
Is the Complement System Dysregulated in Preeclampsia Comorbid with HIV Infection?

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. The Complement System
2.1. Complement Activation
2.2. Complement Regulators
3. The Complement System in HIV Infection
3.1. Complement Activation in HIV Infection
3.2. Enhancement of Complement in HIV Infection
4. The Complement System in Normal Pregnancy
4.1. Immunotolerance in a Healthy Pregnancy
4.2. Complement Regulation in Pregnancy
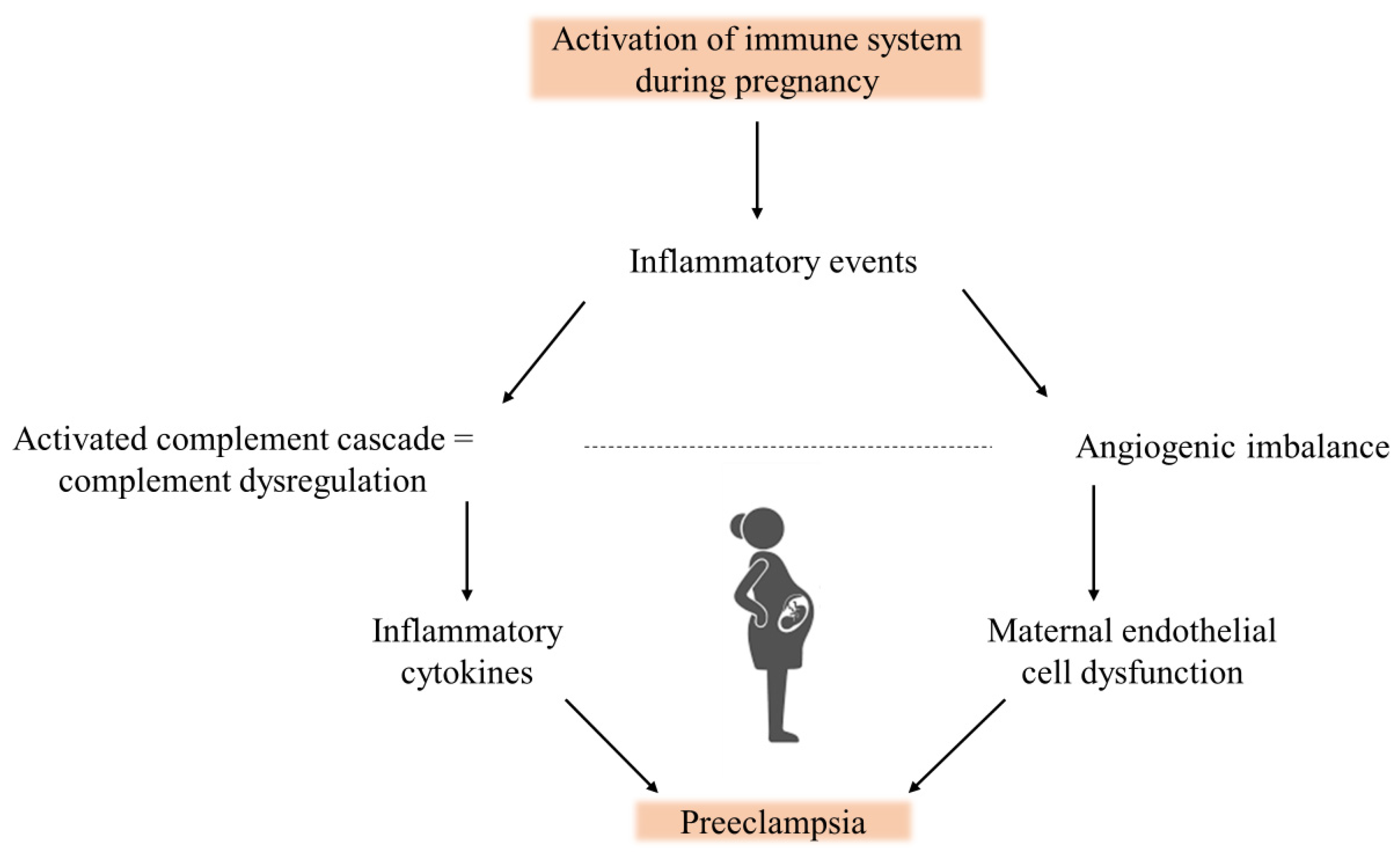
5. The Complement System in Preeclampsia
6. Complement Component C1q
6.1. Structure & Function
6.2. C1q in Pregnancy
6.3. C1q in the Regulation of HIV Infection
7. Complement Component Mannose-Binding Lectin
7.1. Structure and Function
7.2. Preeclampsia and the MBL Pathway
7.3. HIV Infection and the MBL Pathway
8. Complement Component C2
8.1. Structure and Function
8.2. C2 in HIV Infection and Preeclampsia
9. Complement Component C3
9.1. Structure and Function
9.2. C3 in Preeclampsia and HIV Infection
10. Complement Component C4
10.1. Structure and Function
10.2. Preeclampsia and HIV Infection
11. Complement Component C5
11.1. Structure & Function
11.2. C5 and Preeclampsia
11.3. Therapeutic Evidence of C5 in Preeclampsia
11.4. C5 and HIV Infection
12. Complement Component C9 and Membrane Attack Complex (MAC)
12.1. Structure and Function
12.2. C9 and MAC in Preeclampsia and HIV Infection
13. The Complement System in HIV-Associated Preeclampsia
14. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kurjak, A.; Stanojević, M.; Dudenhausen, J. Why maternal mortality in the world remains tragedy in low-income countries and shame for high-income ones: Will sustainable development goals (SDG) help? J. Perinat. Med. 2023, 51, 170–181. [Google Scholar] [CrossRef]
- Bajpai, D.; Popa, C.; Verma, P.; Dumanski, S.; Shah, S. Evaluation and Management of Hypertensive Disorders of Pregnancy. Kidney360 2023, 10, 34067. [Google Scholar] [CrossRef] [PubMed]
- Regal, J.F.; Burwick, R.M.; Fleming, S.D. The complement system and preeclampsia. Curr. Hypertens. Rep. 2017, 19, 87. [Google Scholar] [CrossRef]
- UNAIDS Global AIDS Update 2023. Geneva: Joint United Nations Programme on HIV/AIDS. 2023. Licence: CC BY-NC-SA 3.0 IGO. Available online: https://thepath.unaids.org/wp-content/themes/unaids2023/assets/files/2023_report.pdf (accessed on 1 December 2023).
