
Four Unique Genetic Variants in Three Genes Account for 62.7% of Early-Onset Severe Retinal Dystrophy in Chile: Diagnostic and Therapeutic Consequences

 , ,
, ,  and
and
Abstract
1. Introduction
2. Results
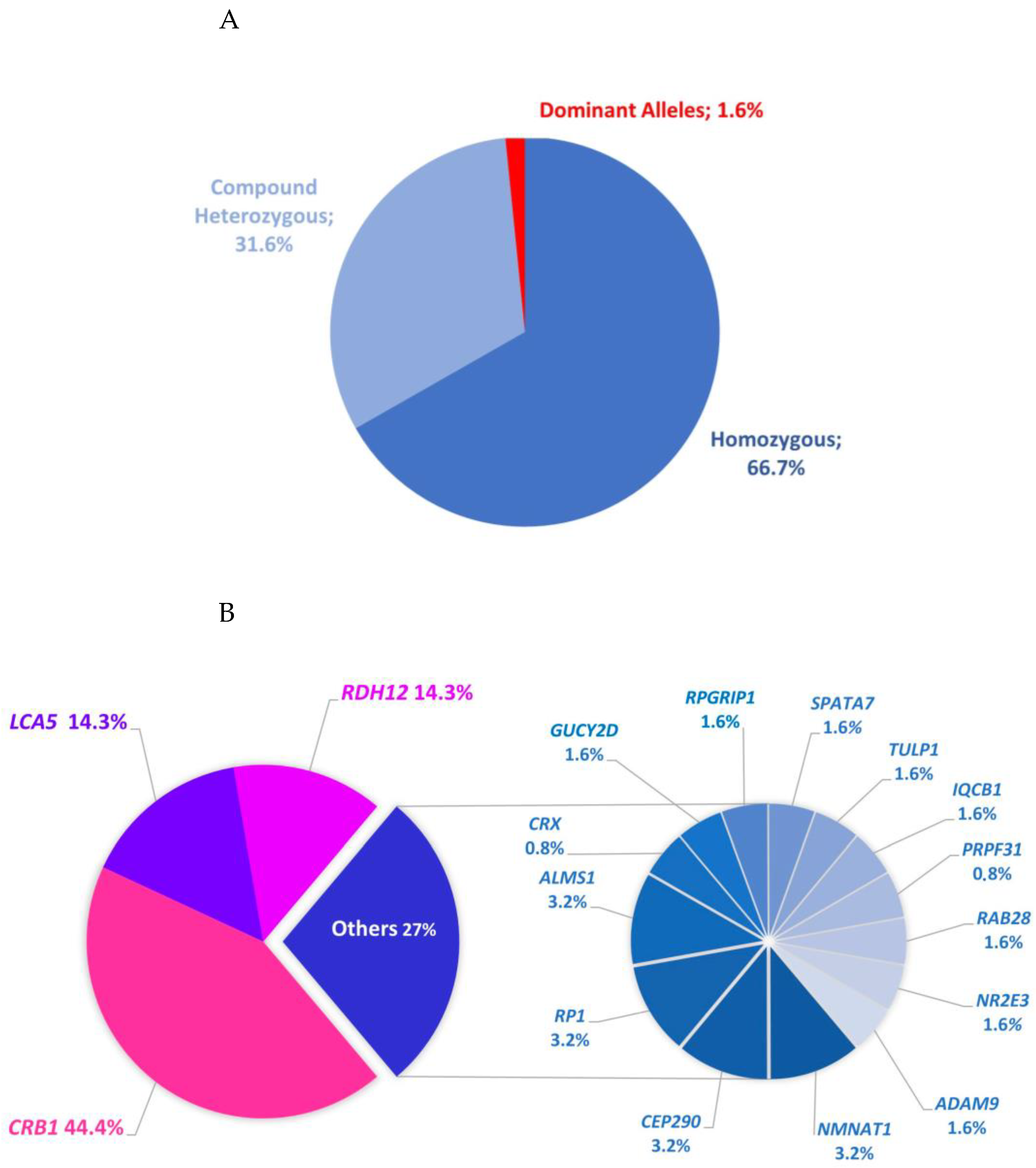
2.1. Data Summary
2.2. Individuals with Mutations in Established Genes Associated with LCA or EOSRD

{kind=link}
| Allele 1 | Allele 2 | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Family | Patient | Gene | MIM# | NM_# | Variant | Parental Origin | Exons | ACMG Category | HGMD Accession (Citation Numbers) | Variant | Parental Origin | Exons | ACMG Category | HGMD |
| 1 | FG313 | ADAM9 | 602713 | 003816.3 | c.333+2_1303del | p | 5–12 | Novel | c.333+2_1303del | (m) | 5–12 | Novel | ||
| 2 | FG297 | ALMS1 | 606844 | 001378454.1 | c.1092del (p.Asp365IlefsTer11) | m | 5 | P | Novel | c.1092del (p.Asp365IlefsTer11) | (p) | 5 | P | Novel |
| FG283 | ALMS1 | 606844 | 001378454.1 | c.1092del (p.Asp365IlefsTer11) | m | 5 | P | Novel | c.1092del (p.Asp365IlefsTer11) | (p) | 5 | P | Novel | |
| 3 | FG277 | CEP290 | 610142 | 025114.4 | c.2991+1655A>G | N.A | Intron 26 | P | CS064383 (31) | c.2991+1655A>G | N.A | Intron 26 | P | CS064383 (31) |
| 4 | FG393 | CEP290 | 610142 | 025114.4 | c.38T>A (p.Val13Asp) | p | 2 | VUS | Novel | c.7341dup (p.Leu2448ThrfsTer8) | (m) | 54 | P | CI062252 (5) |
| 5 | FG50 | CRB1 | 604210 | 201253.3 | c.750T>A (p.Cys250Ter) | m | 3 | P | CM2041497 (1) | c.798_799del (p.Ala267GlnfsTer18) | p | 3 | P | Novel |
| 6 | FG66 | CRB1 | 604210 | 201253.3 | c.2843G>A (p.Cys948Tyr) | m | 9 | P | CM992152 (35) | c.3110_3143dup (p.Ser1049AspfsTer40) | p | 9 | P | CN205417 (1) |
| FG224 | CRB1 | 604210 | 201253.3 | c.2843G>A (p.Cys948Tyr) | m | 9 | P | CM992152 (35) | c.3110_3143dup (p.Ser1049AspfsTer40) | p | 9 | P | CN205417 (1) | |
| 7 | FG112 | CRB1 | 604210 | 201253.3 | c.2843G>A (p.Cys948Tyr) | m | 9 | P | CM992152 (35) | c.3110_3143dup (p.Ser1049AspfsTer40) | p | 9 | P | CN205417 (1) |
| FG113 | CRB1 | 604210 | 201253.3 | c.2843G>A (p.Cys948Tyr) | m | 9 | P | CM992152 (35) | c.3110_3143dup (p.Ser1049AspfsTer40) | p | 9 | P | CN205417 (1) | |
| 8 | FG128 | CRB1 | 604210 | 201253.3 | c.2466G>A (p.Trp822Ter) | NA | 7 | P | Novel | c.2843G>A (p.Cys948Tyr) | N.A | 9 | P | CM992152 (35) |
| 9 | FG239 | CRB1 | 604210 | 201253.3 | c.2843G>A (p.Cys948Tyr) | p | 9 | P | CM992152 (35) | c.2843G>A (p.Cys948Tyr) | (m) | 9 | P | CM992152 (35) |
| 10 | FG362 | CRB1 | 604210 | 201253.3 | c.3110_3143dup (p.Ser1049AspfsTer40) | m | 9 | P | CN205417 (1) | c.3110_3143dup (p.Ser1049AspfsTer40) | p | 9 | P | CN205417 (1) |
| 11 | FG272 | CRB1 | 604210 | 201253.3 | c.3110_3143dup (p.Ser1049AspfsTer40) | NA | 9 | P | CN205417 (1) | c.3110_3143dup (p.Ser1049AspfsTer40) | N.A | 9 | P | CN205417 (1) |
| FG365 | CRB1 | 604210 | 201253.3 | c.3110_3143dup (p.Ser1049AspfsTer40) | NA | 9 | P | CN205417 (1) | c.3110_3143dup (p.Ser1049AspfsTer40) | N.A | 9 | P | CN205417 (1) | |
| FG366 | CRB1 | 604210 | 201253.3 | c.3110_3143dup (p.Ser1049AspfsTer40) | NA | 9 | P | CN205417 (1) | c.3110_3143dup (p.Ser1049AspfsTer40) | N.A | 9 | P | CN205417 (1) | |
| 12 | FG390 | CRB1 | 604210 | 201253.3 | c.3110_3143dup (p.Ser1049AspfsTer40) | m | 9 | P | CN205417 (1) | c.3110_3143dup (p.Ser1049AspfsTer40) | (p) | 9 | P | CN205417 (1) |
| 13 | FG432 | CRB1 | 604210 | 201253.3 | c.2843G>A (p.Cys948Tyr) | m | 9 | P | CM992152 (35) | c.2843G>A (p.Cys948Tyr) | p | 9 | P | CM992152 (35) |
| 14 | FG436 | CRB1 | 604210 | 201253.3 | c.2843G>A (p.Cys948Tyr) | NA | 9 | P | CM992152 (35) | c.2843G>A (p.Cys948Tyr) | N.A | 9 | P | CM992152 (35) |
| 15 | FG444 | CRB1 | 604210 | 201253.3 | c.2843G>A (p.Cys948Tyr) | m | 9 | P | CM992152 (35) | c.2843G>A (p.Cys948Tyr) | p | 9 | P | CM992152 (35) |
| 16 | FG456 | CRB1 | 604210 | 201253.3 | c.2843G>A (p.Cys948Tyr) | NA | 9 | P | CM992152 (35) | c.2843G>A (p.Cys948Tyr) | N.A | 9 | P | CM992152 (35) |
| 17 | FG231 | CRB1 | 604210 | 201253.3 | c.2843G>A (p.Cys948Tyr) | NA | 9 | P | CM992152 (35) | c.2843G>A (p.Cys948Tyr) | N.A | 9 | P | CM992152 (35) |
| 18 | FG395 | CRB1 | 604210 | 201253.3 | c.3110_3143dup (p.Ser1049AspfsTer40) | m | 9 | P | CN205417 (1) | c.750T>A (p.Cys250Ter) | (p) | 3 | LP | CM2041497 (1) |
| 19 | FG399 | CRB1 | 604210 | 201253.3 | c.2843G>A (p.Cys948Tyr) | m | 9 | P | CM992152 (35) | c.2291G>A (p.Arg764His) | p | 7 | P | CM130791 (9) |
| 20 | FG649 | CRB1 | 604210 | 201253.3 | c.653-1G>A | NA | Intron 2 | P | Novel | c.2843G>A (p.Cys948Tyr) | N.A | 9 | P | CM992152 (35) |
| 21 | FG666 | CRB1 | 604210 | 201253.3 | c.2843G>A (p.Cys948Tyr) | NA | 9 | P | CM992152 (35) | c.2843G>A (p.Cys948Tyr) | N.A | 9 | P | CM992152 (35) |
