
NOTCH3 and Pulmonary Arterial Hypertension


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
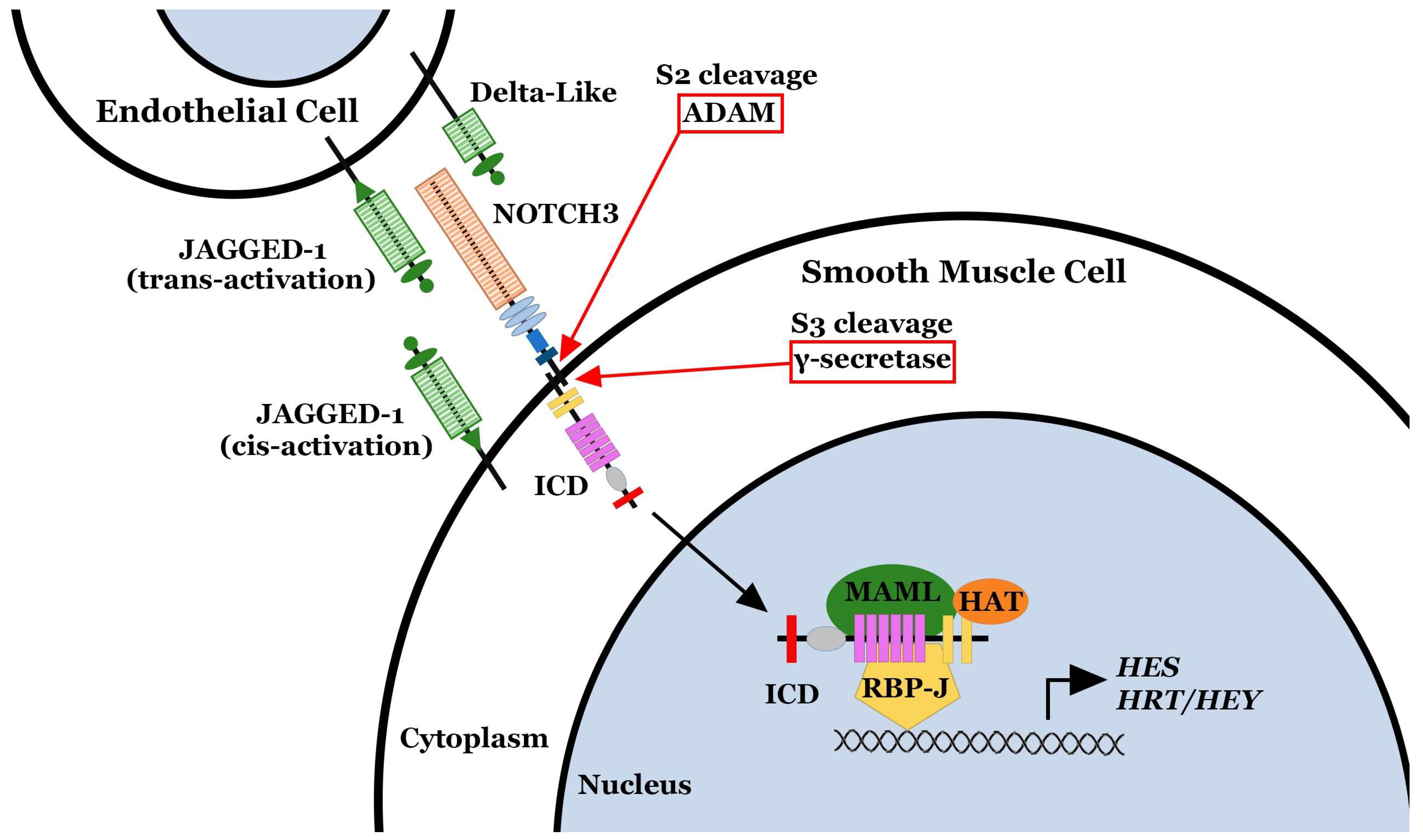
2. NOTCH3 and Pulmonary Arterial Hypertension
3. NOTCH3 Is a Marker of PAH Disease Severity
4. Activation of NOTCH3 via JAG-1 Increases the Proliferation of Small Pulmonary Artery Smooth Muscle Cells
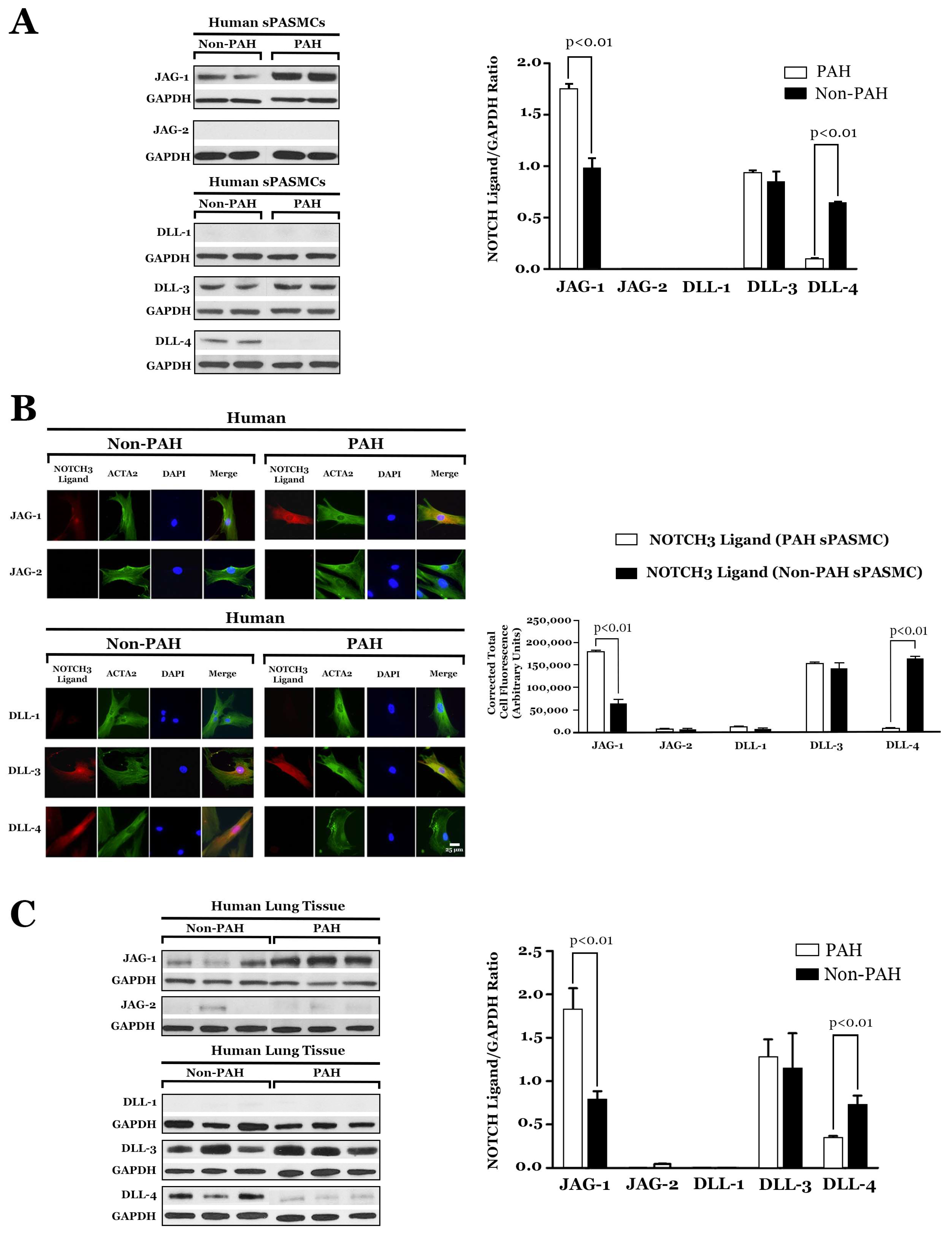
5. JAG-1 and NOTCH3 Signaling Is Constitutive in PAH
6. Role of DLL-4 in the Development of PAH
7. HES-5, a Downstream Effector of NOTCH3 Signaling, Drives the Pulmonary Hypertensive Phenotype
Proliferation of PAH sPASMCs Is Dependent on NOTCH3-HES-5
8. Notch3−/− Mice Do Not Develop PH