- Statistics South Africa 2023. Pretoria: Mid-Year Population Estimates 2022. Statistical Release P0302. Available online: https://www.statssa.gov.za/publications/P0302/P03022022.pdf (accessed on 15 October 2023).
- Derzsy, Z.; Prohászka, Z.; Rigó, J., Jr.; Füst, G.; Molvarec, A. Activation of the complement system in normal pregnancy and preeclampsia. Mol. Immunol. 2010, 47, 1500–1506. [Google Scholar] [CrossRef]
- He, Y.D.; Xu, B.N.; Wang, M.L.; Wang, Y.Q.; Yu, F.; Chen, Q.; Zhao, M.H. Dysregulation of complement system during pregnancy in patients with preeclampsia: A prospective study. Mol. Immunol. 2020, 122, 69–79. [Google Scholar] [CrossRef]
- Saito, S.; Nakashima, A.; Ito, M.; Shima, T. Clinical implication of recent advances in our understanding of IL-17 and reproductive immunology. Expert Rev. Clin. Immunol. 2011, 7, 649–657. [Google Scholar] [CrossRef] [PubMed]
- Laresgoiti-Servitje, E.; Gomez-Lopez, N. The pathophysiology of preeclampsia involves altered levels of angiogenic factors promoted by hypoxia and autoantibody-mediated mechanisms. Biol. Reprod. 2012, 87, 36. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.M.; Liu, B.; Zhao, H.B.; Stone, P.; Chen, Q.; Chamley, L. IL-6, TNFα and TGFβ promote nonapoptotic trophoblast deportation and subsequently causes endothelial cell activation. Placenta 2010, 31, 75–80. [Google Scholar] [CrossRef]
- Clouse, K.; Malope-Kgokong, B.; Bor, J.; Nattey, C.; Mudau, M.; Maskew, M. The South African National HIV Pregnancy Cohort: Evaluating continuity of care among women living with HIV. BMC Public Health 2020, 20, 1662. [Google Scholar] [CrossRef]
- Sikhosana, M.L.; Suchard, M.; Kuonza, L.; Cutland, C.; Slogrove, A.; Otwombe, K.; Motaze, N.V. Association between preeclampsia and HIV: A case-control study in urban South Africa. AJOG Glob. Rep. 2022, 2, 100056. [Google Scholar] [CrossRef]
- Carroll, M.C. The complement system in regulation of adaptive immunity. Nat. Immunol. 2004, 5, 981–986. [Google Scholar] [CrossRef] [PubMed]
- Lokki, A.I.; Heikkinen-Eloranta, J.; Jarva, H.; Saisto, T.; Lokki, M.-L.; Laivuori, H.; Meri, S. Complement activation and regulation in preeclamptic placenta. Front. Immunol. 2014, 5, 312. [Google Scholar] [CrossRef] [PubMed]
- Rossheim, A.E.; Cunningham, T.D.; Hair, P.S.; Shah, T.; Cunnion, K.M.; Troy, S.B. Effects of well-controlled HIV infection on complement activation and function. J. Acquir. Immune Defic. Syn-Dromes 2016, 73, 20. [Google Scholar] [CrossRef] [PubMed]
- Denny, K.J.; Woodruff, T.M.; Taylor, S.M.; Callaway, L.K. Complement in pregnancy: A delicate balance. Am. J. Reprod. Immunol. 2013, 69, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Sarma, J.V.; Ward, P.A. The complement system. Cell Tissue Res. 2011, 343, 227–235. [Google Scholar] [CrossRef] [PubMed]
- Noris, M.; Remuzzi, G. Overview of complement activation and regulation. Semin. Nephrol. 2013, 33, 479–492. [Google Scholar] [CrossRef] [PubMed]
- Teirilä, L.; Heikkinen-Eloranta, J.; Kotimaa, J.; Meri, S.; Lokki, A.I. Regulation of the complement system and immunological tolerance in pregnancy. Semin. Immunol. 2019, 45, 101337. [Google Scholar] [CrossRef]
- Walport, M.J. Complement. Second of two parts. N. Engl. J. Med. 2001, 344, 1140–1144. [Google Scholar] [CrossRef]
- Reid, K.B.; Turner, M.W. Mammalian lectins in activation and clearance mechanisms involving the complement system. Springer Semin. Immunopathol. 1994, 15, 307–326. [Google Scholar] [CrossRef]
- Fujita, T.; Matsushita, M.; Endo, Y. The lectin-complement pathway–its role in innate immunity and evolution. Immunol. Rev. 2004, 198, 185–202. [Google Scholar] [CrossRef]
- Pangburn, M.K. Initiation of the alternative pathway of complement and the history of “tickover”. Immunol. Rev. 2023, 313, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Bexborn, F.; Andersson, P.O.; Chen, H.; Nilsson, B.; Ekdahl, K.N. The tick-over theory revisited: Formation and regulation of the soluble alternative complement C3 convertase (C3 (H2O) Bb). Mol. Immunol. 2008, 45, 2370–2379. [Google Scholar] [CrossRef]
- Harboe, M.; Mollnes, T.E. The alternative complement pathway revisited. J. Cell. Mol. Med. 2008, 12, 1074–1084. [Google Scholar] [CrossRef] [PubMed]
- Janeway, C.A., Jr.; Travers, P.; Walport, M.; Shlomchik, M.J. The complement system and innate immunity. In Immunobiology: The Immune System in Health and Disease, 5th ed.; Garland Science: New York, NY, USA, 2001. [Google Scholar]