| 22 | FG789 | CRB1 | 604210 | 201253.3 | c.2843G>A (p.Cys948Tyr) | NA | 9 | P | CM992152 (35) | c.2843G>A (p.Cys948Tyr) | N.A | 9 | P | CM992152 (35) |
| 23 | FG850 | CRB1 | 604210 | 201253.3 | c.653-1G>A | m | Intron 2 | P | Novel | c.2843G>A (p.Cys948Tyr) | (p) | 9 | P | CM992152 (35) |
| 24 | FG901 | CRB1 | 604210 | 201253.3 | c.2843G>A (p.Cys948Tyr) | NA | 9 | P | CM992152 (35) | c.3110_3143dup (p.Ser1049AspfsTer40) | N.A | 9 | P | CN205417 (1) |
| 25 | FG942 | CRB1 | 604210 | 201253.3 | c.2843G>A (p.Cys948Tyr) | NA | 9 | P | CM992152 (35) | c.2843G>A (p.Cys948Tyr) | N.A | 9 | P | CM992152 (35) |
| 26 | FG979 | CRB1 | 604210 | 201253.3 | c.2843G>A (p.Cys948Tyr) | NA | 9 | P | CM992152 (35) | c.2843G>A (p.Cys948Tyr) | N.A | 9 | P | CM992152 (35) |
| 27 | FG981 | CRB1 | 604210 | 201253.3 | c.2843G>A (p.Cys948Tyr) | NA | 9 | P | CM992152 (35) | c.3827_3828del (p.Glu1276ValfsTer4) | N.A | 10 | P | Novel |
| 28 | FG1004 | CRB1 | 604210 | 201253.3 | c.2843G>A (p.Cys948Tyr) | NA | 9 | P | CM992152 (35) | c.3110_3143dup (p.Ser1049AspfsTer40) | N.A | 9 | P | CN205417 (1) |
| 29 | FG319 | CRX | 602225 | 000554.6 | c.434del (p.Pro145LeufsTer42) | NA | 4 | LP | CD2033314 (1) | - | ||||
| 30 | FG635 | GUCY2D | 600179 | 000180.4 | c.389del (p.Pro130LeufsTer36) | m | 2 | P | CD962030 (4) | c.1343C>A (p.Ser448Ter) | p | 4 | P | CM002036 (5) |
| 31 | FG337 | IQCB1 | 609237 | 001023570.4 | c.1567+2_*2del | NA | 15 | Novel | c.1567+2_*2del | N.A | 15 | Novel | ||
| 32 | FG236 | LCA5 | 611408 | 181714.4 | c.1243G>T (p.Glu415Ter) | NA | 9 | P | CM205420 (1) | c.1569_1582del (p.His523GlnfsTer16) | N.A | 9 | LP | Novel |
| FG237 | LCA5 | 611408 | 181714.4 | c.1243G>T (p.Glu415Ter) | NA | 9 | P | CM205420 (1) | c.1569_1582del (p.His523GlnfsTer16) | N.A | 9 | LP | Novel | |
| 33 | FG360 | LCA5 | 611408 | 181714.4 | c.1243G>T (p.Glu415Ter) | m | 9 | P | CM205420 (1) | c.1243G>T (p.Glu415Ter) | p | 9 | P | CM205420 (1) |
| 34 | FG496 | LCA5 | 611408 | 181714.4 | c.1243G>T (p.Glu415Ter) | m | 9 | P | CM205420 (1) | c.1243G>T (p.Glu415Ter) | p | 9 | P | CM205420 (1) |
| 35 | FG600 | LCA5 | 611408 | 181714.4 | c.1243G>T (p.Glu415Ter) | NA | 9 | P | CM205420 (1) | c.1243G>T (p.Glu415Ter) | N.A | 9 | P | CM205420 (1) |
| 36 | FG659 | LCA5 | 611408 | 181714.4 | c.1243G>T (p.Glu415Ter) | NA | 9 | P | CM205420 (1) | c.1243G>T (p.Glu415Ter) | N.A | 9 | P | CM205420 (1) |
| 37 | FG856 | LCA5 | 611408 | 181714.4 | c.1243G>T (p.Glu415Ter) | NA | 9 | P | CM205420 (1) | c.1243G>T (p.Glu415Ter) | N.A | 9 | P | CM205420 (1) |
| 38 | FG851 | LCA5 | 611408 | 181714.4 | c.1243G>T (p.Glu415Ter) | NA | 9 | P | CM205420 (1) | c.1243G>T (p.Glu415Ter) | N.A | 9 | P | CM205420 (1) |
| 39 | FG1002 | LCA5 | 611408 | 181714.4 | c.1243G>T (p.Glu415Ter) | NA | 9 | P | CM205420 (1) | c.1243G>T (p.Glu415Ter) | N.A | 9 | P | CM205420 (1) |
| 40 | FG465 | NMNAT1 | 608700 | 001297778.1 | c.769G>A (p.Glu257Lys) | m | 5 | LP | CM127755 (33) | c.364del (p.Arg122GlyfsTer20) | p | 4 | P | CD127792 (6) |
| 41 | FG787 | NMNAT1 | 608700 | 001297778.1 | c.769G>A (p.Glu257Lys) | m | 5 | LP | CM127755 (33) | c.507G>A (p.Trp169Ter) | p | 5 | P | CM127758 (9) |
| 42 | FG165 | PRPF31 | 606419 | 015629.4 | c.1060C>T (p.Arg354Ter) | NA | 10 | P | CM1310332 (13) | |||||
| 43 | FG454 | RAB28 | 612994 | 001017979.3 | c.331_333del (p.Val111del) | m | 4 | LP | Novel | c.331_333del (p.Val111del) | N.A | 4 | LP | Novel |
| 44 | FG402 | RDH12 | 608830 | 152443.3 | c.295C>A (p.Leu99Ile) | NA | 5 | P | CM042465 (18) | c.295C>A (p.Leu99Ile) | N.A | 5 | P | CM042465 (18) |
| 45 | FG68 | RDH12 | 608830 | 152443.3 | c.295C>A (p.Leu99Ile) | m | 5 | P | CM042465 (18) | c.295C>A (p.Leu99Ile) | (p) | 5 | P | CM042465 (18) |
| FG69 | RDH12 | 608830 | 152443.3 | c.295C>A (p.Leu99Ile) | m | 5 | P | CM042465 (18) | c.295C>A (p.Leu99Ile) | (p) | 5 | P | CM042465 (18) | |
| 46 | FG383 | RDH12 | 608830 | 152443.3 | c.295C>A (p.Leu99Ile) | NA | 5 | P | CM042465 (18) | c.295C>A (p.Leu99Ile) | N.A | 5 | P | CM042465 (18) |
| 47 | FG429 | RDH12 | 608830 | 152443.3 | c.295C>A (p.Leu99Ile) | NA | 5 | P | CM042465 (18) | c.295C>A (p.Leu99Ile) | N.A | 5 | P | CM042465 (18) |
| 48 | FG612 | RDH12 | 608830 | 152443.3 | c.295C>A (p.Leu99Ile) | NA | 5 | P | CM042465 (18) | c.295C>A (p.Leu99Ile) | N.A | 5 | P | CM042465 (18) |
| 49 | FG667 | RDH12 | 608830 | 152443.3 | c.715dup (p.Arg239ProfsTer34) | NA | 8 | P | CI118737 (2) | c.715dup (p.Arg239ProfsTer34) | N.A | 8 | P | CI118737 (2) |
| 50 | FG694 | RDH12 | 608830 | 152443.3 | c.295C>A (p.Leu99Ile) | m | 5 | P | CM042465 (18) | c.716G>T (p.Arg239Leu) | p | 5 | P | CM205421 (3) |
