
Shedding Light on Dark Chemical Matter: The Discovery of a SARS-CoV-2 Mpro Main Protease Inhibitor through Intensive Virtual Screening and In Vitro Evaluation

 ,
,  , ,
, ,  , , , , and
, , , , and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
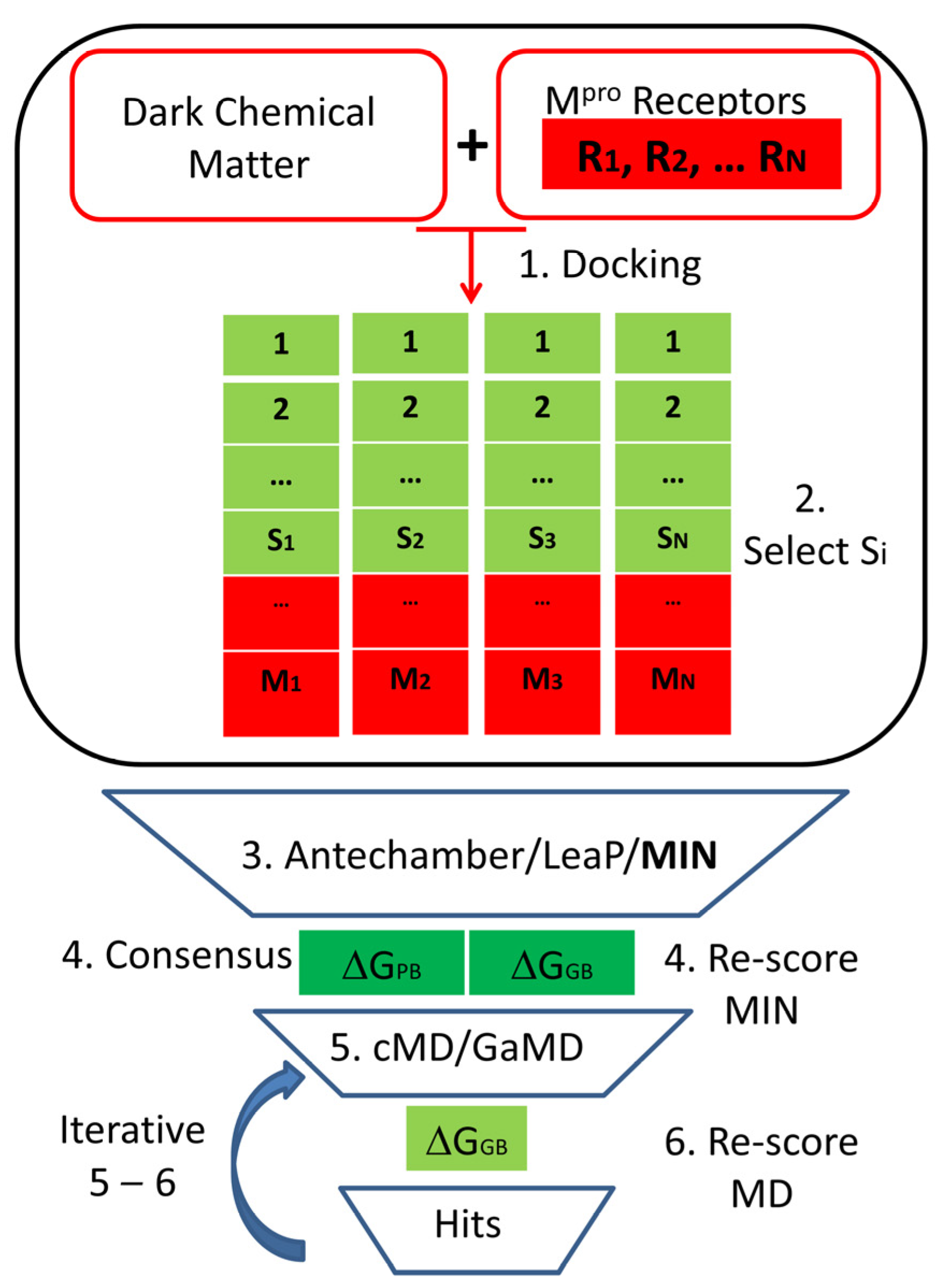
2. Results
2.1. Virtual Screening Targeting the SARS-CoV-2 Mpro Main Protease
2.2. In Vitro Activity Assay of Selected Candidate Compounds
2.3. Binding Analysis of the Active Compound
3. Discussion
4. Materials and Methods
4.1. Computational Studies
4.1.1. SARS-CoV-2 Mpro Protease Representative Structures Selection
4.1.2. Virtual Screening
4.1.3. Binding Free Energy Calculations
4.1.4. Conventional Molecular Dynamics
4.1.5. Gaussian Accelerated Molecular Dynamics
4.2. Experimental Procedure
4.2.1. SARS-CoV-2 Mpro Expression
4.2.2. SARS-CoV-2 Mpro Proteolytic Activity Assay
4.2.3. SARS-CoV-2 Mpro Inhibition Assay
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| Abbreviation | Definition | Page * |
| BL21(DE3) Gold E. Coli | BL21 (DE3) Chemically Competent Escherichia Coli Cells Derivative | 11 |
| ChEMBL | European Molecular Biology Laboratory | 2 |
| cMD | Conventional Molecular Dynamics | 4 |
| COVID-19 | Coronavirus Disease 2019 (2019 novel coronavirus) | 1 |
| DCM | Dark Chemical Matter | 1 |
| DMSO | Dimethyl Sulfoxide | 11 |
| FDA | Food and Drug Administration | 2 |
| FRET | Förster Resonance Energy Transfer | 11 |
| GaMD | Gaussian Accelerated Molecular Dynamics | 4 |
| GAFF2 | General Amber Force Field 2 | 8 |
| GB | Generalized Born | 3 |
| IC50 | Half-Maximal Inhibitory Concentration | 12 |
| IPTG | Isopropyl 1-thio-β-D-galactopyranoside | 11 |
| kcat | Catalytic rate constant | 12 |
| Ki | Substrate Concentration-Independent Inhibition Constant | 1 |
| Kiapp | Apparent Substrate Concentration-Independent Inhibition Constant | 12 |
| Km | Michaelis–Menten Constant | 12 |
| LCPO | Linear Combination of Pairwise Overlaps | 10 |