- Ward, P.A.; Kemper, C. Complement System. In Inflammation: From Molecular and Cellular Mechanisms to the Clinic; Wiley: Hoboken, NJ, USA, 2017; pp. 785–812. [Google Scholar]
- Pierik, E.; Prins, J.R.; Van Goor, H.; Dekker, G.A.; Daha, M.R.; Seelen, M.A.; Scherjon, S.A. Dysregulation of complement activation and placental dysfunction: A potential target to treat preeclampsia? Front. Immunol. 2020, 10, 3098. [Google Scholar] [CrossRef]
- Dunkelberger, J.R.; Song, W.-C. Complement and its role in innate and adaptive immune responses. Cell Res. 2010, 20, 34–50. [Google Scholar] [CrossRef] [PubMed]
- Tincani, A.; Cavazzana, I.; Ziglioli, T.; Lojacono, A.; De Angelis, V.; Meroni, P. Complement activation and pregnancy failure. Clin. Rev. Allergy Immunol. 2010, 39, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Morgan, B.P. The complement system: An overview. In Complement Methods and Protocols; Humana Press: Totowa, NJ, USA, 2000; pp. 1–13. [Google Scholar]
- Davis, A., 3rd. Hereditary and acquired deficiencies of C1 inhibitor. Immunodefic. Rev. 1989, 1, 207–226. [Google Scholar] [PubMed]
- Davis Iii, A.E. C1 inhibitor and hereditary angioneurotic edema. Annu. Rev. Immunol. 1988, 6, 595–628. [Google Scholar] [CrossRef]
- Yu, Q.; Yu, R.; Qin, X. The good and evil of complement activation in HIV-1 infection. Cell. Mol. Immunol. 2010, 7, 334–340. [Google Scholar] [CrossRef]
- Gigli, I.; Fujita, T.; Nussenzweig, V. Modulation of the classical pathway C3 convertase by plasma proteins C4 binding protein and C3b inactivator. Proc. Natl. Acad. Sci. USA 1979, 76, 6596–6600. [Google Scholar] [CrossRef]
- Pangburn, M.K. The alternative pathway. Immunobiol. Complement Syst. 1986, 7, 163. [Google Scholar]
- Posch, W.; Cardinaud, S.; Hamimi, C.; Fletcher, A.; Mühlbacher, A.; Loacker, K.; Eichberger, P.; Dierich, M.P.; Pancino, G.; Lass-Flörl, C. Antibodies attenuate the capacity of dendritic cells to stimulate HIV-specific cyto-toxic T lymphocytes. J. Allergy Clin. Immunol. 2012, 130, 1368–1374.e2. [Google Scholar] [CrossRef] [PubMed]
- De Jong, M.A.; De Witte, L.; Santegoets, S.J.; Fluitsma, D.; Taylor, M.E.; De Gruijl, T.D.; Geijtenbeek, T.B. Mutz-3-derived Langerhans cells are a model to study HIV-1 transmission and potential inhibitors. J. Leukoc. Biol. 2010, 87, 637–643. [Google Scholar] [CrossRef] [PubMed]
- Heesterbeek, D.A.; Angelier, M.L.; Harrison, R.A.; Rooijakkers, S.H. Complement and bacterial infections: From molecular mechanisms to therapeutic applications. J. Innate Immun. 2018, 10, 455–464. [Google Scholar] [CrossRef] [PubMed]
- Ying, H.; Ji, X.; Hart, M.L.; Gupta, K.; Saifuddin, M.; Zariffard, M.R.; Spear, G.T. Interaction of man-nose-binding lectin with HIV type 1 is sufficient for virus opsonization but not neutralization. AIDS Res. Hum. Retroviruses 2004, 20, 327–335. [Google Scholar] [CrossRef] [PubMed]
- Tjomsland, V.; Ellegård, R.; Che, K.; Hinkula, J.; Lifson, J.D.; Larsson, M. Complement opsonization of HIV-1 enhances the uptake by dendritic cells and involves the endocytic lectin and integrin receptor families. PLoS ONE 2011, 6, e23542. [Google Scholar] [CrossRef] [PubMed]
- Crisci, E.; Ellegård, R.; Nyström, S.; Rondahl, E.; Serrander, L.; Bergström, T.; Sjöwall, C.; Eriksson, K.; Larsson, M. Complement opsonization promotes herpes simplex virus 2 infection of human dendritic cells. J. Virol. 2016, 90, 4939–4950. [Google Scholar] [CrossRef] [PubMed]
- Schiela, B.; Bernklau, S.; Malekshahi, Z.; Deutschmann, D.; Koske, I.; Banki, Z.; Thielens, N.M.; Würzner, R.; Speth, C.; Weiss, G. Active human complement reduces the Zika virus load via formation of the membrane-attack complex. Front. Immunol. 2018, 9, 2177. [Google Scholar] [CrossRef] [PubMed]
- Marquez, C.L.; Lau, D.; Walsh, J.; Shah, V.; Mcguinness, C.; Wong, A.; Aggarwal, A.; Parker, M.W.; Jacques, D.A.; Turville, S. Kinetics of HIV-1 capsid uncoating revealed by single-molecule analysis. Elife 2018, 7, e34772. [Google Scholar] [CrossRef] [PubMed]
- Dufloo, J.; Guivel-Benhassine, F.; Buchrieser, J.; Lorin, V.; Grzelak, L.; Dupouy, E.; Mestrallet, G.; Bourdic, K.; Lambotte, O.; Mouquet, H. Anti-HIV-1 antibodies trigger non-lytic complement deposition on infected cells. EMBO Rep. 2020, 21, e49351. [Google Scholar] [CrossRef]