| 51 | FG780 | RDH12 | 608830 | 152443.3 | c.295C>A (p.Leu99Ile) | NA | 5 | P | CM042465 (18) | c.295C>A (p.Leu99Ile) | N.A | 5 | P | CM042465 (18) |
| 52 | FG247 | RP1 | 603937 | 006269.2 | c.5564del (p.Lys1855ArgfsTer42) | m | 4 | P | Novel | c.5564del (p.Lys1855ArgfsTer42) | p | 4 | P | Novel |
| 53 | FG514 | RP1 | 603937 | 006269.2 | c.5564del (p.Lys1855ArgfsTer42) | m | 4 | P | Novel | c.5564del (p.Lys1855ArgfsTer42) | p | 4 | P | Novel |
| 54 | FG487 | RPGRIP1 | 605446 | 020366.4 | c.1077+1_3100-1dup | NA | 10–19 | Novel | c.1077+1_3100-1dup | N.A | 10–19 | Novel | ||
| 55 | FG853 | SPATA7 | 609868 | 018418.5 | c.1171C>T (p.Arg391Ter) | NA | 11 | P | CM1817912 (1) | c.1171C>T (p.Arg391Ter) | N.A | 11 | P | CM1817912 (1) |
| 56 | FG441 | TULP1 | 602280 | 003322.6 | c.1149C>A (p.Asp383Glu) | m | 12 | LP | Novel | c.1149C>A (p.Asp383Glu) | p | 12 | LP | Novel |
| 57 | FG118 | NR2E3 | 604485 | 014249.4 | c.932G>A (p.Arg311Gln) | m | 6 | P | CM000538 (36) | c.932G>A (p.Arg311Gln) | (p) | 6 | P | CM000538 (36) |
| Disease Symptoms | |||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Family | Patient | Gender | YOB | Place of Birth | Consanguinity (F) | Age at Presentation | BCVA RE (Decimal) | BCVA LE (Decimal) | Refraction RE (SE) | Refraction LE (SE) | Nystagmus | Oculodigital Sign | Nyctalopia | Photophobia | ffERG Rods | ffERG Cones | GRABSPDVA | CPA | NRPD | PPRPE | TRMPC | Pseudo-coloboma | Others | Initial Diagnosis | Final Diagnosis |
| 1 | FG313 | M | 1996 | Chillan | 0 | 9 months | lp | lp | N.A | N.A | Yes | N.A | Yes | Yes | N.A | N.A | Yes | No | No | No | No | No | LCA | LCA | |
| 2 | FG297 | M | 1975 | Santiago | 0.0625 | birth | npl | npl | N.A | N.A | Yes | N.A | Yes | No | N.A | N.A | Yes | No | No | No | No | No | Hearing loss, type 2 diabetes mellitus, arterial hypertension, and epileptic seizures | LCA | ALMS |
| FG283 | M | 1973 | Santiago | 0.0625 | birth | lp | lp | N.A | N.A | Yes | N.A | Yes | Yes | N.A | N.A | Yes | No | No | No | No | No | Dense cataract (LE), hearing loss, type 2 diabetes mellitus, arterial hypertension, and epileptic seizures | LCA | ALMS | |
| 3 | FG277 | F | 2013 | Santiago | 0.0625 | 18 months | 0.2 | 0.2 | (+3.25) | (+3.75) | No | No | Yes | No | Rod–Cone | Rod–Cone | No | No | No | No | Yes | No | LCA | LCA | |
| 4 | FG393 | F | 2014 | Santiago | 0 | birth | lp | lp | N.A | N.A | Yes | Yes | Yes | No | N.A | N.A | No | No | No | No | Yes | No | Mental impairment; brother affected with syndactyly | LCA | LCA |
| 5 | FG50 | F | 1972 | Santiago | 0 | 1 year | lp | lp | (+10.5) | (+10.5) | Yes | N.A | Yes | No | N.A | N.A | No | Yes | Yes | Yes | No | Yes | LCA | LCA | |
| 6 | FG66 | F | 2000 | Santiago | 0 | 2 years | 0.05 | 0.05 | 0 | 0 | Yes | No | N.A | N.A | Rod–Cone | Rod–Cone | No | Yes | No | Yes | Yes | Yes | LCA | LCA | |
| FG224 | M | 2010 | Santiago | 0 | birth | 0.05 | 0.05 | (+12) | (+11.25) | Yes | N.A | Yes | Yes | N.A | N.A | No | Yes | Yes | Yes | No | Yes | LCA | LCA | ||
| 7 | FG112 | M | 1990 | Quipue | 0 | birth | lp | npl | N.A | N.A | Yes | Yes | Yes | Yes | N.A | N.A | No | Yes | Yes | Yes | No | Yes | LCA | LCA | |
| FG113 | M | 2002 | Quipue | 0 | birth | lp | lp | N.A | N.A | Yes | Yes | Yes | Yes | N.A | N.A | No | Yes | Yes | Yes | No | Yes | Keratoconus | LCA | LCA | |
| 8 | FG128 | F | 1982 | Coinco | 0 | 3 years | cf | cf | N.A | N.A | Yes | Yes | No | Yes | N.A | N.A | No | Yes | Yes | Yes | No | Yes | Keratoconus | LCA, EOSRD | LCA |
| 9 | FG239 | F | 1986 | Cañete | 0 | birth | lp | lp | N.A | N.A | Yes | Yes | Yes | Yes | N.A | N.A | No | Yes | Yes | Yes | No | Yes | Keratoconus | LCA | LCA |
| 10 | FG362 | M | 1969 | Angol | 0 | <1 year | lp | lp | (+5.0) | (+3) | Yes | N.A | N.A | N.A | Abolished | Abolished | No | Yes | Yes | Yes | No | Yes | Vitreous opacities | LCA | LCA |
| 11 | FG272 | F | 1997 | Santiago | 0 | 1 year | cf | cf | (+3.75) | (+4.0) | Yes | No | Yes | No | N.A | N.A | Yes | Yes | No | No | No | No | LCA | LCA | |
| FG365 | M | 1983 | Santiago | 0 | childhood | lp | lp | impossible | impossible | Yes | Yes | Yes | No | N.A | N.A | N.A | N.A | N.A | N.A | N.A | N.A | White bilateral cataract | LCA | LCA | |
| FG366 | M | 1993 | Santiago | 0 | childhood | hm | hm | N.A | N.A | N.A | N.A | Yes | Yes | N.A | N.A | Yes | Yes | No | No | No | No | Keratoconus | LCA | LCA | |
| 12 | FG390 | M | 1972 | Papudo | 0 | 3 months | npl | lp | N.A | N.A | Yes | Yes | Yes | Yes | N.A | N.A | No | Yes | Yes | No | No | Yes | Keratoconus | LCA | LCA |
| 13 | FG432 | M | 2013 | Coihueco | 0 | 6 months | 0.2 | 0.1 | (+5.5) | (+6.0) | Yes | N.A | Yes | No | N.A | N.A | No | Yes | Yes | Yes | No | Yes | Type 1 diabetes | LCA | LCA |