| LB/ampicillin | Luria Broth/Ampicillin Bacterial Culture Medium | 11 |
| LE | Ligand Efficiency | 9 |
| MMPBSA | Molecular Mechanics Poisson–Boltzmann Surface Area | 3 |
| MMGBSA | Molecular mechanics Generalized Born Surface Area | 3 |
| Mpro | Main Protease | 1 |
| NVT ensemble | Canonical Ensemble: constant particle number (N), constant volume (V) and temperature fluctuating around an equilibrium value (T). | 8 |
| OBC | Onufriev–Bashford–Case | 10 |
| OD | Optical Density | 11 |
| OPC | Four-Point Optimal Point Charge Water Model | 8 |
| PB | Poisson–Boltzmann | 3 |
| PDB | Protein Data Bank | 8 |
| PES | Potential Energy Surface | 10 |
| RNA | Ribonucleic Acid | 2 |
| SASA | Solvent-Accessible Surface Area | 10 |
| SARS-CoV-2 | Severe Acute Respiratory Syndrome Coronavirus 2 | 1 |
| SDS-PAGE | Sodium Dodecyl Sulfate–Polyacrylamide Gel Electrophoresis | 11 |
| SILE | Size-Independent Ligand Efficiency | 9 |
| TIP3P | Transferable Intermolecular Potential with 3 Points | 8 |
| VS | Virtual Screening | 8 |
| ZINC | ZINC is not commercial | 2 |
| * Page number corresponds to the first appearance of the abbreviation in the text. | ||
References
- Xu, X.; Chen, P.; Wang, J.; Feng, J.; Zhou, H.; Li, X.; Zhong, W.; Hao, P. Evolution of the Novel Coronavirus from the Ongoing Wuhan Outbreak and Modeling of Its Spike Protein for Risk of Human Transmission. Sci. China Life Sci. 2020, 63, 457–460. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. General’s Opening Remarks at the Media Briefing on COVID-19-18 March 2020; World Health Organization: Geneva, Switzerland, 2020. [Google Scholar]
- WHO. Statement on the Fifteenth Meeting of the IHR. Emergency Committee on the COVID-19 Pandemic. 2005. Available online: https://www.who.int/news/item/05-05-2023-statement-on-the-fifteenth-meeting-of-the-international-health-regulations-(2005)-emergency-committee-regarding-the-coronavirus-disease-(covid-19)-pandemic (accessed on 7 June 2023).