- Roberts, L.; Passmore, J.-A.S.; Williamson, C.; Little, F.; Bebell, L.M.; Mlisana, K.; Burgers, W.A.; Van Loggerenberg, F.; Walzl, G.; Siawaya, J.F.D. Plasma cytokine levels during acute HIV-1 infection predict HIV disease progression. Aids 2010, 24, 819–831. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Lan, J.; Shepherd, N.; Hu, N.; Xing, Y.; Byrd, D.; Amet, T.; Jewell, C.; Gupta, S.; Kounga, C. Block-age of CD59 function restores activities of neutralizing and nonneutralizing antibodies in triggering anti-body-dependent complement-mediated lysis of HIV-1 virions and provirus-activated latently infected cells. J. Virol. 2015, 89, 9393–9406. [Google Scholar] [CrossRef] [PubMed]
- Stoiber, H.; Kacani, L.; Speth, C.; Würzner, R.; Dierich, M. The supportive role of complement in HIV pathogenesis. Immunol. Rev. 2001, 180, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Girardi, G.; Bulla, R.; Salmon, J.E.; Tedesco, F. The complement system in the pathophysiology of pregnancy. Mol. Immunol. 2006, 43, 68–77. [Google Scholar] [CrossRef] [PubMed]
- Girardi, G. Complement activation, a threat to pregnancy. Semin. Immunopathol. 2018, 40, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Thellin, O.; Coumans, B.; Zorzi, W.; Igout, A.; Heinen, E. Tolerance to the foeto-placental ‘graft’: Ten ways to support a child for nine months. Curr. Opin. Immunol. 2000, 12, 731–737. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Kusama, K.; Bai, R.; Ishikawa, S.; Fukushima, S.; Suda, Y.; Imakawa, K. Increase in complement iC3b is associated with anti-inflammatory cytokine expression during late pregnancy in mice. PLoS ONE 2017, 12, e0178442. [Google Scholar] [CrossRef]
- Burwick, R.M.; Rincon, M.; Beeraka, S.S.; Gupta, M.; Feinberg, B.B. Evaluation of hemolysis as a severe feature of preeclampsia. Hypertension 2018, 72, 460–465. [Google Scholar] [CrossRef] [PubMed]
- Lau, E.Y.; Liu, J.; Archer, E.; Mcdonald, S.M.; Liu, J. Maternal weight gain in pregnancy and risk of obesity among offspring: A systematic review. J. Obes. 2014, 2014, 524939. [Google Scholar] [CrossRef]
- Lokki, A.I.; Heikkinen-Eloranta, J.K.; Laivuori, H. The immunogenetic conundrum of preeclampsia. Front. Immunol. 2018, 9, 2630. [Google Scholar] [CrossRef]
- Hackmon, R.; Pinnaduwage, L.; Zhang, J.; Lye, S.J.; Geraghty, D.E.; Dunk, C.E. Definitive class I human leukocyte antigen expression in gestational placentation: HLA-F, HLA-E, HLA-C, and HLA-G in extravillous trophoblast invasion on placentation, pregnancy, and parturition. Am. J. Reprod. Immunol. 2017, 77, e12643. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Xu, B.; Song, D.; Yu, F.; Chen, Q.; Zhao, M. Correlations between complement system’s activation factors and anti-angiogenesis factors in plasma of patients with early/late-onset severe preeclampsia. Hypertens. Pregnancy 2016, 35, 499–509. [Google Scholar] [CrossRef] [PubMed]
- Parham, P.; Moffett, A. Variable NK cell receptors and their MHC class I ligands in immunity, reproduction and human evolution. Nat. Rev. Immunol. 2013, 13, 133–144. [Google Scholar] [CrossRef]
- Kennedy, P.R.; Chazara, O.; Gardner, L.; Ivarsson, M.A.; Farrell, L.E.; Xiong, S.; Hiby, S.E.; Colucci, F.; Sharkey, A.M.; Moffett, A. Activating KIR2DS4 is expressed by uterine NK cells and contributes to successful pregnancy. J. Immunol. 2016, 197, 4292–4300. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.A.; Hsu, J.L.; Assiri, A.M.; Broering, D.C. Targeted complement inhibition and microvasculature in transplants: A therapeutic perspective. Clin. Exp. Immunol. 2016, 183, 175–186. [Google Scholar] [CrossRef]
- Xu, C.; Mao, D.; Holers, V.M.; Palanca, B.; Cheng, A.M.; Molina, H. A critical role for murine complement regulator crry in fetomaternal tolerance. Science 2000, 287, 498–501. [Google Scholar] [CrossRef]
- Labarrere, C.A.; Dicarlo, H.L.; Bammerlin, E.; Hardin, J.W.; Kim, Y.M.; Chaemsaithong, P.; Haas, D.M.; Kassab, G.S.; Romero, R. Failure of physiologic transformation of spiral arteries, endothelial and trophoblast cell activation, and acute atherosis in the basal plate of the placenta. Am. J. Obstet. Gynecol. 2017, 216, 287.e1–287.e16. [Google Scholar] [CrossRef]
- Jacobsen, D.P.; Lekva, T.; Moe, K.; Fjeldstad, H.E.; Johnsen, G.M.; Sugulle, M.; Staff, A.C. Pregnancy and postpartum levels of circulating maternal sHLA-G in preeclampsia. J. Reprod. Immunol. 2021, 143, 103249. [Google Scholar] [CrossRef]