| 14 | FG436 | M | 2011 | Gorbea | 0 | 3 months | cf | cf | (+8.5) | (+7.5) | Yes | Yes | Yes | Yes | Rod | Cone | No | Yes | No | Yes | No | Yes | LCA | LCA | |
| 15 | FG444 | M | 2014 | Villarica | N.A | 2 months | 0.025 | 0.025 | (+8.0) | (+8.0) | Yes | N.A | Yes | Yes | N.A | N.A | No | Yes | Yes | No | No | Yes | LCA | LCA | |
| 16 | FG456 | M | 1995 | Santiago | 0.0313 | birth | cf | 0.05 | (+5.25) | (+4.0) | Yes | N.A | Yes | No | N.A | N.A | No | Yes | Yes | No | No | Yes | Optic nerve drusen | LCA | LCA |
| 17 | FG231 | F | 2010 | Chillan | 0 | 7 months | 0.15 | 0.1 | (+7.5) | (+7.5) | Yes | No | Yes | No | N.A | N.A | No | Yes | Yes | Yes | No | Yes | LCA | LCA | |
| 18 | FG395 | M | 1989 | Padre Hurtado | 0 | birth | hm | cf | NA | NA | Yes | Yes | Yes | Yes | N.A | N.A | No | Yes | Yes | Yes | No | Yes | LCA | LCA | |
| 19 | FG399 | M | 2009 | Concepcion | 0 | 3 years | 0.05 | 0.15 | (−1.75) | (−1.50) | Yes | No | Yes | No | Abolished | Abolished | No | Yes | Yes | Yes | No | Yes | Mild mental impairment | LCA | LCA |
| 20 | FG649 | M | 2003 | Quillota | 0 | birth | 0.5 | 0.6 | (−1.50) | (−1.50) | Yes | No | Yes | No | N.A | N.A | No | Yes | Yes | No | Yes | No | LCA, EOSRD | LCA | |
| 21 | FG666 | F | 1985 | Colina | 0 | birth | lp | lp | N.A | N.A | No | Yes | Yes | No | N.A | N.A | No | Yes | Yes | No | No | Yes | LCA | LCA | |
| 22 | FG789 | M | 2013 | Rancagua | 0 | 2 years | 0.2 | 0.2 | (+3.00) | (+2.50) | No | No | Yes | No | N.A | N.A | No | Yes | Yes | Yes | No | No | EOSRD | LCA | |
| 23 | FG850 | M | 2002 | Santiago | 0 | 3 years | 0.5 | 0.2 | (−0.50) | NA | No | No | Yes | No | Rod–Cone | Rod–Cone | No | Yes | Yes | Yes | No | No | EOSRD | LCA | |
| 24 | FG901 | F | 1988 | Antofagasta | 0 | birth | 0.08 | 0.04 | (+0.50) | (+0.75) | Yes | Yes | Yes | Yes | N.A | N.A | No | Yes | Yes | No | No | Yes | Optic disc drusen | LCA | LCA |
| 25 | FG942 | F | 1998 | Parral | 0 | 3 years | cf | cf | (+3.75) | (+3.00) | Yes | Yes | Yes | No | Abolished | Abolished | No | Yes | Yes | Yes | No | Yes | LCA | LCA | |
| 26 | FG979 | M | 1969 | Santiago | N.A | 2 years | cf | cf | N.A | N.A | Yes | Yes | Yes | No | N.A | N.A | Yes | Yes | No | No | No | No | LCA, EOSRD | LCA | |
| 27 | FG981 | M | 1976 | Santiago | N.A | birth | cf | cf | (+3.25) | (+2.50) | Yes | No | Yes | Yes | N.A | N.A | No | Yes | Yes | Yes | No | Yes | LCA | LCA | |
| 28 | FG1004 | F | 2018 | Santiago | 0 | birth | 0.05 | 0.05 | (+6.5) | (+6.5) | Yes | No | Yes | Yes | N.A | N.A | No | Yes | Yes | Yes | No | Yes | LCA | LCA | |
| 29 | FG319 | F | 1991 | Santiago | 0 | 4 years | 0.025 | cf | NA | NA | Yes | N.A | Yes | Yes | N.A | N.A | Yes | Yes | No | No | No | No | LCA | LCA | |
| 30 | FG635 | F | 2018 | Santiago | N.A | birth | lp | lp | (+4.00) | (+4.00) | Yes | Yes | Yes | Yes | Normal | Cone | No | No | No | No | No | No | EOSRD | LCA | |
| 31 | FG337 | M | 2002 | Santiago | 0 | 2 months | lp | lp | N.A | N.A | Yes | Yes | No | Yes | N.A | N.A | No | Yes | No | No | No | No | Renal failure at 14 years of age | LCA | SLNS |
| 32 | FG236 | M | 1983 | Santiago | 0 | childhood | hm | hm | N.A | N.A | Yes | N.A | Yes | No | N.A | N.A | Yes | Yes | No | No | No | No | LCA | LCA | |
| FG237 | F | 1997 | Santiago | 0 | 1 year | cf | cf | N.A | N.A | Yes | No | Yes | No | N.A | N.A | Yes | Yes | No | No | No | No | LCA | LCA | ||
| 33 | FG360 | F | 1995 | Santiago | 0.0156 | birth | cf | cf | N.A | N.A | Yes | Yes | Yes | No | N.A | N.A | Yes | Yes | No | No | No | No | LCA | LCA | |
| 34 | FG496 | F | 1964 | Santiago | 0 | birth | 0.05 | 0.1 | (+2.25) | (+2.0) | Yes | Yes | Yes | No | Rod | Cone | No | No | No | No | Yes | No | LCA | LCA | |
| 35 | FG600 | F | 1991 | Santiago | 0 | 6 months | 0.05 | 0.2 | N.A | N.A | Yes | N.A | Yes | Yes | N.A | N.A | Yes | Yes | No | No | No | No | LCA | LCA | |
| 36 | FG659 | M | 1999 | Santiago | 0 | birth | 0.3 | 0.4 | (+0.50) | (+0.25) | No | No | Yes | No | Rod–Cone | Rod–Cone | Yes | No | No | No | No | No | LCA | LCA | |
| 37 | FG856 | F | 1970 | Temuco | 0.0039 | birth | 0.1 | hm | (−0.75) | (−3.25) | No | No | Yes | Yes | N.A | N.A | Yes | Yes | No | No | No | No | LCA | LCA | |
| 38 | FG851 | M | 1989 | Santiago | 0 | birth | 0.05 | 0.05 | (−1.00) | (−0.75) | Yes | No | Yes | No | Abolished | Abolished | Yes | Yes | No | No | No | No | LCA | LCA | |
| 39 | FG1002 | F | 2016 | Iquique | 0 | 3 months | cf | cf | N.A | N.A | Yes | No | Yes | Yes | N.A | N.A | No | No | No | No | Yes | No | LCA | LCA | |
| 40 | FG465 | M | 1988 | Santiago | 0 | birth | lp | lp | N.A | N.A | Yes | Yes | No | No | N.A | N.A | N.A | N.A | N.A | N.A | N.A | N.A | White bilateral cataract | LCA | LCA |