- Rzymski, P.; Pokorska-Śpiewak, M.; Jackowska, T.; Kuchar, E.; Nitsch-Osuch, A.; Pawłowska, M.; Babicki, M.; Jaroszewicz, J.; Szenborn, L.; Wysocki, J.; et al. Key Considerations during the Transition from the Acute Phase of the COVID-19 Pandemic: A Narrative Review. Vaccines 2023, 11, 1502. [Google Scholar] [CrossRef] [PubMed]
- Duan, Y.; Zhou, H.; Liu, X.; Iketani, S.; Lin, M.; Zhang, X.; Bian, Q.; Wang, H.; Sun, H.; Hong, S.J.; et al. Molecular Mechanisms of SARS-CoV-2 Resistance to Nirmatrelvir. Nature 2023, 622, 376–382. [Google Scholar] [CrossRef]
- Hashemian, S.M.R.; Sheida, A.; Taghizadieh, M.; Memar, M.Y.; Hamblin, M.R.; Baghi, H.B.; Nahand, J.S.; Asemi, Z.; Mirzaei, H. Paxlovid (Nirmatrelvir/Ritonavir): A New Approach to COVID-19 Therapy? Biomed. Pharmacother. 2023, 162, 114367. [Google Scholar] [CrossRef]
- Beigel, J.H.; Tomashek, K.M.; Dodd, L.E.; Mehta, A.K.; Zingman, B.S.; Kalil, A.C.; Hohmann, E.; Chu, H.Y.; Luetkemeyer, A.; Kline, S.; et al. Remdesivir for the Treatment of COVID-19-Preliminary Report. N. Engl. J. Med. 2020, 383, 1813–1836. [Google Scholar] [CrossRef] [PubMed]
- Cihlar, T.; Mackman, R.L. Journey of Remdesivir from the Inhibition of Hepatitis C Virus to the Treatment of COVID-19. Antivir. Ther. 2022, 27, 13596535221082773. [Google Scholar] [CrossRef] [PubMed]
- Sheahan, T.P.; Sims, A.C.; Zhou, S.; Graham, R.L.; Pruijssers, A.J.; Agostini, M.L.; Leist, S.R.; Schäfer, A.; Dinnon, K.H., III; Stevens, L.J.; et al. An Orally Bioavailable Broad-Spectrum Antiviral Inhibits SARS-CoV-2 in Human Airway Epithelial Cell Cultures and Multiple Coronaviruses in Mice. Sci. Transl. Med. 2020, 12, Eabb5883. [Google Scholar] [CrossRef]
- Focosi, D. Molnupiravir: From Hope to Epic Fail? Viruses 2022, 14, 2560. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Patel, T.K.; Patel, P.B.; Barvaliya, M.; Saurabh, M.K.; Bhalla, H.L.; Khosla, P.P. Efficacy and Safety of Lopinavir-Ritonavir in COVID-19: A Systematic Review of Randomized Controlled Trials. J. Infect. Public Health 2021, 14, 740–748. [Google Scholar] [CrossRef] [PubMed]
- Kalil, A.C.; Patterson, T.F.; Mehta, A.K.; Tomashek, K.M.; Wolfe, C.R.; Ghazaryan, V.; Marconi, V.C.; Ruiz-Palacios, G.M.; Hsieh, L.; Kline, S.; et al. Baricitinib plus Remdesivir for Hospitalized Adults with COVID-19. N. Engl. J. Med. 2021, 384, 795–807. [Google Scholar] [CrossRef]
- Riva, L.; Yuan, S.; Yin, X.; Martin-Sancho, L.; Matsunaga, N.; Pache, L.; Burgstaller-Muehlbacher, S.; De Jesus, P.D.; Teriete, P.; Hull, M.V.; et al. Discovery of SARS-CoV-2 Antiviral Drugs through Large-Scale Compound Repurposing. Nature 2020, 586, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; De Clercq, E. Therapeutic Options for the 2019 Novel Coronavirus (2019-nCoV). Nat. Rev. Drug Discov. 2020, 19, 149–150. [Google Scholar] [CrossRef] [PubMed]
- Richardson, P.; Griffin, I.; Tucker, C.; Smith, D.; Oechsle, O.; Phelan, A.; Rawling, M.; Savory, E.; Stebbing, J. Baricitinib as Potential Treatment for 2019-nCoV Acute Respiratory Disease. Lancet 2020, 395, 10241. [Google Scholar] [CrossRef]
- Stebbing, J.; Phelan, A.; Griffin, I.; Tucker, C.; Oechsle, O.; Smith, D.; Richardson, P. COVID-19: Combining Antiviral and Anti-Inflammatory Treatments. Lancet Infect. Dis. 2020, 20, 400–402. [Google Scholar] [CrossRef]
