
Investigating Contributions of Canonical Transient Receptor Potential Channel 3 to Hippocampal Hyperexcitability and Seizure-Induced Neuronal Cell Death

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
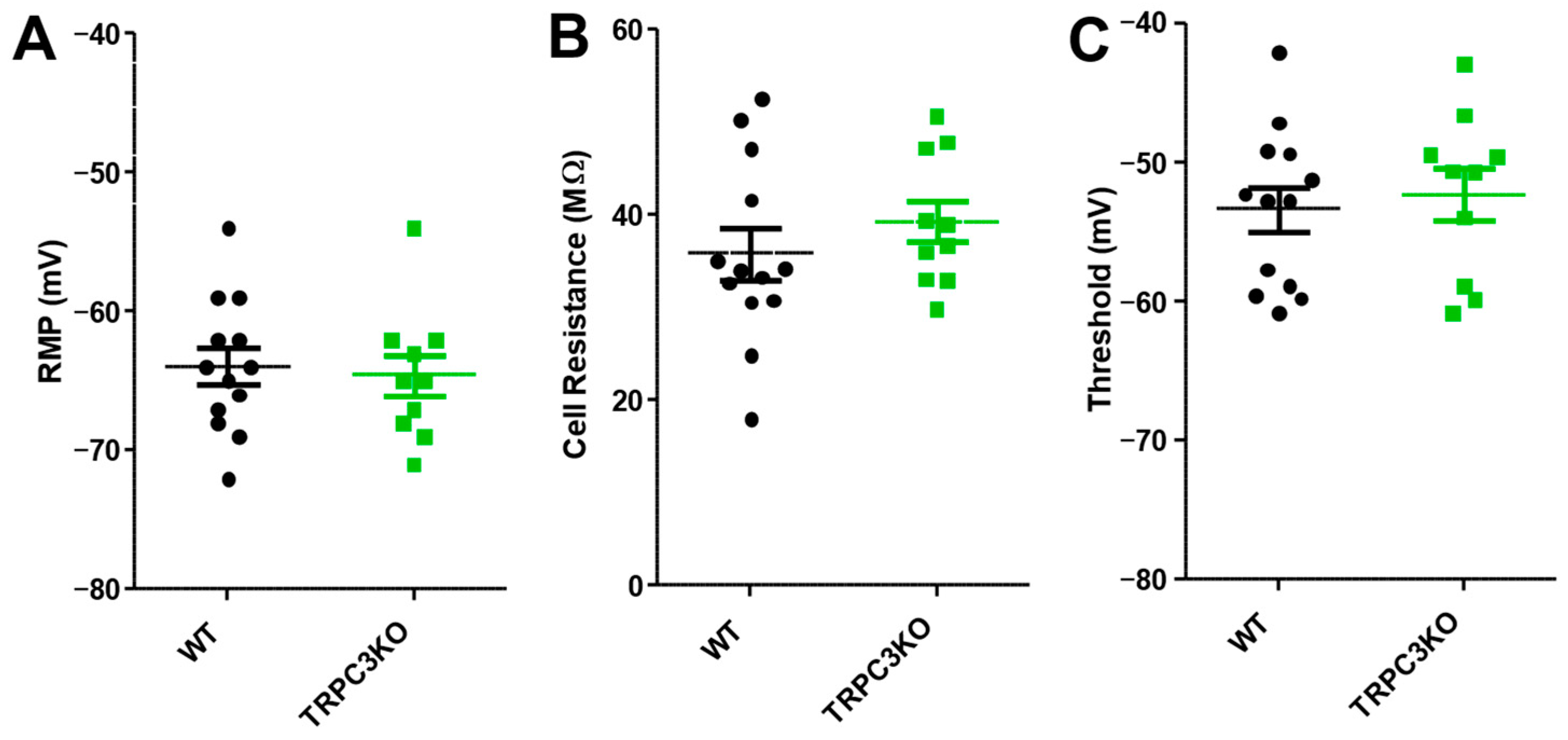
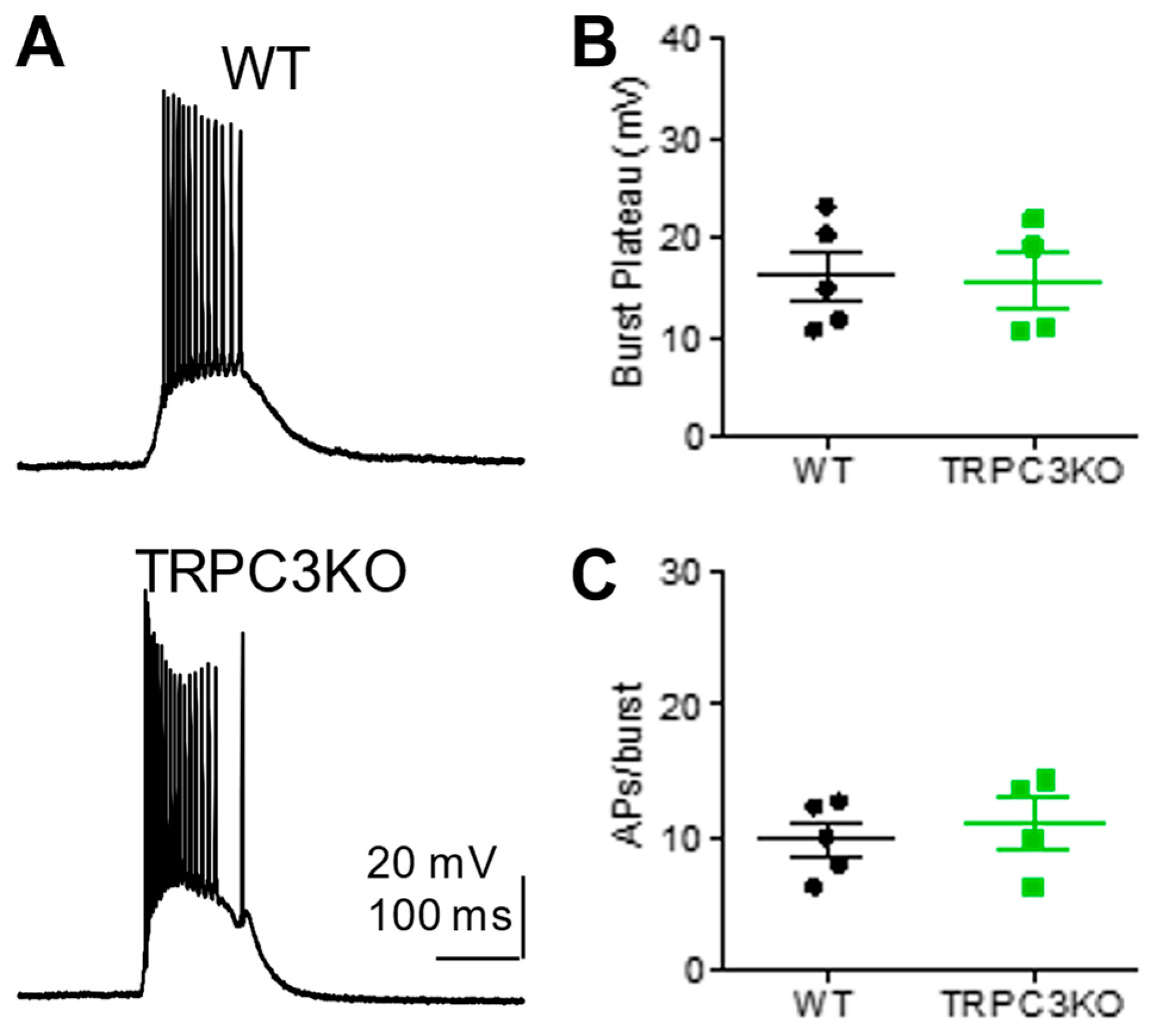
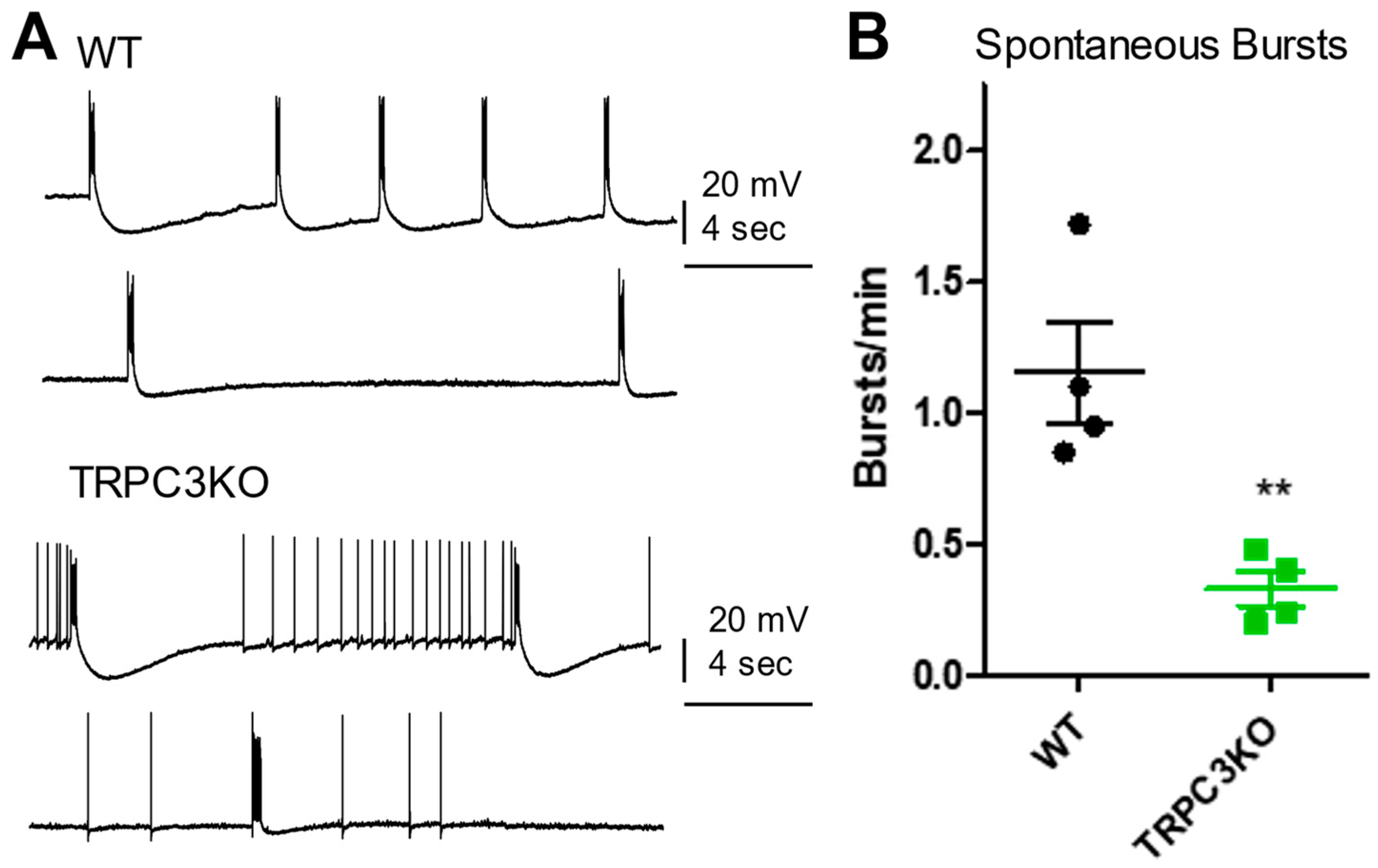
2.1. Comparison of Electrophysiological Properties and Epileptiform Discharges of CA1 Pyramidal Neurons in WT and TRPC3KO Mice
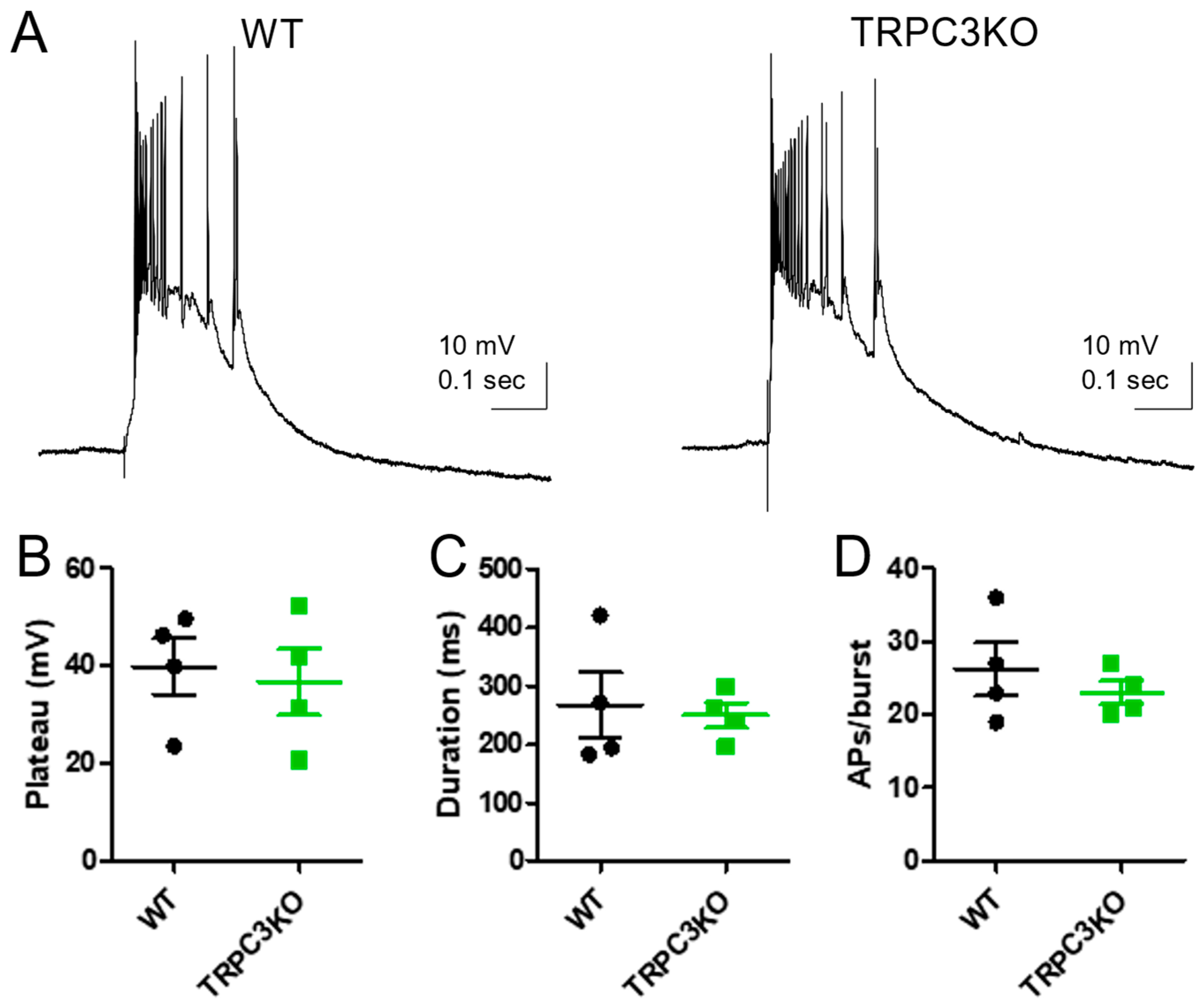
2.2. Comparison of Electrophysiological Properties and Epileptiform Discharges of CA3 Pyramidal Neurons in WT and TRPC3KO Mice
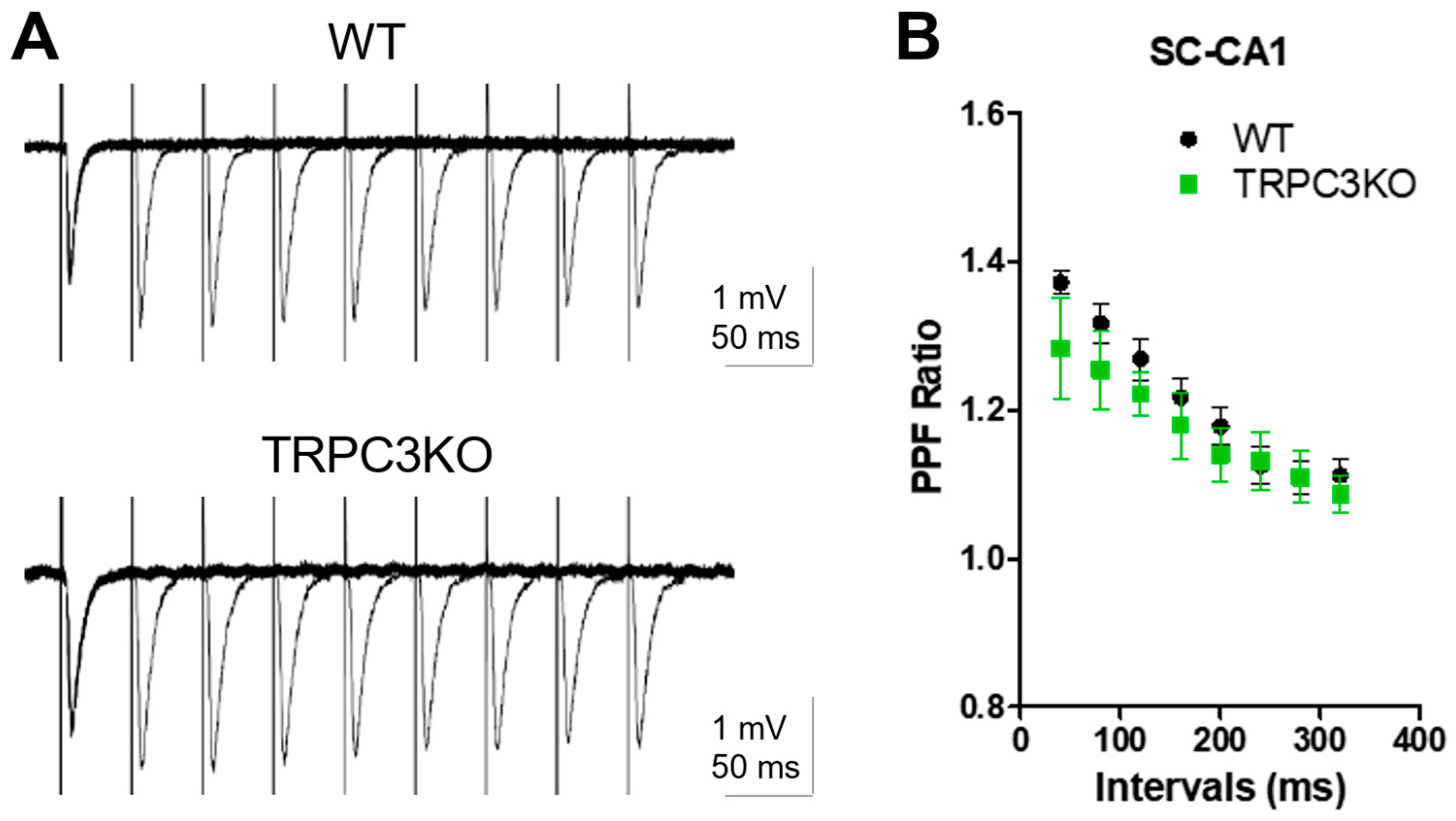
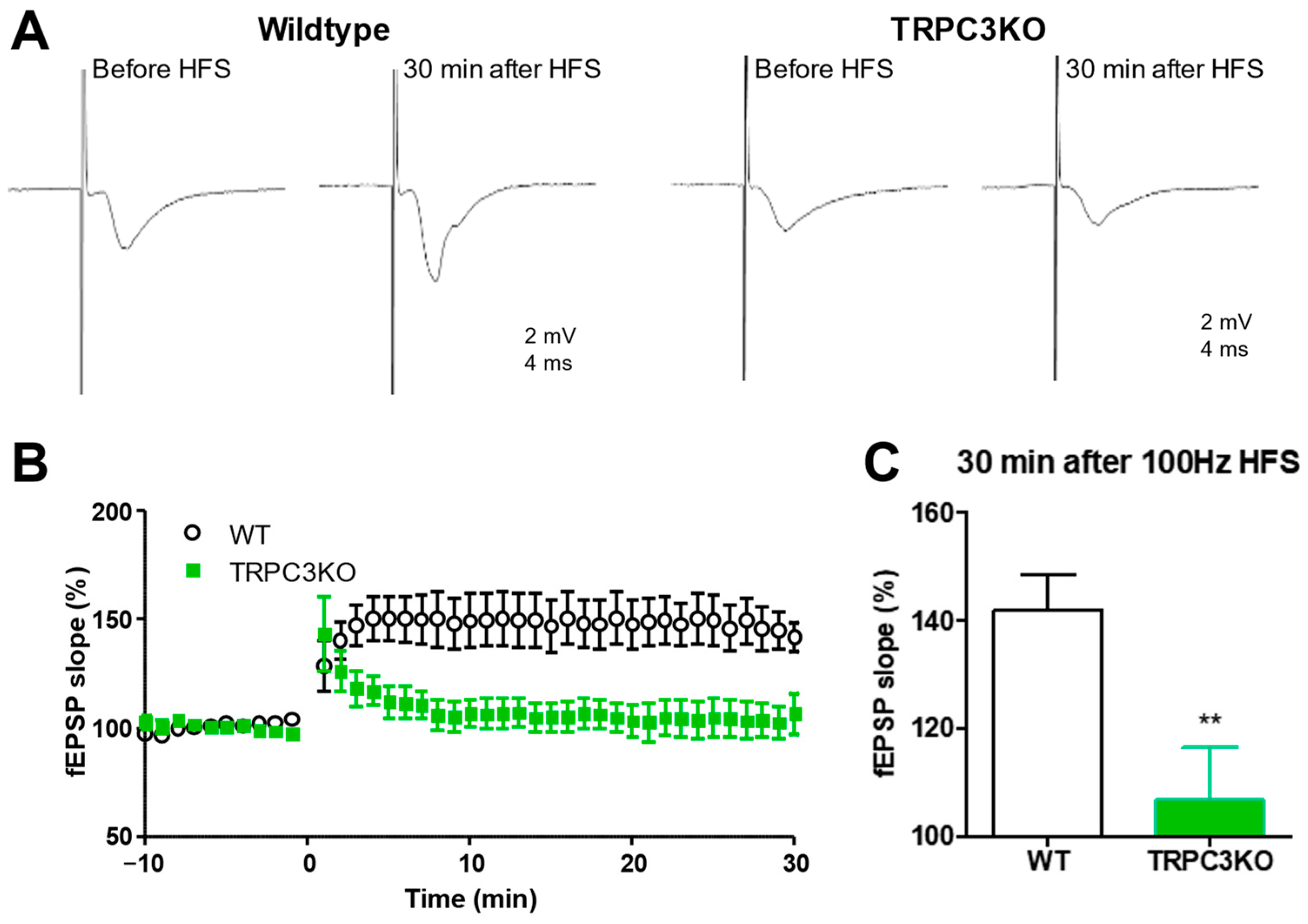
2.3. Comparison of Short-Term and Long-Term Synaptic Plasticity in WT and TRPC3KO Mice
2.4. Comparison of SE-Induced Neuronal Cell Death in WT and TRPC3KO Mice
3. Discussion
4. Materials and Methods
4.1. Electrophysiological Recordings
4.2. Fluoro-Jade C (FJC) Staining
4.3. Nissl Counterstaining and Stereological Analysis of Cell Death
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pedersen, S.F.; Owsianik, G.; Nilius, B. TRP Channels: An Overview. Cell Calcium 2005, 38, 233–252. [Google Scholar] [CrossRef]