| 41 | FG787 | M | 2020 | Talca | N.A | birth | N.A | N.A | N.A | N.A | Yes | Yes | Yes | No | N.A | N.A | No | Yes | No | No | No | Yes | LCA | LCA | |
| 42 | FG165 | F | 1950 | Santiago | 0 | 7 years | lp | lp | N.A | N.A | Yes | N.A | Yes | Yes | N.A | N.A | Yes | Yes | No | No | No | No | LCA | ADRP | |
| 43 | FG454 | F | 1964 | Los Angeles | 0 | 16 months | 0.1 | 0.1 | N.A | N.A | Yes | N.A | No | Yes | N.A | N.A | Yes | Yes | No | No | No | No | LCA | CORD | |
| 44 | FG402 | F | 2000 | Santiago | 0 | birth | 0.2 | 0.2 | (−0.25) | (+0.25) | No | No | Yes | Yes | Abolished | Abolished | Yes | Yes | No | No | No | Yes | Diffuse paravenous pigmentation | LCA | LCA |
| 45 | FG68 | M | 1986 | Requinoa | 0 | N.A | 0.1 | 0.1 | (+0.75) | (+0.75) | No | No | No | Yes | Abolished | Abolished | Yes | Yes | No | No | No | No | EOSRD | LCA | |
| FG69 | M | 1986 | Requinoa | 0 | childhood | 0.1 | 0.1 | (+1.25) | (+1.5) | N.A | N.A | Yes | No | Abolished | Abolished | Yes | Yes | No | No | No | No | EOSRD | LCA | ||
| 46 | FG383 | F | 2009 | Santiago | 0 | 2 years | 0.2 | 0.1 | (+1) | (+1.5) | N.A | N.A | Yes | Yes | N.A | N.A | Yes | Yes | No | Yes | No | No | LCA, EOSRD | LCA | |
| 47 | FG429 | M | 1986 | Santiago | 0 | 5 years | hm | hm | (−3.0) | (−3.75) | No | No | No | Yes | N.A | N.A | Yes | Yes | No | No | No | No | EOSRD | LCA | |
| 48 | FG612 | F | 1988 | Machali | 0 | 7 years | npl | hm | N.A | N.A | No | No | Yes | No | N.A | N.A | Yes | Yes | No | No | No | Yes | EOSRD | LCA | |
| 49 | FG667 | F | 1996 | Santiago | (0.0156) | birth | hm | 0.08 | N.A | N.A | Yes | No | Yes | No | N.A | N.A | Yes | Yes | No | No | No | Yes | LCA | LCA | |
| 50 | FG694 | F | 2011 | Arica | 0 | birth | 0.6 | 0.4 | (+1.25) | (+1.25) | Yes | No | Yes | Yes | Abolished | Cone | Yes | Yes | No | No | No | No | Diffuse paravenous pigmentation | LCA | LCA |
| 51 | FG780 | M | 2016 | Santiago | 0 | 3 years | 0.5 | 0.5 | (+1.25) | (+1.25) | No | No | Yes | Yes | N.A | N.A | No | Yes | No | Yes | Yes | No | Diffuse paravenous atrophy | EOSRD | LCA |
| 52 | FG247 | F | 1988 | Santiago | 0.0625 | childhood | hm | hm | (−9.5) | (−10.5) | Yes | No | Yes | Yes | N.A | N.A | Yes | Yes | No | No | No | No | LCA | LCA | |
| 53 | FG514 | M | 1993 | Santiago | 0* | childhood | 0.33 | hm | (+0.75) | N.A | Yes | No | Yes | No | N.A | N.A | Yes | Yes | No | No | No | No | Optic disc drusen | LCA | LCA |
| 54 | FG487 | M | 1995 | Coquimbo | 0 | 5 years | hm | cf | 0 | (−3.0) | Yes | No | Yes | No | N.A | N.A | Yes | No | No | No | No | No | LCA | LCA | |
| 55 | FG853 | F | 1980 | Santiago | 0 | NA | nlp | lp | N.A | NA | Yes | No | Yes | No | N.A | N.A | Yes | Yes | No | No | No | No | LCA | LCA | |
| 56 | FG441 | F | 2018 | Santiago | (0.0625) | 2 years | cf | hm | (−6.0) | (−5.5) | Yes | No | Yes | Yes | N.A | N.A | Yes | No | No | No | No | No | Persistent ductus arterioso | LCA | LCA |
| 57 | FG118 | F | 2013 | Puerto Montt | 0 | birth | N.A | N.A | N.A | N.A | N.A | N.A | Yes | No | N.A | N.A | No | Yes | Yes | No | Yes | No | Bilateral retinal detachment | LCA | GFS |
2.3. Subjects Carrying Mutations in Other IRD Genes
2.4. Individuals Who Developed Additional Symptoms Consistent with a Syndromic IRD
3. Discussion
4. Families, Materials, and Methods
4.1. Subjects and Clinical Assessment
4.2. Capture Panel Design and Library Preparation
4.3. Bioinformatic Analysis
4.4. Sanger Validation and Segregation Analysis
4.5. Haplotype Analysis
4.6. Assessment of the Potential Shared Ancestry among Individuals with the RP1 c.5564del (p.Lys1855Argfs*42) Variant
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Georgiou, M.; Fujinami, K.; Michaelides, M. Inherited retinal diseases: Therapeutics, clinical trials and end points—A review. Clin. Exp. Ophthalmol. 2021, 49, 270–288. [Google Scholar] [CrossRef] [PubMed]
- Kumaran, N.; Moore, A.T.; Weleber, R.G.; Michaelides, M. Leber congenital amaurosis/earlyonset severe retinal dystrophy: Clinical features, molecular genetics and therapeutic interventions. Br. J. Ophthalmol. 2017, 101, 1147–1154. [Google Scholar] [CrossRef] [PubMed]
- Hasan, S.M.; Azmeh, A.; Mostafa, O.; Megarbane, A. Coat’s like vasculopathy in leber congenital amaurosis secondary to homozygous mutations in CRB1: A case report and discussion of the management options. BMC Res. Notes 2016, 9, 91. [Google Scholar] [CrossRef] [PubMed]
- Porto, F.; Jones, E.; Branch, J.; Soens, Z.; Maia, I.; Sena, I.; Sampaio, S.; Simões, R.; Chen, R. Molecular Screening of 43 Brazilian Families Diagnosed with Leber Congenital Amaurosis or Early-Onset Severe Retinal Dystrophy. Genes 2017, 8, 355. [Google Scholar] [CrossRef] [PubMed]
