
The Importance of Molecular Genetic Testing for Precision Diagnostics, Management, and Genetic Counseling in MODY Patients

 , , ,
, , ,  , , and
, , and
Abstract
:1. Introduction
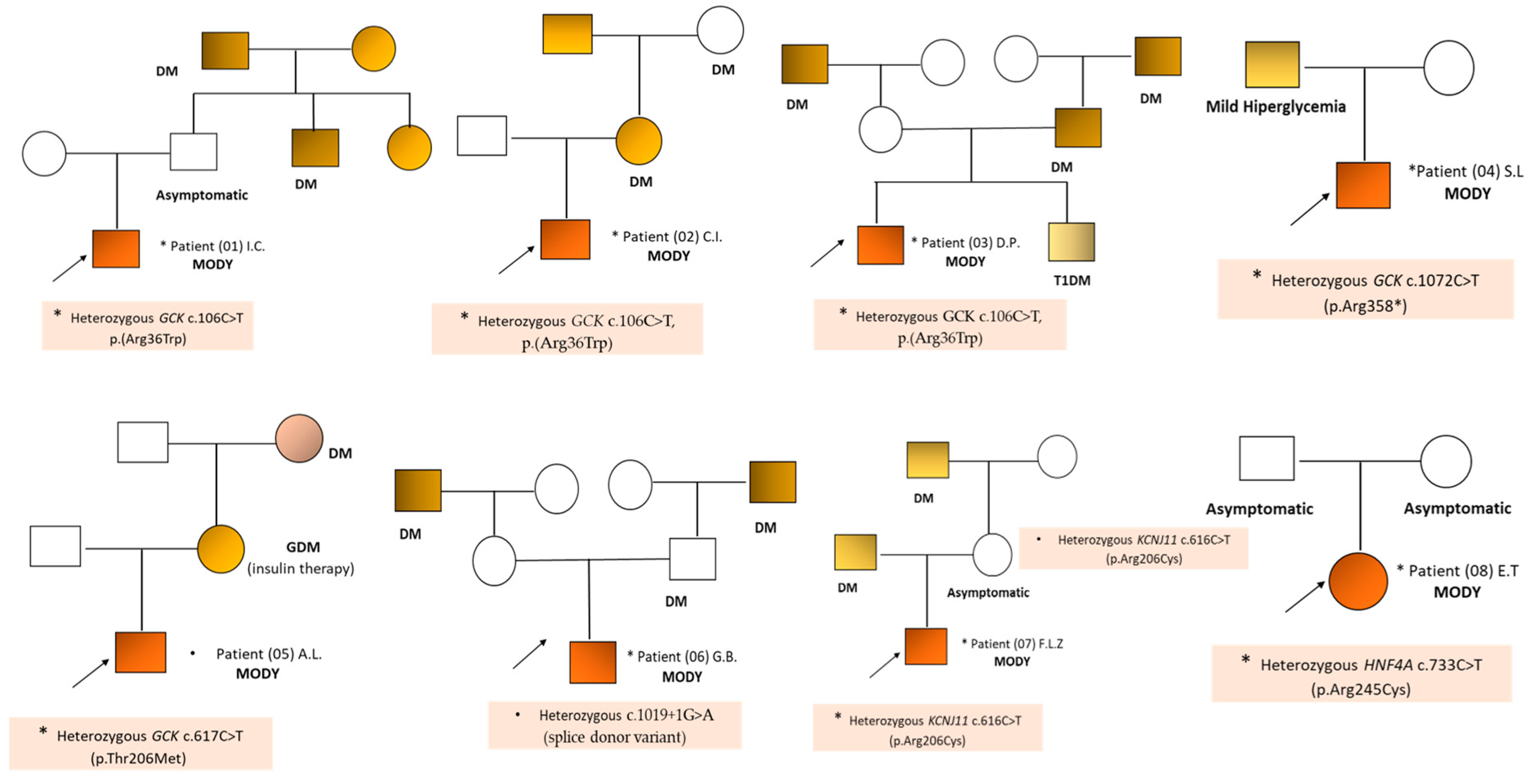
2. Results
3. Discussions
Genetic Counseling in Patients with MODY
4. Material and Methods
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Bonnefond, A.; Unnikrishnan, R.; Doria, A.; Vaxillaire, M.; Kulkarni, R.N.; Mohan, V.; Trischitta, V.; Froguel, P. Monogenic diabetes. Nat. Rev. Dis. Primers 2023, 9, 12. [Google Scholar] [CrossRef] [PubMed]
- Riddle, M.C.; Philipson, L.H.; Rich, S.S.; Carlsson, A.; Franks, P.W.; Greeley, S.A.W.; Nolan, J.J.; Pearson, E.R.; Zeitler, P.S.; Hattersley, A.T. Monogenic Diabetes: From Genetic Insights to Population-Based Precision in Care. Diabetes Care 2020, 43, 3117–3128. [Google Scholar] [PubMed]
- Chung, W.K.; Erion, K.; Florez, J.C.; Hattersley, A.T.; Hivert, M.F.; Lee, C.G.; McCarthy, M.I.; Nolan, J.J.; Norris, J.M.; Pearson, E.R.; et al. Precision Medicine in Diabetes: A Consensus Report From the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetes Care 2020, 43, 1617–1635. [Google Scholar] [CrossRef]
- Sun, H.Y.; Lin, X.Y. Genetic perspectives on childhood monogenic diabetes: Diagnosis, management, and future directions. World J. Diabetes 2023, 14, 1738–1753. [Google Scholar] [CrossRef] [PubMed]
- Kant, R.; Davis, A.; Verma, V. Maturity-Onset Diabetes of the Young: Rapid Evidence Review. Am. Fam. Physician 2022, 105, 162–167. [Google Scholar] [PubMed]
- Nkonge, K.M.; Nkonge, D.K.; Nkonge, T.N. The epidemiology, molecular pathogenesis, diagnosis, and treatment of maturity-onset diabetes of the young (MODY). Clin. Diabetes Endocrinol. 2020, 6, 20. [Google Scholar] [CrossRef] [PubMed]
- American Diabetes Association. 2. Classification and Diagnosis of Diabetes: Standards of Medical Care in Diabetes-2020. Diabetes Care 2020, 43 (Suppl. S1), S14–S31. [Google Scholar] [CrossRef]
- Greeley, S.A.W.; Polak, M.; Njølstad, P.R.; Barbetti, F.; Williams, R.; Castano, L.; Raile, K.; Chi, D.V.; Habeb, A.; Hattersley, A.T.; et al. ISPAD Clinical Practice Consensus Guidelines 2022: The diagnosis and management of monogenic diabetes in children and adolescents. Pediatr. Diabetes 2022, 23, 1188–1211. [Google Scholar] [CrossRef] [PubMed]
- Naylor, R.; Knight Johnson, A.; del Gaudio, D. Maturity-Onset Diabetes of the Young Overview. In GeneReviews® [Internet], 1993–2024; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2018; Available online: https://www.ncbi.nlm.nih.gov/books/NBK500456/, (accessed on 1 April 2024).
- OMIM—Online Mendelian Inheritance in Man. Available online: https://www.omim.org/, (accessed on 1 April 2024).