9. Notch3 Inhibition with a γ-Secretase Inhibitor Reverses PH
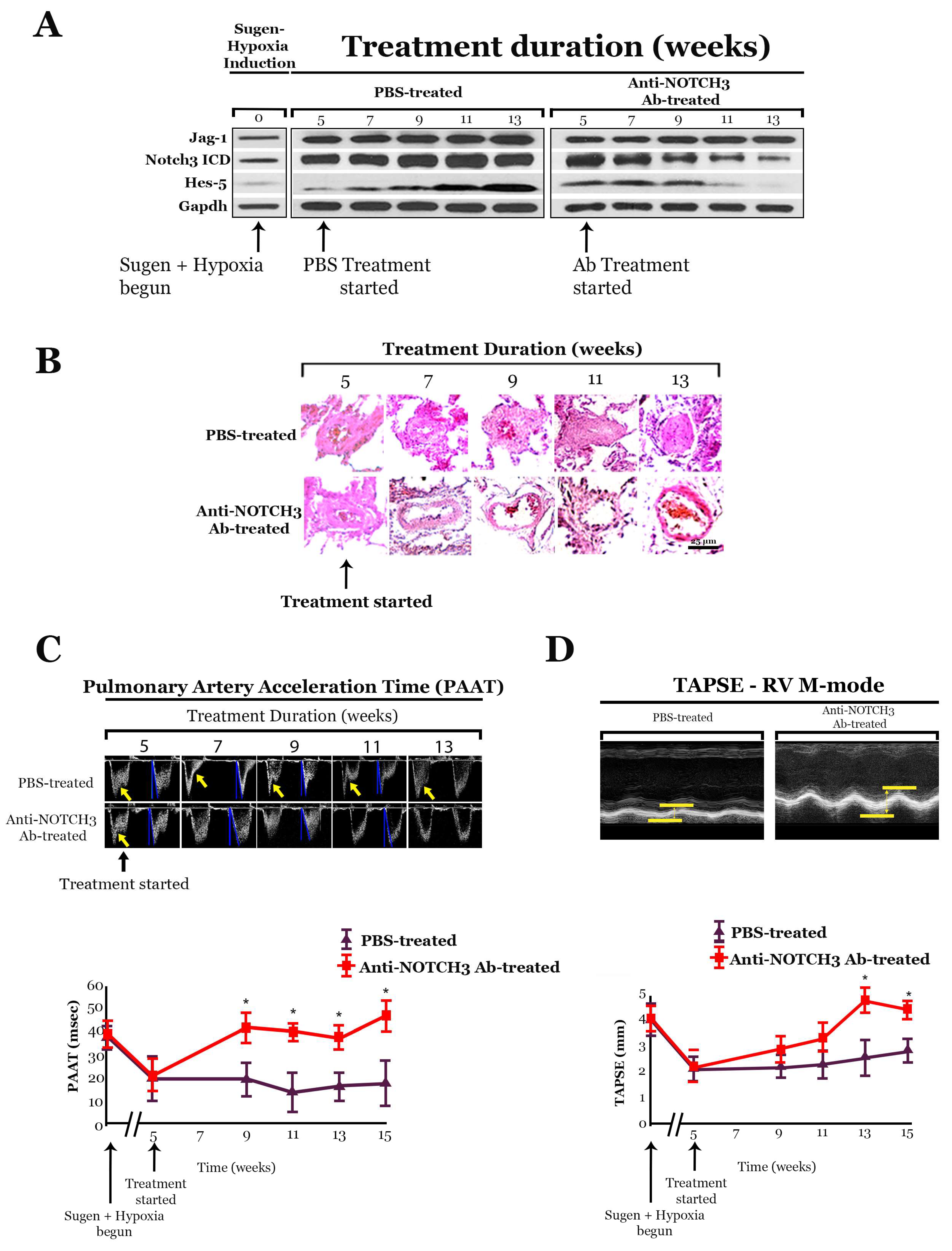
10. Monoclonal Antibody That Blocks JAG-1/NOTCH3 Binding Inhibits NOTCH3 Cleavage
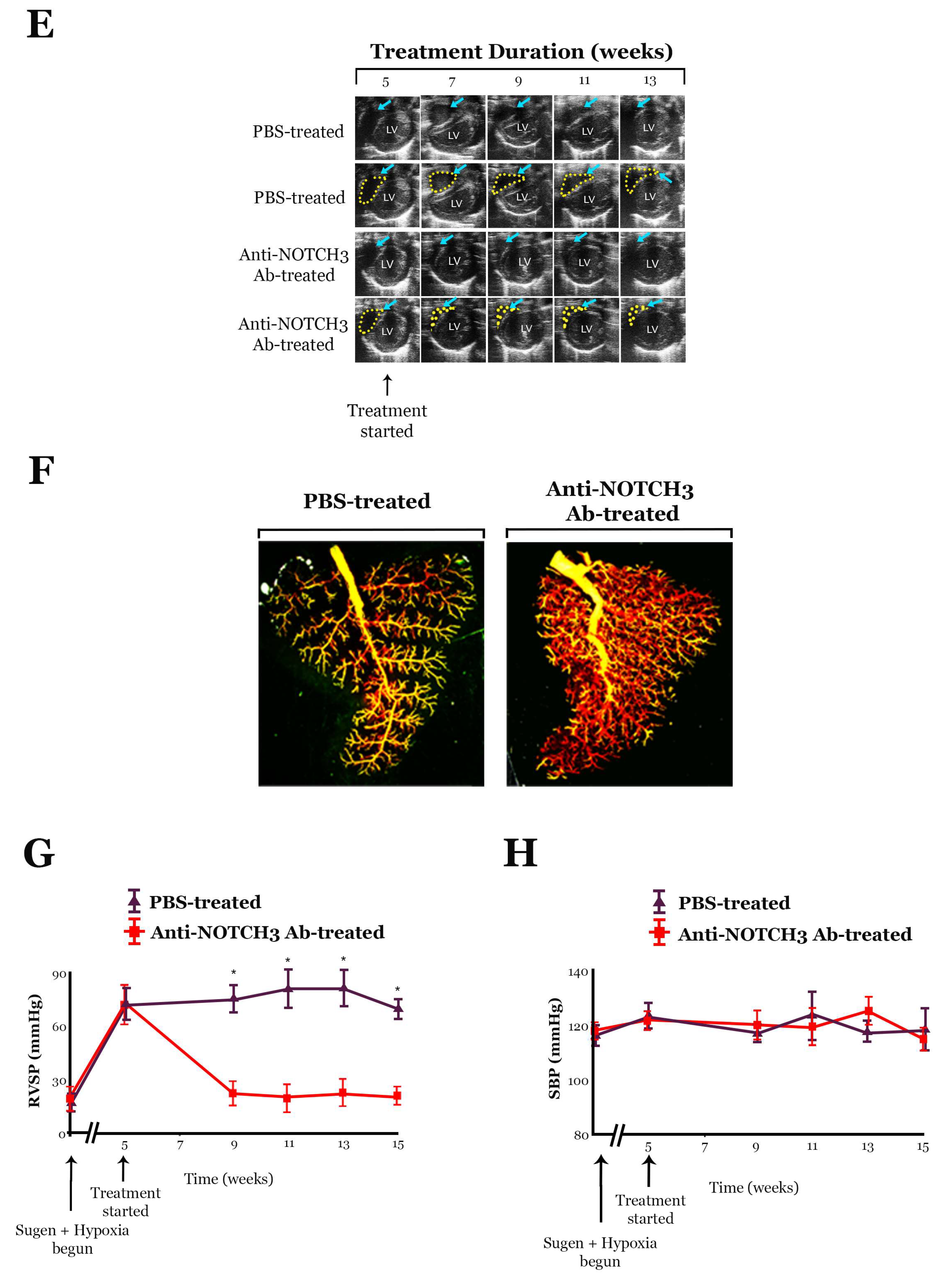
10.1. Anti-NOTCH3 Ab 28042 Treatment Reverses PH in Mice
10.2. Treatment with Anti-NOTCH3 Ab 28042 Reverses PH in Rats
11. Conclusions
- (1)
- Constitutive NOTCH3 ICD expression induces sPASMC proliferation. This notion, coupled with the finding that NOTCH3 is overexpressed at the mRNA and NOTCH3 ICD at the protein level in the lungs of humans with PAH, supports the critical role of NOTCH3 signaling in mediating sPASMC proliferation seen in this disease. Previous studies have