- Waters, A.M.; Beales, P.L. Ciliopathies: An expanding disease spectrum. Pediatr. Nephrol. 2011, 26, 1039–1056. [Google Scholar] [CrossRef] [PubMed]
- Leroy, B.P.; Birch, D.G.; Duncan, J.L.; Lam, B.L.; Koenekoop, R.K.; Porto, F.B.O.; Russell, S.R.; Girach, A. Leber congenital amaurosis due to CEP290 mutations—Severe vision impairment with a high unmet medical need: A review. Retina 2021, 41, 898–907. [Google Scholar] [CrossRef]
- Forsyth, R.; Gunay-Aygun, M. Bardet-Biedl Syndrome Overview. GeneReviews®. University of Washington, Seattle, 2020. Available online: https://www.ncbi.nlm.nih.gov/books/NBK1363/ (accessed on 18 November 2022).
- Marshall, J.D.; Muller, J.; Collin, G.B.; Milan, G.; Kingsmore, S.F.; Dinwiddie, D.; Farrow, E.G.; Miller, N.A.; Favaretto, F.; Maffei, P.; et al. Alström Syndrome: Mutation spectrum of ALMS1. Hum. Mutat. 2015, 36, 660–668. [Google Scholar] [CrossRef] [PubMed]
- Kaur, A.; Dhir, S.K.; Goyal, G.; Mittal, N.; Goyal, R.K. Senior Loken Syndrome. J. Clin. Diagn. Res. 2016, 10, SD03–SD04. [Google Scholar] [CrossRef]
- Garafalo, A.V.; Cideciyan, A.V.; Heon, E.; Sheplock, R.; Pearson, A.; WeiYang Yu, C.; Sumaroka, A.; Aguirre, G.D.; Jacobson, S.G. Progress in treating inherited retinal diseases: Early subretinal gene therapy clinical trials and candidates for future initiatives. Prog. Retin. Eye Res. 2020, 77, 100827. [Google Scholar] [CrossRef] [PubMed]
- Fujinami-Yokokawa, Y.; Fujinami, K.; Kuniyoshi, K.; Hayashi, T.; Ueno, S.; Mizota, A.; Shinoda, K.; Arno, G.; Pontikos, N.; Yang, L.; et al. Clinical and Genetic Characteristics of 18 Patients from 13 Japanese Families with CRX associated retinal disorder: Identification of Genotype-phenotype Association. Sci. Rep. 2020, 10, 9531. [Google Scholar] [CrossRef]
- Stone, E.M.; Andorf, J.L.; Whitmore, S.S.; DeLuca, A.P.; Giacalone, J.C.; Streb, L.M.; Braun, T.A.; Mullins, R.F.; Scheetz, T.E.; Sheffield, V.C.; et al. Clinically Focused Molecular Investigation of 1000 Consecutive Families with Inherited Retinal Disease. Ophthalmology 2017, 124, 1314–1331. [Google Scholar] [CrossRef] [PubMed]
- Corton, M.; Tatu, S.D.; Avila-Fernandez, A.; Vallespín, E.; Tapias, I.; Cantalapiedra, D.; Blanco-Kelly, F.; Riveiro-Alvarez, R.; Bernal, S.; García-Sandoval, B.; et al. High frequency of CRB1 mutations as cause of Early-Onset Retinal Dystrophies in the Spanish population. Orphanet J. Rare Dis. 2013, 8, 20. [Google Scholar] [CrossRef]
- Hanany, M.; Rivolta, C.; Sharon, D. Worldwide carrier frequency and genetic prevalence of autosomal recessive inherited retinal diseases. Proc. Natl. Acad. Sci. USA 2020, 117, 2710–2716. [Google Scholar] [CrossRef] [PubMed]
- Aleman, T.S.; Uyhazi, K.E.; Serrano, L.W.; Vasireddy, V.; Bowman, S.J.; Ammar, M.J.; Pearson, D.J.; Maguire, A.M.; Bennett, J. RDH12 Mutations Cause a Severe Retinal Degeneration With Relatively Spared Rod Function. Investig. Opthalmol. Vis. Sci. 2018, 59, 5225–5236. [Google Scholar] [CrossRef] [PubMed]
- Coppieters, F.; Lefever, S.; Leroy, B.P.; De Baere, E. CEP290, a gene with many faces: Mutation overview and presentation of CEP290base. Hum. Mutat. 2010, 31, 1097–1108. [Google Scholar] [CrossRef] [PubMed]
- Feldhaus, B.; Weisschuh, N.; Nasser, F.; den Hollander, A.I.; Cremers, F.P.M.; Zrenner, E.; Kohl, S.; Zobor, D. CEP290 Mutation Spectrum and Delineation of the Associated Phenotype in a Large German Cohort: A Monocentric Study. Am. J. Ophthalmol. 2020, 211, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Perrault, I.; Hanein, S.; Zanlonghi, X.; Serre, V.; Nicouleau, M.; Defoort-Delhemmes, S.; Delphin, N.; Fares-Taie, L.; Gerber, S.; Xerri, O.; et al. Mutations in NMNAT1 cause Leber congenital amaurosis with early-onset severe macular and optic atrophy. Nat. Genet. 2012, 44, 975–977. [Google Scholar] [CrossRef] [PubMed]
- Eblimit, A.; Zaneveld, S.A.; Liu, W.; Thomas, K.; Wang, K.; Li, Y.; Mardon, G.; Chen, R. NMNAT1 E257K variant, associated with Leber Congenital Amaurosis (LCA9), causes a mild retinal degeneration phenotype. Exp. Eye Res. 2018, 173, 32–43. [Google Scholar] [CrossRef] [PubMed]
- Sohocki, M.M.; Sullivan, L.S.; Mintz-Hittner, H.A.; Birch, D.; Heckenlively, J.R.; Freund, C.L.; McInnes, R.R.; Daiger, S.P. A range of clinical phenotypes associated with mutations in CRX. a photoreceptor transcription-factor gene. Am. J. Hum. Genet. 1998, 63, 1307–1315. [Google Scholar] [CrossRef]
- Patel, N.; Alkuraya, H.; Alzahrani, S.S.; Nowailaty, S.R.; Seidahmed, M.Z.; Alhemidan, A.; Ben-Omran, T.; Ghazi, N.; Al-Aqeel, A.; Al-Owain, M.; et al. Mutations in known disease genes account for the majority of autosomal recessive retinal dystrophies. Clin. Genet. 2018, 94, 554–563. [Google Scholar] [CrossRef]