- Jang, K.M. Maturity-onset diabetes of the young: Update and perspectives on diagnosis and treatment. Yeungnam Univ. J. Med. 2020, 37, 13–21. [Google Scholar] [CrossRef]
- Zhang, H.; Colclough, K.; Gloyn, A.L.; Pollin, T.I. Monogenic diabetes: A gateway to precision medicine in diabetes. J. Clin. Investig. 2021, 131, e142244. [Google Scholar] [CrossRef]
- Shepherd, M.; Ellis, I.; Ahmad, A.M.; Todd, P.J.; Bowen-Jones, D.; Mannion, G.; Ellard, S.; Sparkes, A.C.; Hattersley, A.T. Predictive genetic testing in maturity-onset diabetes of the young (MODY). Diabet Med. 2001, 18, 417–421. [Google Scholar] [CrossRef]
- Delvecchio, M.; Pastore, C.; Giordano, P. Treatment options for MODY patients: A systematic review of literature. Diabetes Ther. 2020, 11, 1667–1685. [Google Scholar] [CrossRef] [PubMed]
- Urakami, T. Maturity-onset diabetes of the young (MODY): Current perspectives on diagnosis and treatment. Diabetes Metab. Syndr. Obes. 2019, 12, 1047–1056. [Google Scholar] [CrossRef] [PubMed]
- Hager, J.; Blanché, H.; Sun, F.; Vaxillaire, N.V.; Poller, W.; Cohen, D.; Czernichow, P.; Velho, G.; Robert, J.J.; Cohen, N.; et al. Six mutations in the glucokinase gene identified in MODY by using a nonradioactive sensitive screening technique. Diabetes 1994, 43, 730–733. [Google Scholar] [CrossRef]
- Abreu, G.M.; Tarantino, R.M.; da Fonseca, A.C.P.; Andrade, J.R.F.O.; de Souza, R.B.; Soares, C.A.P.D.; Cambraia, A.; Cabello, P.H.; Rodacki, M.; Zajdenverg, L.; et al. Identification of Variants Responsible for Monogenic Forms of Diabetes in Brazil. Front. Endocrinol. 2022, 13, 827325. [Google Scholar] [CrossRef] [PubMed]
- Ivanoshchuk, D.E.; Shakhtshneider, E.V.; Rymar, O.D.; Ovsyannikova, A.K.; Mikhailova, S.V.; Fishman, V.S.; Valeev, E.S.; Orlov, P.S.; Voevoda, M.I. The Mutation Spectrum of Maturity Onset Diabetes of the Young (MODY)-Associated Genes among Western Siberia Patients. J. Pers. Med. 2021, 11, 57. [Google Scholar] [CrossRef]
- Bennett, J.T.; Vasta, V.; Zhang, M.; Narayanan, J.; Gerrits, P.; Hahn, S.H. Molecular genetic testing of patients with monogenic diabetes and hyperinsulinism. Mol. Genet. Metab. 2015, 114, 451–458. [Google Scholar] [CrossRef]
- Giuffrida, F.M.A.; Moises, R.S.; Weinert, L.S.; Calliari, L.E.; Manna, T.D.; Dotto, R.P.; Franco, L.F.; Caetano, L.A.; Teles, M.G.; Lima, R.A.; et al. Brazilian Monogenic Diabetes Study Group (BRASMOD). Maturity-onset diabetes of the young (MODY) in Brazil: Establishment of a national registry and appraisal of available genetic and clinical data. Diabetes Res. Clin. Pract. 2017, 123, 134–142. [Google Scholar] [CrossRef]
- Fendler, W.; Borowiec, M.; Antosik, K.; Szadkowska, A.; Deja, G.; Jarosz-Chobot, P.; Mysliwiec, M.; Wyka, K.; Pietrzak, I.; Skupien, J.; et al. HDL cholesterol as a diagnostic tool for clinical differentiation of GCK-MODY from HNF1A-MODY and type 1 diabetes in children and young adults. Clin. Endocrinol. 2011, 75, 321–327. [Google Scholar] [CrossRef]