- Antonopoulou, I.; Sapountzaki, E.; Rova, U.; Christakopoulos, P. Inhibition of the Main Protease of SARS-CoV-2 (Mpro) by Repurposing/Designing Drug-like Substances and Utilizing Nature’s Toolbox of Bioactive Compounds. Comput. Struct. Biotechnol. J. 2022, 20, 1306–1344. [Google Scholar] [CrossRef] [PubMed]
- Avilés-Alía, A.I.; Zulaica, J.; Perez, J.J.; Rubio-Martínez, J.; Geller, R.; Granadino-Roldán, J.M. The Discovery of Inhibitors of the SARS-CoV-2 S Protein through Computational Drug Repurposing. Comput. Biol. Med. 2024, 171, 108163. [Google Scholar] [CrossRef]
- Kouznetsova, V.L.; Zhang, A.; Miller, M.A.; Tatineni, M.; Greenberg, J.P.; Tsigelny, I.F. Potential SARS-CoV-2 Spike Protein-ACE2 Interface Inhibitors: Repurposing FDAapproved Drugs. J. Expl. Res. Pharm. 2022, 7, 17–29. [Google Scholar] [CrossRef]
- Rubio-Martínez, J.; Jiménez-Alesanco, A.; Ceballos-Laita, L.; Ortega-Alarcón, D.; Vega, S.; Calvo, C.; Benítez, C.; Abian, O.; Velázquez-Campoy, A.; Thomson, T.M.; et al. Discovery of Diverse Natural Products as Inhibitors of SARS-CoV-2 M pro Protease through Virtual Screening. J. Chem. Inf. Model. 2021, 61, 6094–6106. [Google Scholar] [CrossRef] [PubMed]
- Peralta-Moreno, M.N.; Anton-Muñoz, V.; Ortega-Alarcon, D.; Jimenez-Alesanco, A.; Vega, S.; Abian, O.; Velazquez-Campoy, A.; Thomson, T.M.; Granadino-Roldán, J.M.; Machicado, C.; et al. Autochthonous Peruvian Natural Plants as Potential SARS-CoV-2 Mpro Main Protease Inhibitors. Pharmaceuticals 2023, 16, 585. [Google Scholar] [CrossRef]
- Wen, C.-C.; Kuo, Y.-H.; Jan, J.-T.; Liang, P.-H.; Wang, S.-Y.; Liu, H.-G.; Lee, C.-K.; Chang, S.-T.; Kuo, C.-J.; Lee, S.-S.; et al. Specific Plant Terpenoids and Lignoids Possess Potent Antiviral Activities against Severe Acute Respiratory Syndrome Coronavirus. J. Med. Chem. 2007, 50, 4087–4095. [Google Scholar] [CrossRef]
- Jo, S.; Kim, S.; Shin, D.H.; Kim, M.-S. Inhibition of SARS-CoV 3CL Protease by Flavonoids. J. Enzym. Inh. Med. Chem. 2020, 35, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Khamto, N.; Utama, K.; Tateing, S.; Sangthong, P.; Rithchumpon, P.; Cheechana, N.; Saiai, A.; Semakul, N.; Punyodom, W.; Meepowpan, P. Discovery of Natural Bisbenzylisoquinoline Analogs from the Library of Thai Traditional Plants as SARS-CoV-2 3CLPro Inhibitors: In Silico Molecular Docking, Molecular Dynamics, and In Vitro Enzymatic Activity. J. Chem. Inf. Model. 2023, 63, 2104–2121. [Google Scholar] [CrossRef]
- Sadybekov, A.V.; Katritch, V. Computational Approaches Streamlining Drug Discovery. Nature 2023, 616, 673–685. [Google Scholar] [CrossRef] [PubMed]
- Zdrazil, B.; Felix, E.; Hunter, F.; Manners, E.J.; Blackshaw, J.; Corbett, S.; de Veij, M.; Ioannidis, H.; Lopez, D.M.; Mosquera, J.F.; et al. The ChEMBL Database in 2023: A Drug Discovery Platform Spanning Multiple Bioactivity Data Types and Time Periods. Nucleic Acids Res. 2024, 52, D1180–D1192. [Google Scholar] [CrossRef] [PubMed]
- Irwin, J.J.; Shoichet, B.K. ZINC—A Free Database of Commercially Available Compounds for Virtual Screening. J. Chem. Inf. Model. 2005, 45, 177–182. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Tingle, B.I.; Tang, K.G.; Castanon, M.; Gutierrez, J.J.; Khurelbaatar, M.; Dandarchuluun, C.; Moroz, Y.S.; Irwin, J.J. ZINC-22—A Free Multi-Billion-Scale Database of Tangible Compounds for Ligand Discovery. J. Chem. Inf. Model. 2023, 63, 1166–1176. [Google Scholar] [CrossRef] [PubMed]