- Cunningham, M.W., Jr.; Castillo, J.; Ibrahim, T.; Cornelius, D.C.; Campbell, N.; Amaral, L.; Vaka, V.R.; Usry, N.; Williams, J.M.; Lamarca, B. AT1-AA (angiotensin II type 1 receptor agonistic autoantibody) blockade prevents preeclamptic symptoms in placental ischemic rats. Hypertension 2018, 71, 886–893. [Google Scholar] [CrossRef]
- Lillegard, K.E. The Role of Complement System Activation in Placental Ischemia-Induced Hypertension. Ph.D. Thesis, University of Minnesota, Minneapolis, MN, USA, 2013. [Google Scholar]
- Szalai, G.; Xu, Y.; Romero, R.; Chaiworapongsa, T.; Xu, Z.; Chiang, P.J.; Ahn, H.; Sundell, B.; Plazyo, O.; Jiang, Y. In vivo experiments reveal the good, the bad and the ugly faces of sFlt-1 in pregnancy. PLoS ONE 2014, 9, e110867. [Google Scholar] [CrossRef]
- Banadakoppa, M.; Balakrishnan, M.; Yallampalli, C. Upregulation and release of soluble fms-like tyrosine kinase receptor 1 mediated by complement activation in human syncytiotrophoblast cells. Am. J. Reproduct. Immunol. 2018, 80, e13033. [Google Scholar] [CrossRef]
- Ito, Y.; Matsuoka, K.; Uesato, T.; Sago, H.; Okamoto, A.; Nakazawa, A.; Hata, K. Increased expression of perforin, granzyme B, and C5b-9 in villitis of unknown etiology. Placenta 2015, 36, 531–537. [Google Scholar] [CrossRef]
- Huppertz, B. An updated view on the origin and use of angiogenic biomarkers for preeclampsia. Expert Rev. Mol. Diagn. 2018, 18, 1053–1061. [Google Scholar] [CrossRef] [PubMed]
- Niewold, P.; Dijkstra, D.J.; Cai, Y.; Goletti, D.; Palmieri, F.; Van Meijgaarden, K.E.; Verreck, F.A.; Akkerman, O.W.; Hofland, R.W.; Delemarre, E.M. Identification of circulating monocytes as producers of tuberculosis disease biomarker C1q. Sci. Rep. 2023, 13, 11617. [Google Scholar] [CrossRef]
- Schumaker, V.N.; Hanson, D.C.; Kilchherr, E.; Phillips, M.L.; Poon, P.H. A molecular mechanism for the activation of the first component of complement by immune complexes. Mol. Immunol. 1986, 23, 557–565. [Google Scholar] [CrossRef] [PubMed]
- Potlukova, E.; Kralikova, P. Complement component c1q and anti-c1q antibodies in theory and in clinical practice. Scand. J. Immunol. 2008, 67, 423–430. [Google Scholar] [CrossRef] [PubMed]
- Agostinis, C.; Zito, G.; Toffoli, M.; Balduit, A.; Mangogna, A.; Belmonte, B.; Romano, F.; Stampalija, T.; Salviato, T.; Defendi, F. Evaluation of levels, specificity and pathophysiology of anti-C1q autoantibodies in pregnancy. J. Reprod. Immunol. 2023, 159, 104081. [Google Scholar] [CrossRef]
- Agostinis, C.; Bulla, R.; Tripodo, C.; Gismondi, A.; Stabile, H.; Bossi, F.; Guarnotta, C.; Garlanda, C.; De Seta, F.; Spessotto, P. An alternative role of C1q in cell migration and tissue remodeling: Contribution to trophoblast invasion and placental development. J. Immunol. 2010, 185, 4420–4429. [Google Scholar] [CrossRef] [PubMed]
- Singh, J.; Ahmed, A.; Girardi, G. Role of complement component C1q in the onset of preeclampsia in mice. Hypertension 2011, 58, 716–724. [Google Scholar] [CrossRef]
- Agostinis, C.; Mangogna, A.; Balduit, A.; Kishore, U.; Bulla, R. A non-redundant role of complement protein C1q in normal and adverse pregnancy. Explor. Immunol. 2022, 2, 622–636. [Google Scholar] [CrossRef]
- Agostinis, C.; Stampalija, T.; Tannetta, D.; Loganes, C.; Vecchi Brumatti, L.; De Seta, F.; Celeghini, C.; Radillo, O.; Sargent, I.; Tedesco, F. Complement component C1q as potential diagnostic but not predictive marker of preeclampsia. Am. J. Reprod. Immunol. 2016, 76, 475–481. [Google Scholar] [CrossRef]
- Stoiber, H.; Ebenbichler, C.; Dierich, M.P.; Thiele, N.M.; Arlaud, G.J. The envelope glycoprotein of HIV-1 gp120 and human complement protein C1q bind to the same peptides derived from three different regions of gp41, the transmembrane glycoprotein of HIV-1, and share antigenic homology. Eur. J. Immunol. 1994, 24, 294–300. [Google Scholar] [CrossRef] [PubMed]
- Thielens, N.; Bally, I.; Ebenbichler, C.; Dierich, M.; Arlaud, G. Interaction of C1 with HIV-1. Behring Inst. Mitteilungen 1994, 93, 165–170. [Google Scholar]
- Dobó, J.; Szakács, D.; Oroszlán, G.; Kortvely, E.; Kiss, B.; Boros, E.; Szász, R.; Závodszky, P.; Gál, P.; Pál, G. MASP-3 is the exclusive pro-factor D activator in resting blood: The lectin and the alternative complement path-ways are fundamentally linked. Sci. Rep. 2016, 6, 31877. [Google Scholar] [CrossRef] [PubMed]