- Valentínová, L.; Beer, N.L.; Staník, J.; Tribble, N.D.; van de Bunt, M.; Hučková, M.; Barrett, A.; Klimeš, I.; Gašperíková, D.; Gloyn, A.L. Identification and functional characterisation of novel glucokinase mutations causing maturity-onset diabetes of the young in Slovakia. PLoS ONE 2012, 7, e34541. [Google Scholar] [CrossRef]
- Miller, S.P.; Anand, G.R.; Karschnia, E.J.; Bell, G.I.; LaPorte, D.C.; Lange, A.J. Characterization of glucokinase mutations associated with maturity-onset diabetes of the young type 2 (MODY-2): Different glucokinase defects lead to a common phenotype. Diabetes 1999, 48, 1645–1651. [Google Scholar] [CrossRef] [PubMed]
- Costa, A.; Bescós, M.; Velho, G.; Chêvre, J.; Vidal, J.; Sesmilo, G.; Bellanné-Chantelot, C.; Froguel, P.; Casamitjana, R.; Rivera-Fillat, F.; et al. Genetic and clinical characterisation of maturity-onset diabetes of the young in Spanish families. Eur. J. Endocrinol. 2000, 142, 380–386. [Google Scholar] [CrossRef] [PubMed]
- Stanik, J.; Dusatkova, P.; Cinek, O.; Valentinova, L.; Huckova, M.; Skopkova, M.; Dusatkova, L.; Stanikova, D.; Pura, M.; Klimes, I.; et al. De novo mutations of GCK, HNF1A and HNF4A may be more frequent in MODY than previously assumed. Diabetologia 2014, 57, 480–484. [Google Scholar] [CrossRef] [PubMed]
- Raimondo, A.; Chakera, A.J.; Thomsen, S.K.; Colclough, K.; Barrett, A.; De Franco, E.; Chatelas, A.; Demirbilek, H.; Akcay, T.; Alawneh, H.; et al. Phenotypic severity of homozygous GCK mutations causing neonatal or childhood-onset diabetes is primarily mediated through effects on protein stability. Hum. Mol. Genet. 2014, 23, 6432–6440. [Google Scholar] [PubMed]
- Njølstad, P.R.; Sagen, J.V.; Bjørkhaug, L.; Odili, S.; Shehadeh, N.; Bakry, D.; Sarici, S.U.; Alpay, F.; Molnes, J.; Molven, A.; et al. Permanent neonatal diabetes caused by glucokinase deficiency: Inborn error of the glucose-insulin signaling pathway. Diabetes 2003, 52, 2854–2860. [Google Scholar] [CrossRef] [PubMed]
- Gaál, Z.; Szűcs, Z.; Kántor, I.; Luczay, A.; Tóth-Heyn, P.; Benn, O.; Felszeghy, E.; Karádi, Z.; Madar, L.; Balogh, I.A. Comprehensive Analysis of Hungarian MODY Patients-Part II: Glucokinase MODY Is the Most Prevalent Subtype Responsible for about 70% of Confirmed Cases. Life 2021, 11, 771. [Google Scholar] [CrossRef]
- Ming-Qiang, Z.; Yang-Li, D.; Ke, H.; Wei, W.; Jun-Fen, F.; Chao-Chun, Z.; Guan-Ping, D. Maturity onset diabetes of the young (MODY) in Chinese children: Genes and clinical phenotypes. J. Pediatr. Endocrinol. Metab. 2019, 32, 759–765. [Google Scholar] [CrossRef] [PubMed]