- Beryozkin, A.; Aweidah, H.; Carrero Valenzuela, R.D.; Berman, M.; Iguzquiza, O.; Cremers, F.P.M.; Khan, M.I.; Swaroop, A.; Amer, R.; Khateb, S.; et al. Retinal Degeneration Associated With RPGRIP1: A Review of Natural History, Mutation Spectrum, and Genotype–Phenotype Correlation in 228 Patients. Front. Cell Dev. Biol. 2021, 9, 746781. [Google Scholar] [CrossRef] [PubMed]
- Martin-Merida, I.; Avila-Fernandez, A.; Del Pozo-Valero, M.; Blanco-Kelly, F.; Zurita, O.; Perez-Carro, R.; Aguilera-Garcia, D.; Riveiro-Alvarez, R.; Arteche, A.; Trujillo-Tiebas, M.J.; et al. Genomic Landscape of Sporadic Retinitis Pigmentosa: Findings from 877 Spanish Cases. Ophthalmology 2019, 126, 1181–1188. [Google Scholar] [CrossRef]
- Jia, D.; Gao, P.; Lv, Y.; Huang, Y.; Reilly, J.; Sun, K.; Han, Y.; Hu, H.; Chen, X.; Zhang, Z.; et al. Tulp1 deficiency causes early-onset retinal degeneration through affecting ciliogenesis and activating ferroptosis in zebrafish. Cell Death Dis. 2022, 13, 962. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting Functional Effect of Human Missense Mutations Using PolyPhen-2. Curr. Protoc. Hum. Genet. 2013, 76, 7–20. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Henikoff, S.; Ng, P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009, 4, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, J.M.; Cooper, D.N.; Schuelke, M.; Seelow, D. MutationTaster2: Mutation prediction for the deep-sequencing age. Nat. Methods 2014, 11, 361–362. [Google Scholar] [CrossRef] [PubMed]
- Parry, D.A.; Toomes, C.; Bida, L.; Danciger, M.; Towns, K.V.; McKibbin, M.; Jacobson, S.G.; Logan, C.V.; Ali, M.; Bond, J.; et al. Loss of the metalloprotease ADAM9 leads to cone-rod dystrophy in humans and retinal degeneration in mice. Am. J. Hum. Genet. 2009, 84, 683–691. [Google Scholar] [CrossRef]
- Wang, J.; Xiao, X.; Li, S.; Wang, P.; Sun, W.; Zhang, Q. Dominant RP in the Middle While Recessive in Both the N- and C-Terminals Due to RP1 Truncations: Confirmation, Refinement, and Questions. Front. Cell Dev. Biol. 2021, 9, 634478. [Google Scholar] [CrossRef] [PubMed]
- Iarossi, G.; Marino, V.; Maltese, P.E.; Colombo, L.; D’Esposito, F.; Manara, E.; Dhuli, K.; Modarelli, A.M.; Cennamo, G.; Magli, A.; et al. Expanding the Clinical and Genetic Spectrum of RAB28-Related Cone-Rod Dystrophy: Pathogenicity of Novel Variants in Italian Families. Int. J. Mol. Sci. 2020, 22, 381. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, J.; Chen, N.; Wang, L.; Zhang, F.; Ma, Z.; Li, G.; Yang, L. Application of Whole Exome and Targeted Panel Sequencing in the Clinical Molecular Diagnosis of 319 Chinese Families with Inherited Retinal Dystrophy and Comparison Study. Genes 2018, 9, 360. [Google Scholar] [CrossRef]
- Liu, X.; Tao, T.; Zhao, L.; Li, G.; Yang, L. Molecular diagnosis based on comprehensive genetic testing in 800 Chinese families with non-syndromic inherited retinal dystrophies. Clin. Exp. Ophthalmol. 2021, 49, 46–59. [Google Scholar] [CrossRef] [PubMed]
- Homburger, J.R.; Moreno-Estrada, A.; Gignoux, C.R.; Nelson, D.; Sanchez, E.; Ortiz-Tello, P.; Pons-Estel, B.A.; Acevedo-Vasquez, E.; Miranda, P.; Langefeld, C.D.; et al. Genomic Insights into the Ancestry and Demographic History of South America. PLoS Genet. 2015, 11, e1005602. [Google Scholar] [CrossRef] [PubMed]
- Gil, F.G. El Sistema Político de Chile; Andres Bello: Santiago, Chile, 1969. [Google Scholar]
- Zanolli, M.; Oporto, J.I.; Verdaguer, J.I.; López, J.P.; Zacharías, S.; Romero, P.; Ossandón, D.; Denk, O.; Acuña, O.; López, J.M.; et al. Genetic testing for inherited ocular conditions in a developing country. Ophthalmic Genet. 2020, 41, 36–40. [Google Scholar] [CrossRef]
- Valverde, D.; Pereiro, I.; Vallespín, E.; Ayuso, C.; Borrego, S.; Baiget, M. Complexity of Phenotype–Genotype Correlations in Spanish Patients with RDH12 Mutations. Investig. Ophthalmol. Vis. Sci. 2009, 50, 1065–1068. [Google Scholar] [CrossRef] [PubMed]
- Corton, M.; Avila-Fernandez, A.; Vallespín, E.; López-Molina, M.I.; Almoguera, B.; Martín-Garrido, E.; Tatu, S.D.; Khan, M.I.; Blanco-Kelly, F.; Riveiro-Alvarez, R.; et al. Involvement of LCA5 in Leber congenital amaurosis and retinitis pigmentosa in the Spanish population. Ophthalmology 2014, 121, 399–407. [Google Scholar] [CrossRef] [PubMed]
- Perrault, I.; Rozet, J.M.; Calvas, P.; Gerber, S.; Camuzat, A.; Dollfus, H.; Châtelin, S.; Souied, E.; Ghazi, I.; Leowski, C.; et al. Retinal-specific guanylate cyclase gene mutations in Leber’s congenital amaurosis. Nat. Genet. 1996, 14, 461–464. [Google Scholar] [CrossRef] [PubMed]