- Nørgaard-Pedersen, C.; Rom, L.; Steffensen, R.; Kesmodel, U.; Christiansen, O. Plasma level of man-nose-binding lectin is associated with the risk of recurrent pregnancy loss but not pregnancy outcome after the diagnosis. Hum. Reprod. Open 2022, 2022, hoac024. [Google Scholar] [CrossRef] [PubMed]
- Than, N.G.; Romero, R.; Erez, O.; Kusanovic, J.P.; Tarca, A.L.; Edwin, S.S.; Kim, J.S.; Hassan, S.S.; Espinoza, J.; Mittal, P. A role for mannose-binding lectin, a component of the innate immune system in pre-eclampsia. Am. J. Reprod. Immunol. 2008, 60, 333–345. [Google Scholar] [CrossRef]
- Celik, N.; Ozan, H. Maternal serum mannose-binding lectin in severe preeclampsia. Clin. Experiment. Obstet. Gynecol. 2008, 35, 179–182. [Google Scholar]
- Sziller, I.; Babula, O.; Hupuczi, P.; Nagy, B.; Rigo, B.; Szabo, G.; Papp, Z.; Linhares, I.; Witkin, S. Man-nose-binding lectin (MBL) codon 54 gene polymorphism protects against development of pre-eclampsia, HELLP syndrome and pre-eclampsia-associated intrauterine growth restriction. MHR Basic Sci. Reprod. Med. 2007, 13, 281–285. [Google Scholar] [CrossRef] [PubMed]
- Giannubilo, S.R.; Landi, B.; Ciavattini, A. Preeclampsia: What could happen in a subsequent pregnancy? Obstet. Gynecol. Surv. 2014, 69, 747–762. [Google Scholar] [CrossRef]
- Jin, S.; Wu, C.; Zhang, Y. Complement in structure and immune homeostasis in placenta. Authorea Prepr. 2021. [Google Scholar] [CrossRef]
- Kitzmiller, J.L.; Watt, N.; Driscoll, S.G. Decidual arteriopathy in hypertension and diabetes in pregnancy: Immunofluorescent studies. Am. J. Obstet. Gynecol. 1981, 141, 773–779. [Google Scholar] [CrossRef] [PubMed]
- Girardi, G.; Yarilin, D.; Thurman, J.M.; Holers, V.M.; Salmon, J.E. Complement activation induces dysregulation of angiogenic factors and causes fetal rejection and growth restriction. J. Exp. Med. 2006, 203, 2165–2175. [Google Scholar] [CrossRef] [PubMed]
- Ballegaard, V.; Haugaard, A.; Garred, P.; Nielsen, S.; Munthe-Fog, L. The lectin pathway of complement: Advantage or disadvantage in HIV pathogenesis? Clin. Immunol. 2014, 154, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Thielens, N.M.; Tacnet-Delorme, P.; Arlaud, G.J. Interaction of C1q and mannan-binding lectin with viruses. Immunobiology 2002, 205, 563–574. [Google Scholar] [CrossRef]
- Eisen, S.; Dzwonek, A.; Klein, N.J. Mannose-binding lectin in HIV infection. Future Virol. 2008, 3, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Turner, M.W. Mannose-binding lectin: The pluripotent molecule of the innate immune system. Immunol. Today 1996, 17, 532–540. [Google Scholar] [CrossRef] [PubMed]
- Heggelund, L.; Mollnes, T.E.; Ueland, T.; Christophersen, B.; Aukrust, P.; Frøland, S.S. Mannose-binding lectin in HIV infection: Relation to disease progression and highly active antiretroviral therapy. J. Acquir. Immune Defic. Syndr. 2003, 32, 354–361. [Google Scholar] [CrossRef] [PubMed]
- Wallis, R.; Dodds, A.W.; Mitchell, D.A.; Sim, R.B.; Reid, K.B.; Schwaeble, W.J. Molecular interactions between MASP-2, C4, and C2 and their activation fragments leading to complement activation via the lectin path-way. J. Biol. Chem. 2007, 282, 7844–7851. [Google Scholar] [CrossRef] [PubMed]
- Bentley, D.R. Primary structure of human complement component C2 Homology to two unrelated protein families. Biochem. J. 1986, 239, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Johnson, U.; Gustavii, B. Complement components in normal pregnancy. Acta Pathol. Microbiol. Scand. Ser. C Immunol. 1987, 95, 97–99. [Google Scholar] [CrossRef]
- Richardson, A.J.; Islam, F.A.; Guymer, R.H.; Baird, P.N. Analysis of rare variants in the complement component 2 (C2) and factor B (BF) genes refine association for age-related macular degeneration (AMD). Investig. Ophthalmol. Vis. Sci. 2009, 50, 540–543. [Google Scholar] [CrossRef]
- Lintner, K.E.; Wu, Y.L.; Yang, Y.; Spencer, C.H.; Hauptmann, G.; Hebert, L.A.; Atkinson, J.P.; Yu, C.Y. Early components of the complement classical activation pathway in human systemic autoimmune diseases. Front. Immunol. 2016, 7, 36. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, R.L. The complement system: Its importance in the host response to viral infection. Microbiol. Rev. 1982, 46, 71–85. [Google Scholar] [CrossRef] [PubMed]
- Huson, M.A.; Kalkman, R.; Stolp, S.M.; Janssen, S.; Alabi, A.S.; Beyeme, J.O.; Van Der Poll, T.; Grobusch, M.P.J.I. The impact of HIV on presentation and outcome of bacterial sepsis and other causes of acute febrile illness in Gabon. Infection 2015, 43, 443–451. [Google Scholar] [CrossRef] [PubMed]