- Aloi, C.; Salina, A.; Minuto, N.; Tallone, R.; Lugani, F.; Mascagni, A.; Mazza, O.; Cassanello, M.; Maghnie, M.; d’Annunzio, G. Glucokinase mutations in pediatric patients with impaired fasting glucose. Acta Diabetol. 2017, 54, 913–923. [Google Scholar] [CrossRef] [PubMed]
- Kavvoura, F.K.; Raimondo, A.; Thanabalasingham, G.; Barrett, A.; Webster, A.L.; Shears, D.; Mann, N.P.; Ellard, S.; Gloyn, A.L.; Owen, K.R. Reclassification of diabetes etiology in a family with multiple diabetes phenotypes. J. Clin. Endocrinol. Metab. 2014, 99, E1067–E1071. [Google Scholar] [CrossRef]
- Galán, M.; Vincent, O.; Roncero, I.; Azriel, S.; Boix-Pallares, P.; Delgado-Alvarez, E.; Díaz-Cadórniga, F.; Blázquez, E.; Navas, M.A. Effects of novel maturity-onset diabetes of the young (MODY)-associated mutations on glucokinase activity and protein stability. Biochem. J. 2006, 393 Pt 1, 389–396. [Google Scholar] [CrossRef]
- Osbak, K.K.; Colclough, K.; Saint-Martin, C.; Beer, N.L.; Bellanné-Chantelot, C.; Ellard, S.; Gloyn, A.L. Update on mutations in glucokinase (GCK), which cause maturity-onset diabetes of the young, permanent neonatal diabetes, and hyperinsulinemic hypoglycemia. Hum. Mutat. 2009, 30, 1512–1526. [Google Scholar] [CrossRef] [PubMed]
- Zmysłowska, A.; Jakiel, P.; Gadzalska, K.; Majos, A.; Płoszaj, T.; Ben-Skowronek, I.; Deja, G.; Glowinska-Olszewska, B.; Jarosz-Chobot, P.; Klonowska, B.; et al. Next- generation sequencing is an effective method for diagnosing patients with different forms of monogenic diabetes. Diabetes Res. Clin. Pract. 2022, 183, 109154. [Google Scholar] [CrossRef] [PubMed]
- Mirshahi, U.L.; Colclough, K.; Wright, C.F.; Wood, A.R.; Beaumont, R.N.; Tyrrell, J.; Laver, T.W.; Stahl, R.; Golden, A.; Goehringer, J.M.; et al. Reduced penetrance of MODY-associated HNF1A/HNF4A variants but not GCK variants in clinically unselected cohorts. Am. J. Hum. Genet. 2022, 109, 2018–2028. [Google Scholar] [CrossRef] [PubMed]
- Thomson, K.L.; Gloyn, A.L.; Colclough, K.; Batten, M.; Allen, L.I.; Beards, F.; Hattersley, A.T.; Ellard, S. Identification of 21 novel glucokinase (GCK) mutations in UK and European Caucasians with maturity-onset diabetes of the young (MODY). Hum. Mutat. 2003, 22, 417. [Google Scholar] [CrossRef]
- Johansen, A.; Ek, J.; Mortensen, H.B.; Pedersen, O.; Hansen, T. Half of clinically defined maturity-onset diabetes of the young patients in Denmark do not have mutations in HNF4A, GCK, and TCF1. J. Clin. Endocrinol. Metab. 2005, 90, 4607–4614. [Google Scholar] [CrossRef] [PubMed]
- Jurgens, S.J.; Choi, S.H.; Morrill, V.N.; Chaffin, M.; Pirruccello, J.P.; Halford, J.L.; Weng, L.C.; Nauffal, V.; Roselli, C.; Hall, A.W.; et al. Analysis of rare genetic variation underlying cardiometabolic diseases and traits among 200,000 individuals in the UK Biobank. Nat. Genet. 2022, 54, 240–250. [Google Scholar] [CrossRef] [PubMed]