also established a link between NOTCH3 signaling and the coordinated regulation of HES-5 effector expression in the context of sPASMC proliferation. It has been found that siRNA inhibition of HES5 expression causes a decrease in sPASMC proliferation and a shift in gene expression in vSMCs toward a more differentiated phenotype.
- (2)
- Human PAH vasculopathy is characterized by high steady-state levels of NOTCH3 and the downstream effector, HES-5, in vSMCs lining small precapillary pulmonary arteries. Additionally, there is a strong correlation between NOTCH3 signaling (protein levels of NOTCH3 ICD) and the magnitude of PAH in humans and PH in animals. NOTCH3 ICD protein levels in lung tissue can serve as a specific molecular marker of PAH severity in humans and PH in rodents.
- (3)
- Notch3 signaling is required for the development of hypoxic PH in rodents. Notch3−/− mice are resistant to the development of PH and are unable to generate a medial hyperplastic response to hypoxia because Notch3-mediated proliferative and anti-apoptotic effects on sPASMC are required for the development of pulmonary vascular medial thickening.
- (4)
- Other forms of rodent PH, including Sugen PH, can be effectively treated by blocking Notch3 cleavage with the γ-secretase inhibitor, DAPT, or a monoclonal anti-NOTCH3-specific antibody that blocks JAG-1 binding to NOTCH3.