- Clapham, D.E.; Runnels, L.W.; Strübing, C. The TRP Ion Channel Family. Nat. Rev. Neurosci. 2001, 2, 387–396. [Google Scholar] [CrossRef]
- Birnbaumer, L. The TRPC Class of Ion Channels: A Critical Review of Their Roles in Slow, Sustained Increases in Intracellular Ca2+ Concentrations. Annu. Rev. Pharmacol. Toxicol. 2009, 49, 395–426. [Google Scholar] [CrossRef]
- Zhu, X.; Jiang, M.; Peyton, M.; Boulay, G.; Hurst, R.; Stefani, E.; Birnbaumer, L. Trp, a Novel Mammalian Gene Family Essential for Agonist-Activated Capacitative Ca2+ Entry. Cell 1996, 85, 661–671. [Google Scholar] [CrossRef]
- Montell, C. Drosophila TRP Channels. Pflügers Arch.-Eur. J. Physiol. 2005, 451, 19–28. [Google Scholar] [CrossRef]
- Vazquez, G.; Wedel, B.J.; Aziz, O.; Trebak, M.; Putney, J.W. The Mammalian TRPC Cation Channels. Biochim. Biophys. Acta-Mol. Cell Res. 2004, 1742, 21–36. [Google Scholar] [CrossRef]
- Yildirim, E.; Birnbaumer, L. TRPC2: Molecular Biology and Functional Importance. Handb. Exp. Pharmacol. 2007, 179, 53–75. [Google Scholar] [CrossRef]