- Huson, M.A.; Wouters, D.; Van Mierlo, G.; Grobusch, M.P.; Zeerleder, S.S.; Van Der Poll, T. HIV coinfection enhances complement activation during sepsis. J. Infect. Dis. 2015, 212, 474–483. [Google Scholar] [CrossRef] [PubMed]
- Muller-Eberhard, H.; Nilsson, U. Relation of a ß1-glycoprotein of human serum to the complement system. J. Exp. Med. 1960, 111, 217–234. [Google Scholar] [CrossRef] [PubMed]
- Elvington, M.; Liszewski, M.K.; Bertram, P.; Kulkarni, H.S.; Atkinson, J.P. A C3 (H2O) recycling pathway is a component of the intracellular complement system. J. Clin. Investig. 2017, 127, 970–981. [Google Scholar] [CrossRef]
- Ricklin, D.; Reis, E.S.; Mastellos, D.C.; Gros, P.; Lambris, J.D. Complement component C3—The “Swiss Army Knife” of innate immunity and host defense. Immunol. Rev. 2016, 274, 33–58. [Google Scholar] [CrossRef]
- Daigo, K.; Inforzato, A.; Barajon, I.; Garlanda, C.; Bottazzi, B.; Meri, S.; Mantovani, A. Pentraxins in the activation and regulation of innate immunity. Immunol. Rev. 2016, 274, 202–217. [Google Scholar] [CrossRef]
- Sjöberg, A.; Trouw, L.; Blom, A. Complement activation and inhibition: A delicate balance. Trends Immunol. 2009, 30, 83–90. [Google Scholar] [CrossRef]
- Lokki, A.I.; Kaartokallio, T.; Holmberg, V.; Onkamo, P.; Koskinen, L.L.; Saavalainen, P.; Heinonen, S.; Kajantie, E.; Kere, J.; Kivinen, K. Analysis of complement C3 gene reveals susceptibility to severe preeclampsia. Front. Immunol. 2017, 8, 589. [Google Scholar] [CrossRef] [PubMed]
- Girardi, G.; Lingo, J.J.; Fleming, S.D.; Regal, J.F. Essential role of complement in pregnancy: From implantation to parturition and beyond. Front. Immunol. 2020, 11, 1681. [Google Scholar] [CrossRef]
- Kennelly, M.A.; Killeen, S.L.; Phillips, C.M.; Alberdi, G.; Lindsay, K.L.; Mehegan, J.; Cronin, M.; Mcauliffe, F.M. Maternal C3 complement and C-reactive protein and pregnancy and fetal outcomes: A secondary analysis of the PEARS RCT-An mHealth-supported, lifestyle intervention among pregnant women with overweight and obesity. Cytokine 2022, 149, 155748. [Google Scholar] [CrossRef]
- Talagrand-Reboul, E.; Raffetin, A.; Zachary, P.; Jaulhac, B.; Eldin, C. Immunoserological diagnosis of human borrelioses: Current knowledge and perspectives. Front. Cell. Infect. Microbiol. 2020, 10, 241. [Google Scholar] [CrossRef]
- Boteva, L.; Morris, D.L.; Cortés-Hernández, J.; Martin, J.; Vyse, T.J.; Fernando, M.M. Genetically determined partial complement C4 deficiency states are not independent risk factors for SLE in UK and Spanish populations. Am. J. Hum. Genet. 2012, 90, 445–456. [Google Scholar] [CrossRef] [PubMed]
- Mortensen, S.; Kidmose, R.T.; Petersen, S.V.; Szilágyi, Á.; Prohászka, Z.; Andersen, G.R. Structural Basis for the Function of Complement Component C4 within the Classical and Lectin Pathways of Complement. J. Immunol. 2015, 194, 5488–5496. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Liu, M. Complement C4, Infections, and Autoimmune Diseases. Front. Immunol. 2021, 14, 694928. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Dai, S.; Gordon, J.; Qin, X. Complement and HIV-1 infection/HIV-associated neurocognitive disorders. J. Neurovirol. 2014, 20, 184–198. [Google Scholar] [CrossRef]
- Holers, V.M. Complement and its receptors: New insights into human disease. Annu. Rev. Immunol. 2014, 32, 433–459. [Google Scholar] [CrossRef]
- Merle, N.S.; Church, S.E.; Fremeaux-Bacchi, V.; Roumenina, L.T. Complement system part I–molecular mechanisms of activation and regulation. Front. Immunol. 2015, 6, 262. [Google Scholar] [CrossRef]
- Girardi, G.; Prohászka, Z.; Bulla, R.; Tedesco, F.; Scherjon, S. Complement activation in animal and human pregnancies as a model for immunological recognition. Mol. Immunol. 2011, 48, 1621–1630. [Google Scholar] [CrossRef] [PubMed]
- Levine, R.J.; Maynard, S.E.; Qian, C.; Lim, K.-H.; England, L.J.; Yu, K.F.; Schisterman, E.F.; Thadhani, R.; Sachs, B.P.; Epstein, F.H. Circulating angiogenic factors and the risk of preeclampsia. N. Engl. J. Med. 2004, 350, 672–683. [Google Scholar] [CrossRef] [PubMed]
- Rana, S.; Hacker, M.R.; Modest, A.M.; Salahuddin, S.; Lim, K.-H.; Verlohren, S.; Perschel, F.H.; Karumanchi, S.A. Circulating angiogenic factors and risk of adverse maternal and perinatal outcomes in twin pregnancies with suspected preeclampsia. Hypertension 2012, 60, 451–458. [Google Scholar] [CrossRef] [PubMed]