Author Contributions
Funding
Conflicts of Interest
References
- Badagliacca, R.; Poscia, R.; Pezzuto, B.; Nocioni, M.; Mezzapesa, M.; Francone, M.; Giannetta, E.; Papa, S.; Gambardella, C.; Sciomer, S.; et al. Right ventricular remodeling in idiopathic pulmonary arterial hypertension: Adaptive versus maladaptive morphology. J. Heart Lung Transplant. 2015, 34, 395–403. [Google Scholar] [CrossRef] [PubMed]
- Hardegree, E.L.; Sachdev, A.; Fenstad, E.R.; Villarraga, H.R.; Frantz, R.P.; McGoon, M.D.; Oh, J.K.; Ammash, N.M.; Connolly, H.M.; Eidem, B.W.; et al. Impaired left ventricular mechanics in pulmonary arterial hypertension: Identification of a cohort at high risk. Circ. Heart Fail. 2013, 6, 748–755. [Google Scholar] [CrossRef] [PubMed]
- Farber, H.W.; Loscalzo, J. Pulmonary arterial hypertension. N. Engl. J. Med. 2004, 351, 1655–1665. [Google Scholar] [CrossRef] [PubMed]
- Steffes, L.C.; Froistad, A.A.; Andruska, A.; Boehm, M.; McGlynn, M.; Zhang, F.; Zhang, W.; Hou, D.; Tian, X.; Miquerol, L.; et al. A Notch3-Marked Subpopulation of Vascular Smooth Muscle Cells Is the Cell of Origin for Occlusive Pulmonary Vascular Lesions. Circulation 2020, 142, 1545–1561. [Google Scholar] [CrossRef] [PubMed]
- Humbert, M.; Guignabert, C.; Bonnet, S.; Dorfmüller, P.; Klinger, J.R.; Nicolls, M.R.; Olschewski, A.J.; Pullamsetti, S.S.; Schermuly, R.T.; Stenmark, K.R.; et al. Pathology and pathobiology of pulmonary hypertension: State of the art and research perspectives. Eur. Respir. J. 2019, 53, 1801887. [Google Scholar] [CrossRef] [PubMed]
- Rabinovitch, M.; Guignabert, C.; Humbert, M.; Nicolls, M.R. Inflammation and immunity in the pathogenesis of pulmonary arterial hypertension. Circ. Res. 2014, 115, 165–175. [Google Scholar] [CrossRef]
- Morris, H.E.; Neves, K.B.; Montezano, A.C.; MacLean, M.R.; Touyz, R.M. Notch3 signalling and vascular remodelling in pulmonary arterial hypertension. Clin. Sci. 2019, 133, 2481–2498. [Google Scholar] [CrossRef]
- Li, X.; Zhang, X.; Leathers, R.; Makino, A.; Huang, C.; Parsa, P.; Macias, J.; Yuan, J.X.; Jamieson, S.W.; Thistlethwaite, P.A. Notch3 signaling promotes the development of pulmonary arterial hypertension. Nat. Med. 2009, 15, 1289–1297. [Google Scholar] [CrossRef] [PubMed]
- Hosseini-Alghaderi, S.; Baron, M. Notch3 in Development, Health and Disease. Biomolecules 2020, 10, 485. [Google Scholar] [CrossRef]
- Joutel, A.; Andreux, F.; Gaulis, S.; Domenga, V.; Cecillon, M.; Battail, N.; Piga, N.; Chapon, F.; Godfrain, C.; Tournier-Lasserve, E. The ectodomain of the Notch3 receptor accumulates within the cerebrovasculature of CADASIL patients. J. Clin. Investig. 2000, 105, 597–605. [Google Scholar] [CrossRef] [PubMed]
- Kopan, R.; Ilagan, M.X.G. The Canonical Notch Signaling Pathway: Unfolding the Activation Mechanism. Cell 2009, 137, 216–233. [Google Scholar] [CrossRef]
- Seib, E.; Klein, T. The role of ligand endocytosis in notch signalling. Biol. Cell 2021, 113, 401–418. [Google Scholar] [CrossRef]
- Nandagopal, N.; Santat, L.A.; Elowitz, M.B. Cis-activation in the Notch signaling pathway. eLife 2019, 8, e37880. [Google Scholar] [CrossRef]
- Zhang, Y.; Hernandez, M.; Gower, J.; Winicki, N.; Morataya, X.; Alvarez, S.; Yuan, J.X.; Shyy, J.; Thistlethwaite, P.A. JAGGED-NOTCH3 signaling in vascular remodeling in pulmonary arterial hypertension. Sci. Transl. Med. 2022, 14, eabl5471. [Google Scholar] [CrossRef]
- Andersson, E.R.; Sandberg, R.; Lendahl, U. Notch signaling: Simplicity in design, versatility in function. Development 2011, 138, 3593–3612. [Google Scholar] [CrossRef]