- Kollewe, A.; Schwarz, Y.; Oleinikov, K.; Raza, A.; Haupt, A.; Wartenberg, P.; Wyatt, A.; Boehm, U.; Ectors, F.; Bildl, W.; et al. Subunit Composition, Molecular Environment, and Activation of Native TRPC Channels Encoded by Their Interactomes. Neuron 2022, 110, 4162–4175.e7. [Google Scholar] [CrossRef]
- Putney, J.W. Physiological Mechanisms of TRPC Activation. Pflugers Arch. 2005, 451, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Zheng, F.; Phelan, K.D. The Role of Canonical Transient Receptor Potential Channels in Seizure and Excitotoxicity. Cells 2014, 3, 288–303. [Google Scholar] [CrossRef] [PubMed]
- Fan, C.; Choi, W.; Sun, W.; Du, J.; Lu, W. Structure of the Human Lipid-Gated Cation Channel TRPC3. eLife 2018, 7, e36852. [Google Scholar] [CrossRef]
- Bon, R.S.; Beech, D.J. In Pursuit of Small Molecule Chemistry for Calcium-Permeable Non-Selective TRPC Channels—Mirage or Pot of Gold? Br. J. Pharmacol. 2013, 170, 459–474. [Google Scholar] [CrossRef]
- Bon, R.S.; Wright, D.J.; Beech, D.J.; Sukumar, P. Pharmacology of TRPC Channels and Its Potential in Cardiovascular and Metabolic Medicine. Annu. Rev. Pharmacol. Toxicol. 2022, 62, 427–446. [Google Scholar] [CrossRef]
- Cole, B.A.; Becker, E.B.E. Modulation and Regulation of Canonical Transient Receptor Potential 3 (TRPC3) Channels. Cells 2023, 12, 2215. [Google Scholar] [CrossRef]