- Wassermann, A.M.; Lounkine, E.; Hoepfner, D.; Le Goff, G.; King, F.J.; Studer, C.; Peltier, J.M.; Grippo, M.L.; Prindle, V.; Tao, J.; et al. Dark Chemical Matter as a Promising Starting Point for Drug Lead Discovery. Nat. Chem. Biol. 2015, 11, 958–966. [Google Scholar] [CrossRef] [PubMed]
- Bray, N. Shedding Light on Dark Chemical Matter. Nat. Rev. Drug Discov. 2015, 14, 817. [Google Scholar] [CrossRef]
- Santibáñez-Morán, M.G.; López-López, E.; Prieto-Martínez, F.D.; Sánchez-Cruz, N.; Medina-Franco, J.L. Consensus Virtual Screening of Dark Chemical Matter and Food Chemicals Uncover Potential Inhibitors of SARS-CoV-2 Main Protease. RSC Adv. 2020, 10, 25089–25099. [Google Scholar] [CrossRef]
- Ballante, F.; Rudling, A.; Zeifman, A.; Luttens, A.; Vo, D.D.; Irwin, J.J.; Kihlberg, J.; Brea, J.; Loza, M.I.; Carlsson, J. Docking Finds GPCR Ligands in Dark Chemical Matter. J. Med. Chem. 2020, 63, 613–620. [Google Scholar] [CrossRef]
- Alhossary, A.; Handoko, S.D.; Mu, Y.; Kwoh, C.K. Fast, Accurate, and Reliable Molecular Docking with QuickVina 2. Bioinformatics 2015, 31, 2214–2216. [Google Scholar] [CrossRef] [PubMed]
- Nissink, J.W.M. Simple Size-Independent Measure of Ligand Efficiency. J. Chem. Inf. Model. 2009, 49, 1617–1622. [Google Scholar] [CrossRef] [PubMed]
- Kollman, P.A.; Massova, I.; Reyes, C.; Kuhn, B.; Huo, S.; Chong, L.; Lee, M.; Lee, T.; Duan, Y.; Wang, W.; et al. Calculating Structures and Free Energies of Complex Molecules: Combining Molecular Mechanics and Continuum Models. Acc. Chem. Res. 2000, 33, 889–897. [Google Scholar] [CrossRef] [PubMed]
- JiJin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of Mpro from SARS-CoV-2 and Discovery of Its Inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, J.C.; Fadl, S.; Villanueva, A.J.; Rabeh, W.M. Catalytic Dyad Residues His41 and Cys145 Impact the Catalytic Activity and Overall Conformational Fold of the Main SARS-CoV-2 Protease 3-Chymotrypsin-Like Protease. Front. Chem. 2021, 9, 692168. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Fernandes, H.S.; Sousa, S.F.; Cerqueira, N.M.F.S.A. New Insights into the Catalytic Mechanism of the SARS-CoV-2 Main Protease: An ONIOM QM/MM Approach. Mol. Divers 2022, 26, 1373–1381. [Google Scholar] [CrossRef] [PubMed]
- Kovalevsky, A.; Aniana, A.; Coates, L.; Bonnesen, P.V.; Nashed, N.T.; Louis, J.M. Contribution of the Catalytic Dyad of SARS-CoV-2 Main Protease to Binding Covalent and Noncovalent Inhibitors. J. Biol. Chem. 2023, 299, 104886. [Google Scholar] [CrossRef] [PubMed]
- Stoddard, S.V.; Stoddard, S.D.; Oelkers, B.K.; Fitts, K.; Whalum, K.; Whalum, K.; Hemphill, A.D.; Manikonda, J.; Martinez, L.M.; Riley, E.G.; et al. Optimization Rules for SARS-CoV-2 Mpro Antivirals: Ensemble Docking and Exploration of the Coronavirus Protease Active Site. Viruses 2020, 12, 942. [Google Scholar] [CrossRef]
- Izadi, S.; Anandakrishnan, R.; Onufriev, A.V. Building Water Models: A Different Approach. J. Phys. Chem. Lett. 2014, 5, 3863–3871. [Google Scholar] [CrossRef]
- Case, D.; Ben-Shalom, I.Y.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.E., III; Cruzeiro, V.W.D.; Darden, T.; Duke, R.E.; Ghoreishi, D.; Gohlke, H.; et al. AMBER, v18; University of California: San Francisco, CA, USA, 2018.