- Fukuzawa, T.; Sampei, Z.; Haraya, K.; Ruike, Y.; Shida-Kawazoe, M.; Shimizu, Y.; Gan, S.W.; Irie, M.; Tsuboi, Y.; Tai, H. Long lasting neutralization of C5 by SKY59, a novel recycling antibody, is a potential therapy for complement-mediated diseases. Sci. Rep. 2017, 7, 1080. [Google Scholar] [CrossRef]
- Alrahmani, L.; Willrich, M.A.V. The complement alternative pathway and preeclampsia. Curr. Hypertens. Rep. 2018, 20, 40. [Google Scholar] [CrossRef]
- Kelly, R.J.; Höchsmann, B.; Szer, J.; Kulasekararaj, A.; De Guibert, S.; Röth, A.; Weitz, I.C.; Armstrong, E.; Risitano, A.M.; Patriquin, C.J. Eculizumab in pregnant patients with paroxysmal nocturnal hemoglobinuria. N. Engl. J. Med. 2015, 373, 1032–1039. [Google Scholar] [CrossRef]
- Guilhaudis, L.; Jacobs, A.; Caffrey, M. Solution structure of the HIV gp120 C5 domain. Eur. J. Biochem. 2002, 269, 4860–4867. [Google Scholar] [CrossRef]
- Sullivan, B.L.; Takefman, D.M.; Spear, G.T. Complement can neutralize HIV-1 plasma virus by a C5-independent mechanism. Virology 1998, 248, 173–181. [Google Scholar] [CrossRef]
- Wagner, E.; Frank, M.M. Therapeutic potential of complement modulation. Nat. Rev. Drug Discov. 2010, 9, 43–56. [Google Scholar] [CrossRef]
- Joller, N.; Hafler, J.P.; Brynedal, B.; Kassam, N.; Spoerl, S.; Levin, S.D.; Sharpe, A.H.; Kuchroo, V.K. Cutting edge: TIGIT has T cell-intrinsic inhibitory functions. J. Immunol. 2011, 186, 1338–1342. [Google Scholar] [CrossRef]
- Dudkina, N.V.; Spicer, B.A.; Reboul, C.F.; Conroy, P.J.; Lukoyanova, N.; Elmlund, H.; Law, R.H.; Ekkel, S.M.; Kondos, S.C.; Goode, R.J. Structure of the poly-C9 component of the complement membrane attack complex. Nat. Commun. 2016, 7, 10588. [Google Scholar] [CrossRef] [PubMed]
- Spicer, C.D.; Jumeaux, C.; Gupta, B.; Stevens, M.M. Peptide and protein nanoparticle conjugates: Versatile platforms for biomedical applications. Chem. Soc. Rev. 2018, 47, 3574–3620. [Google Scholar] [CrossRef] [PubMed]
- Vlaicu, S.I.; Tegla, C.A.; Cudrici, C.D.; Danoff, J.; Madani, H.; Sugarman, A.; Niculescu, F.; Mircea, P.A.; Rus, V.; Rus, H. Role of C5b-9 complement complex and response gene to complement-32 (RGC-32) in cancer. Immunol. Res. 2013, 56, 109–121. [Google Scholar] [CrossRef] [PubMed]
- Brewster, J.A.; Orsi, N.M.; Gopichandran, N.; Mcshane, P.; Ekbote, U.V.; Walker, J.J. Gestational effects on host inflammatory response in normal and pre-eclamptic pregnancies. Eur. J. Obstet. Gynecol. Reprod. Biol. 2008, 140, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.-J.; Xu, G.-F.; Lv, M.; Zhou, H.; Huang, H.-F.; Luo, Q. Predictive Value of Maternal Serum Biomarkers for Preeclampsia and Birth Weight: A Case–Control Study in Chinese Pregnant Women. J. Women’s Health 2018, 27, 1519–1524. [Google Scholar] [CrossRef]
- Kalumba, V.; Moodley, J.; Naidoo, T. Is the prevalence of pre-eclampsia affected by HIV/AIDS? A retro-spective case-control study: Cardiovascular topics. Cardiovasc. J. Afr. 2013, 24, 24–27. [Google Scholar] [CrossRef]
- Jiang, T.T.; Chaturvedi, V.; Ertelt, J.M.; Kinder, J.M.; Clark, D.R.; Valent, A.M.; Xin, L.; Way, S.S. Regula-tory T cells: A new key for further unlocking the enigma of fetal tolerance and pregnancy complications. J. Immunol. 2014, 192, 4949–4956. [Google Scholar] [CrossRef]
- Lazarus, J.V.; Safreed-Harmon, K.; Barton, S.E.; Costagliola, D.; Dedes, N.; Del Amo Valero, J.; Gatell, J.M.; Baptista-Leite, R.; Mendão, L.; Porter, K. Beyond viral suppression of HIV–the new quality of life frontier. BMC Med. 2016, 14, 94. [Google Scholar] [CrossRef]











Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Govender, S.; David, M.; Naicker, T. Is the Complement System Dysregulated in Preeclampsia Comorbid with HIV Infection? Int. J. Mol. Sci. 2024, 25, 6232. https://doi.org/10.3390/ijms25116232
Govender S, David M, Naicker T. Is the Complement System Dysregulated in Preeclampsia Comorbid with HIV Infection? International Journal of Molecular Sciences. 2024; 25(11):6232. https://doi.org/10.3390/ijms25116232
Chicago/Turabian StyleGovender, Sumeshree, Mikyle David, and Thajasvarie Naicker. 2024. "Is the Complement System Dysregulated in Preeclampsia Comorbid with HIV Infection?" International Journal of Molecular Sciences 25, no. 11: 6232. https://doi.org/10.3390/ijms25116232