- Vallespin, E.; Lopez-Martinez, M.-A.; Cantalapiedra, D.; Riveiro-Alvarez, R.; Aguirre-Lamban, J.; Avila-Fernandez, A.; Villaverde, C.; Trujillo-Tiebas, M.-J.; Ayuso, C. Frequency of CEP290 c.2991_1655A>G mutation in 175 Spanish families affected with Leber congenital amaurosis and early-onset retinitis pigmentosa. Mol. Vis. 2007, 13, 2160–2162. [Google Scholar] [PubMed]
- Vallespin, E.; Cantalapiedra, D.; Riveiro-Alvarez, R.; Wilke, R.; Aguirre-Lamban, J.; Avila-Fernandez, A.; Lopez-Martinez, M.A.; Gimenez, A.; Trujillo-Tiebas, M.J.; Ramos, C.; et al. Mutation Screening of 299 Spanish Families with Retinal Dystrophies by Leber Congenital Amaurosis Genotyping Microarray. Investig. Ophthalmol. Vis. Sci. 2007, 48, 5653–5661. [Google Scholar] [CrossRef]
- Hull, S.; Arno, G.; Plagnol, V.; Robson, A.; Webster, A.R.; Moore, A.T. Exome sequencing reveals ADAM9 mutations in a child with cone-rod dystrophy. Acta Ophthalmol. 2015, 93, e392–e393. [Google Scholar] [CrossRef]
- Georgiou, M.; Ali, N.; Yang, E.; Grewal, P.S.; Rotsos, T.; Pontikos, N.; Robson, A.G.; Michaelides, M. Extending the phenotypic spectrum of PRPF8, PRPH2, RP1 and RPGR, and the genotypic spectrum of early-onset severe retinal dystrophy. Orphanet J. Rare Dis. 2021, 16, 128. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- DePristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; del Angel, G.; Rivas, M.A.; Hanna, M.; et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011, 43, 491–498. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Charbit-Henrion, F.; Parlato, M.; Hanein, S.; Duclaux-Loras, R.; Nowak, J.; Begue, B.; Rakotobe, S.; Bruneau, J.; Fourrage, C.; Alibeu, O.; et al. Diagnostic Yield of Next-generation Sequencing in Very Early-onset Inflammatory Bowel Diseases: A Multicentre Study. J. Crohn’s Colitis 2018, 12, 1104–1112. [Google Scholar] [CrossRef]
- Goossens, D.; Moens, L.N.; Nelis, E.; Lenaerts, A.-S.; Glassee, W.; Kalbe, A.; Frey, B.; Kopal, G.; De Jonghe, P.; De Rijk, P.; et al. Simultaneous mutation and copy number variation (CNV) detection by multiplex PCR-based GS-FLX sequencing. Hum. Mutat. 2009, 30, 472–476. [Google Scholar] [CrossRef] [PubMed]
- Kopanos, C.; Tsiolkas, V.; Kouris, A.; Chapple, C.E.; Albarca Aguilera, M.; Meyer, R.; Massouras, A. VarSome: The human genomic variant search engine. Bioinformatics 2019, 35, 1978–1980. [Google Scholar] [CrossRef] [PubMed]
- Delaneau, O.; Howie, B.; Cox, A.J.; Zagury, J.-F.; Marchini, J. Haplotype estimation using sequencing reads. Am. J. Hum. Genet. 2013, 93, 687–696. [Google Scholar] [CrossRef]
- Genin, E.; Tullio-Pelet, A.; Begeot, F.; Lyonnet, S.; Abel, L. Estimating the age of rare disease mutations: The example of Triple-A syndrome. J. Med. Genet. 2004, 41, 445–449. [Google Scholar] [CrossRef]
- Auton, A.; Abecasis, G.R.; Altshuler, D.M.; Durbin, R.M.; Abecasis, G.R.; Bentley, D.R.; Chakravarti, A.; Clark, A.G.; Donnelly, P.; Eichler, E.E.; et al. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar]
- Manichaikul, A.; Mychaleckyj, J.C.; Rich, S.S.; Daly, K.; Sale, M.; Chen, W.-M. Robust relationship inference in genome-wide association studies. Bioinformatics 2010, 26, 2867–2873. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moya, R.; Angée, C.; Hanein, S.; Jabot-Hanin, F.; Kaplan, J.; Perrault, I.; Rozet, J.-M.; Fares Taie, L. Four Unique Genetic Variants in Three Genes Account for 62.7% of Early-Onset Severe Retinal Dystrophy in Chile: Diagnostic and Therapeutic Consequences. Int. J. Mol. Sci. 2024, 25, 6151. https://doi.org/10.3390/ijms25116151
Moya R, Angée C, Hanein S, Jabot-Hanin F, Kaplan J, Perrault I, Rozet J-M, Fares Taie L. Four Unique Genetic Variants in Three Genes Account for 62.7% of Early-Onset Severe Retinal Dystrophy in Chile: Diagnostic and Therapeutic Consequences. International Journal of Molecular Sciences. 2024; 25(11):6151. https://doi.org/10.3390/ijms25116151
Chicago/Turabian StyleMoya, Rene, Clémentine Angée, Sylvain Hanein, Fabienne Jabot-Hanin, Josseline Kaplan, Isabelle Perrault, Jean-Michel Rozet, and Lucas Fares Taie. 2024. "Four Unique Genetic Variants in Three Genes Account for 62.7% of Early-Onset Severe Retinal Dystrophy in Chile: Diagnostic and Therapeutic Consequences" International Journal of Molecular Sciences 25, no. 11: 6151. https://doi.org/10.3390/ijms25116151
APA StyleMoya, R., Angée, C., Hanein, S., Jabot-Hanin, F., Kaplan, J., Perrault, I., Rozet, J.-M., & Fares Taie, L. (2024). Four Unique Genetic Variants in Three Genes Account for 62.7% of Early-Onset Severe Retinal Dystrophy in Chile: Diagnostic and Therapeutic Consequences. International Journal of Molecular Sciences, 25(11), 6151. https://doi.org/10.3390/ijms25116151