- Tian, C.; Kasavajhala, K.; Belfon, K.A.; Raguette, L.; Huang, H.; Migues, A.N.; Bickel, J.; Wang, Y.; Pincay, J.; Wu, Q.; et al. ff19SB: Amino-Acid-Specific Protein Backbone Parameters Trained against Quantum Mechanics Energy Surfaces in Solution. J. Chem. Theory Comput. 2020, 16, 528–552. [Google Scholar] [CrossRef]
- Privat, C.; Granadino-Roldan, J.M.; Bonet, J.; Tomas, M.S.; Perez, J.J.; RubioMartinez, J. Fragment Dissolved Molecular Dynamics: A Systematic and Efficient Method to Locate Binding Sites. Phys. Chem. Chem. Phys. 2021, 23, 3123–3134. [Google Scholar] [CrossRef]
- Perez, J.J.; Tomas, M.S.; Rubio-Martinez, J. Assessment of the Sampling Performance of Multiple-Copy Dynamics versus a Unique Trajectory. J. Chem. Inf. Model 2016, 56, 1950–1962. [Google Scholar] [CrossRef]
- Rokach, L.; Maimon, O. (Eds.) Data Mining and Knowledge Discovery Handbook; Clustering Methods; Springer: Boston, MA, USA, 2005; pp. 321–352. [Google Scholar]
- Roe, D.R.; Cheatham, T.E. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An Open Chemical Toolbox. J. Cheminformatics 2011, 3, 33. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Automatic Atom Type and Bond Type Perception in Molecular Mechanical Calculations. J. Mol. Graph. 2006, 25, 247260. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and Testing of a General AMBER Force Field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, B.; Gerber, P.; Shulz-Gasch, T.; Stahl, M. Validation and Use of the MM-PBSA Approach for Drug Discovery. J. Med. Chem. 2005, 48, 4040–4048. [Google Scholar] [CrossRef]
- Gohlke, H.; Case, D.A. Converging Free Energy Estimates: MMPB(GB)SA Studies on the Protein-Protein Complex Ras-Raf. J. Comput. Chem. 2004, 25, 238–250. [Google Scholar] [CrossRef]
- Reynolds, C.H.; Tounge, B.A.; Bembenek, S.D. Ligand Binding Efficiency: Trends, Physical Basis, and Implications. J. Med. Chem. 2008, 51, 2432–2438. [Google Scholar] [CrossRef] [PubMed]
- Kuntz, I.D.; Chen, K.; Sharp, K.A.; Kollman, P.A. The Maximal Affinity of Ligands. Proc. Natl. Acad. Sci. USA 1999, 96, 9997–10002. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hajduk, P.J. Fragment-Based Drug Design: How Big Is Too Big? J. Med. Chem. 2006, 49, 6972–6976. [Google Scholar] [CrossRef] [PubMed]
- Luo, R.; David, L.; Gilson, M.K. Accelerated Poisson-Boltzmann Calculations for Static and Dynamic Systems. J. Comput. Chem. 2002, 23, 1244–1253. [Google Scholar] [CrossRef] [PubMed]
- Tsui, V.; Case, D.A. Theory and Applications of the Generalized Born Solvation Model in Macromolecular Simulations. Biopolymers 2000, 56, 275–291. [Google Scholar] [CrossRef] [PubMed]
- Onufriev, A.; Bashford, D.; Case, D.A. Exploring Protein Native States and Large-Scale Conformational Changes with a Modified Generalized Born Model. Proteins 2004, 55, 383–394. [Google Scholar] [CrossRef] [PubMed]
- Weiser, J.; Shenkin, P.S.; Still, W.C. Approximate Solvent-Accessible Surface Areas from Tetrahedrally Directed Neighbour Densities. Biopolymers 1999, 50, 373–380. [Google Scholar] [CrossRef]
- Miller, B.R.; McGee, T.D.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA.Py: An Efficient Program for End-State Free Energy Calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef]
- Gohlke, H.; Kiel, C.; Case, D.A. Insights into Protein-Protein Binding by Binding Free Energy Calculation and Free Energy Decomposition for the Ras-Raf and Ras-RalGDS Complexes. J. Mol. Biol. 2003, 330, 891–913. [Google Scholar] [CrossRef]
- Case, D.A.; Belfon, K.; Ben-Shalom, I.Y.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.E., III; Cruzeiro, V.W.D.; Darden, T.A.; Duke, R.E.; Giambasu, G.; et al. AMBER, v2020; University of California: San Francisco, CA, USA, 2020.