- Dietrich, A.; Mederos y Schnitzler, M.; Emmel, J.; Kalwa, H.; Hofmann, T.; Gudermann, T. N-Linked Protein Glycosylation Is a Major Determinant for Basal TRPC3 and TRPC6 Channel Activity. J. Biol. Chem. 2003, 278, 47842–47852. [Google Scholar] [CrossRef]
- Hofmann, T.; Obukhov, A.G.; Schaefer, M.; Harteneck, C.; Gudermann, T.; Schultz, G. Direct Activation of Human TRPC6 and TRPC3 Channels by Diacylglycerol. Nature 1999, 397, 259–263. [Google Scholar] [CrossRef]
- Kawasaki, B.T.; Liao, Y.; Birnbaumer, L. Role of Src in C3 Transient Receptor Potential Channel Function and Evidence for a Heterogeneous Makeup of Receptor- and Store-Operated Ca2+ Entry Channels. Proc. Natl. Acad. Sci. USA 2006, 103, 335–340. [Google Scholar] [CrossRef]
- Yao, X.; Garland, C.J. Recent Developments in Vascular Endothelial Cell Transient Receptor Potential Channels. Circ. Res. 2005, 97, 853–863. [Google Scholar] [CrossRef]
- Hartmann, J.; Dragicevic, E.; Adelsberger, H.; Henning, H.A.; Sumser, M.; Abramowitz, J.; Blum, R.; Dietrich, A.; Freichel, M.; Flockerzi, V.; et al. TRPC3 Channels Are Required for Synaptic Transmission and Motor Coordination. Neuron 2008, 59, 392–398. [Google Scholar] [CrossRef]