- Boucher, J.; Gridley, T.; Liaw, L. Molecular pathways of notch signaling in vascular smooth muscle cells. Front. Physiol. 2012, 3, 81. [Google Scholar] [CrossRef]
- Sprinzak, D.; Blacklow, S.C. Biophysics of Notch Signaling. Annu. Rev. Biophys. 2021, 50, 157–189. [Google Scholar] [CrossRef]
- Morris, H.E.; Neves, K.B.; Nilsen, M.; Montezano, A.C.; MacLean, M.R.; Touyz, R.M. Notch3/Hes5 Induces Vascular Dysfunction in Hypoxia-Induced Pulmonary Hypertension Through ER Stress and Redox-Sensitive Pathways. Hypertension 2023, 80, 1683–1696. [Google Scholar] [CrossRef]
- Hounjet, J.; Vooijs, M. The Role of Intracellular Trafficking of Notch Receptors in Ligand-Independent Notch Activation. Biomolecules 2021, 11, 1369. [Google Scholar] [CrossRef]
- Ryan, J.; Bloch, K.; Archer, S.L. Rodent models of pulmonary hypertension: Harmonisation with the world health organisation’s categorisation of human PH. Int. J. Clin. Pract. 2011, 65, 15–34. [Google Scholar] [CrossRef]
- Ryan, J.J.; Marsboom, G.; Archer, S.L. Rodent models of group 1 pulmonary hypertension. In Pharmacotherapy of Pulmonary Hypertension; Springer: Berlin/Heidelberg, Germany, 2013; pp. 105–149. [Google Scholar]
- Voelkel, N.F.; Bogaard, H.J. Sugen, hypoxia and the lung circulation. Pulm. Circ. 2021, 11, 20458940211051188. [Google Scholar] [CrossRef]
- Tobal, R.; Potjewijd, J.; Empel, V.P.M.V.; Ysermans, R.; Schurgers, L.J.; Reutelingsperger, C.P.; Damoiseaux, J.G.M.C.; Paassen, P.V. Vascular Remodeling in Pulmonary Arterial Hypertension: The Potential Involvement of Innate and Adaptive Immunity. Review. Front. Med. 2021, 8, 806899. [Google Scholar] [CrossRef]
- Lesage, F.; Deng, Y.; Renesme, L.; Sauvestre, F.; Ben Fadel, N.; Zhong, S.; Vadivel, A.; Jankov, R.P.; Stewart, D.J.; Thébaud, B. Characterization of a New Monocrotaline Rat Model to Study Chronic Neonatal Pulmonary Hypertension. Am. J. Respir. Cell Mol. Biol. 2021, 65, 331–334. [Google Scholar] [CrossRef]
- Spyropoulos, F.; Vitali, S.H.; Touma, M.; Rose, C.D.; Petty, C.R.; Levy, P.; Kourembanas, S.; Christou, H. Echocardiographic markers of pulmonary hemodynamics and right ventricular hypertrophy in rat models of pulmonary hypertension. Pulm. Circ. 2020, 10, 2045894020910976. [Google Scholar] [CrossRef]
- Yamamura, H.; Yamamura, A.; Ko, E.A.; Pohl, N.M.; Smith, K.A.; Zeifman, A.; Powell, F.L.; Thistlethwaite, P.A.; Yuan, J.X. Activation of Notch signaling by short-term treatment with Jagged-1 enhances store-operated Ca2+ entry in human pulmonary arterial smooth muscle cells. Am. J. Physiol. Cell Physiol. 2014, 306, C871–C878. [Google Scholar] [CrossRef]
- Zhou, B.; Lin, W.; Long, Y.; Yang, Y.; Zhang, H.; Wu, K.; Chu, Q. Notch signaling pathway: Architecture, disease, and therapeutics. Signal Transduct. Target. Ther. 2022, 7, 95. [Google Scholar] [CrossRef]
- Yuan, K.; Agarwal, S.; Chakraborty, A.; Condon, D.F.; Patel, H.; Zhang, S.; Huang, F.; Mello, S.A.; Kirk, O.I.; Vasquez, R.; et al. Lung Pericytes in Pulmonary Vascular Physiology and Pathophysiology. Compr. Physiol. 2021, 11, 2227–2247. [Google Scholar] [CrossRef]
- McKeage, M.J.; Kotasek, D.; Markman, B.; Hidalgo, M.; Millward, M.J.; Jameson, M.B.; Harris, D.L.; Stagg, R.J.; Kapoun, A.M.; Xu, L.; et al. Phase IB Trial of the Anti-Cancer Stem Cell DLL4-Binding Agent Demcizumab with Pemetrexed and Carboplatin as First-Line Treatment of Metastatic Non-Squamous NSCLC. Target. Oncol. 2018, 13, 89–98. [Google Scholar] [CrossRef]
