genomic Characterization of Preclinical Prostate Cancer Cell Line Models. Int. J. Mol. Sci. 2024, 25, 6111. https://doi.org/10.3390/ijms25116111
Beatson EL, Risdon EN, Napoli GC, Price DK, Chau CH, Figg WD. Genomic Characterization of Preclinical Prostate Cancer Cell Line Models. International Journal of Molecular Sciences. 2024; 25(11):6111. https://doi.org/10.3390/ijms25116111
Chicago/Turabian StyleBeatson, Erica L., Emily N. Risdon, Giulia C. Napoli, Douglas K. Price, Cindy H. Chau, and William D. Figg. 2024. "Genomic Characterization of Preclinical Prostate Cancer Cell Line Models" International Journal of Molecular Sciences 25, no. 11: 6111. https://doi.org/10.3390/ijms25116111