- Pitsch, J.; Opitz, T.; Borm, V.; Woitecki, A.; Staniek, M.; Beck, H.; Becker, A.J.; Schoch, S. The Presynaptic Active Zone Protein RIM1 Controls Epileptogenesis Following Status Epilepticus. J. Neurosci. 2012, 32, 12384–12395. [Google Scholar] [CrossRef]
- Li, H.S.; Xu, X.Z.; Montell, C. Activation of a TRPC3-Dependent Cation Current through the Neurotrophin BDNF. Neuron 1999, 24, 261–273. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Calfa, G.; Inoue, T.; Amaral, M.D.; Pozzo-Miller, L. Activity-Dependent Release of Endogenous BDNF from Mossy Fibers Evokes a TRPC3 Current and Ca2+ Elevations in CA3 Pyramidal Neurons. J. Neurophysiol. 2010, 103, 2846–2856. [Google Scholar] [CrossRef] [PubMed]
- Amaral, M.D.; Pozzo-Miller, L. TRPC3 Channels Are Necessary for Brain-Derived Neurotrophic Factor to Activate a Nonselective Cationic Current and to Induce Dendritic Spine Formation. J. Neurosci. 2007, 27, 5179–5189. [Google Scholar] [CrossRef] [PubMed]
- Leal, G.; Afonso, P.M.; Salazar, I.L.; Duarte, C.B. Regulation of Hippocampal Synaptic Plasticity by BDNF. Brain Res. 2015, 1621, 82–101. [Google Scholar] [CrossRef] [PubMed]