- Coleman, R.L.; Handley, K.F.; Burger, R.; Molin, G.Z.D.; Stagg, R.; Sood, A.K.; Moore, K.N. Demcizumab combined with paclitaxel for platinum-resistant ovarian, primary peritoneal, and fallopian tube cancer: The SIERRA open-label phase Ib trial. Gynecol. Oncol. 2020, 157, 386–391. [Google Scholar] [CrossRef]
- Chiorean, E.G.; LoRusso, P.; Strother, R.M.; Diamond, J.R.; Younger, A.; Messersmith, W.A.; Adriaens, L.; Liu, L.; Kao, R.J.; DiCioccio, A.T.; et al. A Phase I First-in-Human Study of Enoticumab (REGN421), a Fully Human Delta-like Ligand 4 (Dll4) Monoclonal Antibody in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2015, 21, 2695–2703. [Google Scholar] [CrossRef]
- Lee, J.; Kim, S.; Hong, J.; Park, Y.; Park, J.; Kang, W.; Lee, K.-W.; Kim, J.W.; Kim, J.-W.; Kim, S.H.; et al. Abstract P02-03: Phase Ia/Ib dose-escalation study of ABL001 (CTX-009, bispecific antibody targeting DLL4 and VEGF-A) as a single agent in patients with advanced solid tumors. Mol. Cancer Ther. 2021, 20 (Suppl. S12), P02-03. [Google Scholar] [CrossRef]
- Fu, S.; Corr, B.R.; Culm-Merdek, K.; Mockbee, C.; Youssoufian, H.; Stagg, R.; Naumann, R.W.; Wenham, R.M.; Rosengarten, R.D.; Benjamin, L.; et al. Phase Ib Study of Navicixizumab Plus Paclitaxel in Patients With Platinum-Resistant Ovarian, Primary Peritoneal, or Fallopian Tube Cancer. J. Clin. Oncol. 2022, 40, 2568–2577. [Google Scholar] [CrossRef]
- Gordon, M.S.; Nemunaitis, J.; Barve, M.; Wainberg, Z.A.; Hamilton, E.P.; Ramanathan, R.K.; Sledge, G.W., Jr.; Yue, H.; Morgan-Lappe, S.E.; Blaney, M.; et al. Phase I Open-Label Study Evaluating the Safety, Pharmacokinetics, and Preliminary Efficacy of Dilpacimab in Patients with Advanced Solid Tumors. Mol. Cancer Ther. 2021, 20, 1988–1995. [Google Scholar] [CrossRef]
- Jimeno, A.; Moore, K.N.; Gordon, M.; Chugh, R.; Diamond, J.R.; Aljumaily, R.; Mendelson, D.; Kapoun, A.M.; Xu, L.; Stagg, R.; et al. A first-in-human phase 1a study of the bispecific anti-DLL4/anti-VEGF antibody navicixizumab (OMP-305B83) in patients with previously treated solid tumors. Investig. New Drugs 2019, 37, 461–472. [Google Scholar] [CrossRef]
- You, W.-K.; Schuetz, T.J.; Lee, S.H. Targeting the DLL/Notch signaling pathway in cancer: Challenges and advances in clinical development. Mol. Cancer Ther. 2023, 22, 3–11. [Google Scholar] [CrossRef]
- Wang, S.; Zhu, G.; Jiang, D.; Rhen, J.; Li, X.; Liu, H.; Lyu, Y.; Tsai, P.; Rose, Y.; Nguyen, T.; et al. Reduced Notch1 Cleavage Promotes the Development of Pulmonary Hypertension. Hypertension 2022, 79, 79–92. [Google Scholar] [CrossRef]
- Gallardo-Vara, E.; Ntokou, A.; Dave, J.M.; Jovin, D.G.; Saddouk, F.Z.; Greif, D.M. Vascular pathobiology of pulmonary hypertension. J. Heart Lung Transplant. 2023, 42, 544–552. [Google Scholar] [CrossRef]
- Fisher, S.A. Smooth muscle diversity in the vascular system. In The Vasculome from Many, One; Elsevier: Amsterdam, The Netherlands, 2022; pp. 45–55. [Google Scholar]
- Popper, H.; Murer, B.; Popper, H.; Murer, B. Hypertension and Vasculopathies. In Pulmonary Pathology: A Practical Guide; Springer: Berlin/Heidelberg, Germany, 2020; pp. 565–580. [Google Scholar]
- Pagliaro, L.; Sorrentino, C.; Roti, G. Targeting Notch trafficking and processing in cancers. Cells 2020, 9, 2212. [Google Scholar] [CrossRef]
- Rajagopal, S.; Yen-Rei, A.Y. The pathobiology of pulmonary arterial hypertension. Cardiol. Clin. 2022, 40, 1–12. [Google Scholar] [CrossRef]