- Miao, Y.; Feher, V.A.; McCammon, J.A. Gaussian Accelerated Molecular Dynamics: Unconstrained Enhanced Sampling and Free Energy Calculation. J. Chem. Theory Comput. 2015, 11, 3584–3595. [Google Scholar] [CrossRef]
- Abian, O.; Ortega-Alarcon, D.; Jimenez-Alesanco, A.; Ceballos-Laita, L.; Vega, S.; Reyburn, H.T.; Rizzuti, B.; Velazquez-Campoy, A. Structural Stability of SARS-CoV-2 3CLpro and Identification of Quercetin as an Inhibitor by Experimental Screening. Int. J. Biol. Macromol. 2020, 164, 1693–1703. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peralta-Moreno, M.N.; Mena, Y.; Ortega-Alarcon, D.; Jimenez-Alesanco, A.; Vega, S.; Abian, O.; Velazquez-Campoy, A.; Thomson, T.M.; Pinto, M.; Granadino-Roldán, J.M.; et al. Shedding Light on Dark Chemical Matter: The Discovery of a SARS-CoV-2 Mpro Main Protease Inhibitor through Intensive Virtual Screening and In Vitro Evaluation. Int. J. Mol. Sci. 2024, 25, 6119. https://doi.org/10.3390/ijms25116119
Peralta-Moreno MN, Mena Y, Ortega-Alarcon D, Jimenez-Alesanco A, Vega S, Abian O, Velazquez-Campoy A, Thomson TM, Pinto M, Granadino-Roldán JM, et al. Shedding Light on Dark Chemical Matter: The Discovery of a SARS-CoV-2 Mpro Main Protease Inhibitor through Intensive Virtual Screening and In Vitro Evaluation. International Journal of Molecular Sciences. 2024; 25(11):6119. https://doi.org/10.3390/ijms25116119
Chicago/Turabian StylePeralta-Moreno, Maria Nuria, Yago Mena, David Ortega-Alarcon, Ana Jimenez-Alesanco, Sonia Vega, Olga Abian, Adrian Velazquez-Campoy, Timothy M. Thomson, Marta Pinto, José M. Granadino-Roldán, and et al. 2024. "Shedding Light on Dark Chemical Matter: The Discovery of a SARS-CoV-2 Mpro Main Protease Inhibitor through Intensive Virtual Screening and In Vitro Evaluation" International Journal of Molecular Sciences 25, no. 11: 6119. https://doi.org/10.3390/ijms25116119
APA StylePeralta-Moreno, M. N., Mena, Y., Ortega-Alarcon, D., Jimenez-Alesanco, A., Vega, S., Abian, O., Velazquez-Campoy, A., Thomson, T. M., Pinto, M., Granadino-Roldán, J. M., Santos Tomas, M., Perez, J. J., & Rubio-Martinez, J. (2024). Shedding Light on Dark Chemical Matter: The Discovery of a SARS-CoV-2 Mpro Main Protease Inhibitor through Intensive Virtual Screening and In Vitro Evaluation. International Journal of Molecular Sciences, 25(11), 6119. https://doi.org/10.3390/ijms25116119