- Binder, D.K.; Croll, S.D.; Gall, C.M.; Scharfman, H.E. BDNF and Epilepsy: Too Much of a Good Thing? Trends Neurosci. 2001, 24, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Bathina, S.; Das, U.N. Brain-Derived Neurotrophic Factor and Its Clinical Implications. Arch. Med. Sci. 2015, 11, 1164–1178. [Google Scholar] [CrossRef] [PubMed]
- Phelan, K.D.; Shwe, U.T.; Cozart, M.A.; Wu, H.; Mock, M.M.; Abramowitz, J.; Birnbaumer, L.; Zheng, F. TRPC3 Channels Play a Critical Role in the Theta Component of Pilocarpine-Induced Status Epilepticus in Mice. Epilepsia 2017, 58, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Phelan, K.D.; Shwe, U.T.; Abramowitz, J.; Wu, H.; Rhee, S.W.; Howell, M.D.; Gottschall, P.E.; Freichel, M.; Flockerzi, V.; Birnbaumer, L.; et al. Canonical Transient Receptor Channel 5 (TRPC5) and TRPC1/4 Contribute to Seizure and Excitotoxicity by Distinct Cellular Mechanisms. Mol. Pharmacol. 2013, 83, 429–438. [Google Scholar] [CrossRef] [PubMed]
- Phelan, K.D.; Shwe, U.T.; Abramowitz, J.; Birnbaumer, L.; Zheng, F. Critical Role of Canonical Transient Receptor Potential Channel 7 in Initiation of Seizures. Proc. Natl. Acad. Sci. USA 2014, 111, 11533–11538. [Google Scholar] [CrossRef] [PubMed]