- Vladar, E.K.; Kunimoto, K.; Rojas-Hernandez, L.S.; Spano, J.M.; Sellers, Z.M.; Joo, N.S.; Cooney, R.A.; Axelrod, J.D.; Milla, C.E. Notch signaling inactivation by small molecule γ-secretase inhibitors restores the multiciliated cell population in the airway epithelium. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2023, 324, L771–L782. [Google Scholar] [CrossRef]
- Nie, P.; Vartak, A.; Li, Y.-M. γ-Secretase inhibitors and modulators: Mechanistic insights into the function and regulation of γ-Secretase. In Seminars in Cell & Developmental Biology; Elsevier: Amsterdam, The Netherlands, 2020; pp. 43–53. [Google Scholar]
- Zhang, Y.; Xie, X.; Zhu, Y.; Liu, L.; Feng, W.; Pan, Y.; Zhai, C.; Ke, R.; Li, S.; Song, Y.; et al. Inhibition of Notch3 prevents monocrotaline-induced pulmonary arterial hypertension. Exp. Lung Res. 2015, 41, 435–443. [Google Scholar] [CrossRef] [PubMed]
- Majumder, S.; Crabtree, J.S.; Golde, T.E.; Minter, L.M.; Osborne, B.A.; Miele, L. Targeting Notch in oncology: The path forward. Nat. Rev. Drug Discov. 2021, 20, 125–144. [Google Scholar] [CrossRef] [PubMed]
- Hahn, L.D.; Papamatheakis, D.G.; Fernandes, T.M.; Poch, D.S.; Yang, J.; Shen, J.; Hoh, C.K.; Hsiao, A.; Kerr, K.M.; Pretorius, V. Multidisciplinary approach to chronic thromboembolic pulmonary hypertension: Role of radiologists. Radiographics 2022, 43, e220078. [Google Scholar] [CrossRef] [PubMed]
- Czerwonka, A.; Kałafut, J.; Nees, M. Modulation of Notch Signaling by Small-Molecular Compounds and Its Potential in Anticancer Studies. Cancers 2023, 15, 4563. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.H.; Ma, J.L.; Ding, D.; Ma, Y.J.; Wei, Y.P.; Jing, Z.C. Experimental animal models of pulmonary hypertension: Development and challenges. Anim. Models Exp. Med. 2022, 5, 207–216. [Google Scholar] [CrossRef]
- Dou, Y.; Pizarro, T.; Zhou, L. Organoids as a model system for studying notch signaling in intestinal epithelial homeostasis and intestinal cancer. Am. J. Pathol. 2022, 192, 1347–1357. [Google Scholar] [CrossRef] [PubMed]
- Zhao, A.; Qin, H.; Sun, M.; Tang, M.; Mei, J.; Ma, K.; Fu, X. Chemical conversion of human epidermal stem cells into intestinal goblet cells for modeling mucus-microbe interaction and therapy. Sci. Adv. 2021, 7, eabb2213. [Google Scholar] [CrossRef]
- Gounder, M.; Ratan, R.; Alcindor, T.; Schöffski, P.; Graaf, W.T.v.d.; Wilky, B.A.; Riedel, R.F.; Lim, A.; Smith, L.M.; Moody, S.; et al. Nirogacestat, a γ-Secretase Inhibitor for Desmoid Tumors. N. Engl. J. Med. 2023, 388, 898–912. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Winicki, N.M.; Puerta, C.; Besse, C.E.; Zhang, Y.; Thistlethwaite, P.A. NOTCH3 and Pulmonary Arterial Hypertension. Int. J. Mol. Sci. 2024, 25, 6248. https://doi.org/10.3390/ijms25116248
Winicki NM, Puerta C, Besse CE, Zhang Y, Thistlethwaite PA. NOTCH3 and Pulmonary Arterial Hypertension. International Journal of Molecular Sciences. 2024; 25(11):6248. https://doi.org/10.3390/ijms25116248
Chicago/Turabian StyleWinicki, Nolan M., Cristian Puerta, Casandra E. Besse, Yu Zhang, and Patricia A. Thistlethwaite. 2024. "NOTCH3 and Pulmonary Arterial Hypertension" International Journal of Molecular Sciences 25, no. 11: 6248. https://doi.org/10.3390/ijms25116248
APA StyleWinicki, N. M., Puerta, C., Besse, C. E., Zhang, Y., & Thistlethwaite, P. A. (2024). NOTCH3 and Pulmonary Arterial Hypertension. International Journal of Molecular Sciences, 25(11), 6248. https://doi.org/10.3390/ijms25116248