- Zakharenko, S.S.; Patterson, S.L.; Dragatsis, I.; Zeitlin, S.O.; Siegelbaum, S.A.; Kandel, E.R.; Morozov, A. Presynaptic BDNF Required for a Presynaptic but Not Postsynaptic Component of LTP at Hippocampal CA1-CA3 Synapses. Neuron 2003, 39, 975–990. [Google Scholar] [CrossRef]
- Zhou, F.-W.; Roper, S.N. TRPC3 Mediates Hyperexcitability and Epileptiform Activity in Immature Cortex and Experimental Cortical Dysplasia. J. Neurophysiol. 2014, 111, 1227–1237. [Google Scholar] [CrossRef] [PubMed]
- Scharfman, H.E. Hyperexcitability in Combined Entorhinal/Hippocampal Slices of Adult Rat after Exposure to Brain-Derived Neurotrophic Factor. J. Neurophysiol. 1997, 78, 1082–1095. [Google Scholar] [CrossRef] [PubMed]
- Gärtner, A.; Polnau, D.G.; Staiger, V.; Sciarretta, C.; Minichiello, L.; Thoenen, H.; Bonhoeffer, T.; Korte, M. Hippocampal Long-Term Potentiation Is Supported by Presynaptic and Postsynaptic Tyrosine Receptor Kinase B-Mediated Phospholipase Cgamma Signaling. J. Neurosci. 2006, 26, 3496–3504. [Google Scholar] [CrossRef]
- Phelan, K.D.; Mock, M.M.; Kretz, O.; Shwe, U.T.; Kozhemyakin, M.; Greenfield, L.J.; Dietrich, A.; Birnbaumer, L.; Freichel, M.; Flockerzi, V.; et al. Heteromeric Canonical Transient Receptor Potential 1 and 4 Channels Play a Critical Role in Epileptiform Burst Firing and Seizure-Induced Neurodegeneration. Mol. Pharmacol. 2012, 81, 384–392. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Phelan, K.D.; Shwe, U.T.; Wu, H.; Zheng, F. Investigating Contributions of Canonical Transient Receptor Potential Channel 3 to Hippocampal Hyperexcitability and Seizure-Induced Neuronal Cell Death. Int. J. Mol. Sci. 2024, 25, 6260. https://doi.org/10.3390/ijms25116260
Phelan KD, Shwe UT, Wu H, Zheng F. Investigating Contributions of Canonical Transient Receptor Potential Channel 3 to Hippocampal Hyperexcitability and Seizure-Induced Neuronal Cell Death. International Journal of Molecular Sciences. 2024; 25(11):6260. https://doi.org/10.3390/ijms25116260
Chicago/Turabian StylePhelan, Kevin D., U Thaung Shwe, Hong Wu, and Fang Zheng. 2024. "Investigating Contributions of Canonical Transient Receptor Potential Channel 3 to Hippocampal Hyperexcitability and Seizure-Induced Neuronal Cell Death" International Journal of Molecular Sciences 25, no. 11: 6260. https://doi.org/10.3390/ijms25116260