- Caswell, R.C.; Snowsill, T.; Houghton, J.A.L.; Chakera, A.J.; Shepherd, M.H.; Laver, T.W.; Knight, B.A.; Wright, D.; Hattersley, A.T.; Ellard, S. Noninvasive Fetal Genotyping by Droplet Digital PCR to Identify Maternally Inherited Monogenic Diabetes Variants. Clin. Chem. 2020, 66, 958–965. [Google Scholar] [CrossRef] [PubMed]
- Tiulpakov, A.; Zubkova, N.; Makretskaya, N.; Krasnova, T.S.; Melnikova, A.I.; Fedyaeva, A.S.; Vasilyev, E.; Petrov, V.M.; Rubtsov, P.M. Minigene splicing assessment of 20 novel synonymous and intronic glucokinase gene variants identified in patients with maturity-onset diabetes of the young. Hum. Mutat. 2020, 41, 129–132. [Google Scholar] [CrossRef] [PubMed]
- Lorini, R.; Klersy, C.; d’Annunzio, G.; Massa, O.; Minuto, N.; Iafusco, D.; Bellannè-Chantelot, C.; Frongia, A.P.; Toni, S.; Meschi, F.; et al. Italian Society of Pediatric Endocrinology and Diabetology (ISPED) Study Group. Maturity-onset diabetes of the young in children with incidental hyperglycemia: A multicenter Italian study of 172 families. Diabetes Care 2009, 32, 1864–1866. [Google Scholar] [CrossRef]
- Glotov, O.S.; Serebryakova, E.A.; Turkunova, M.E.; Efimova, O.A.; Glotov, A.S.; Barbitoff, Y.A.; Nasykhova, Y.A.; Predeus, A.V.; Polev, D.E.; Fedyakov, A.; et al. Whole exome sequencing in Russian children with non-type 1 diabetes mellitus reveals a wide spectrum of genetic variants in MODY related and unrelated genes. Mol. Med. Rep. 2019, 20, 4905–4914. [Google Scholar] [CrossRef]
- Aykut, A.; Karaca, E.; Onay, H.; Gökşen, D.; Çetinkalp, Ş.; Eren, E.; Ersoy, B.; Çakır, E.P.; Büyükinan, M.; Kara, C.; et al. Analysis of the GCK gene in 79 MODY type 2 patients: A multicenter Turkish study, mutation profile and description of twenty novel mutations. Gene 2018, 641, 186–189. [Google Scholar] [CrossRef]
- Salomon-Estebanez, M.; Flanagan, S.E.; Ellard, S.; Rigby, L.; Bowden, L.; Mohamed, Z.; Nicholson, J.; Skae, M.; Hall, C.; Craigie, R.; et al. Conservatively treated Congenital Hyperinsulinism (CHI) due to K-ATP channel gene mutations: Reducing severity over time. Orphanet J. Rare Dis. 2016, 11, 163. [Google Scholar] [CrossRef]
- Huerta-Saenz, L.; Saunders, C.; Yan, Y. Challenging diagnosis of congenital hyperinsulinism in two infants of diabetic mothers with rare pathogenic KCNJ11 and HNF4A gene variants. Int. J. Pediatr. Endocrinol. 2018, 2018, 5. [Google Scholar] [CrossRef]
- Ribalet, B.; John, S.A.; Weiss, J.N. Molecular basis for Kir6.2 channel inhibition by adenine nucleotides. Biophys. J. 2003, 84, 266–276. [Google Scholar] [CrossRef]
- Boodhansingh, K.E.; Kandasamy, B.; Mitteer, L.; Givler, S.; De Leon, D.D.; Shyng, S.L.; Ganguly, A.; Stanley, C.A. Novel dominant KATP channel mutations in infants with congenital hyperinsulinism: Validation by in vitro expression studies and in vivo carrier phenotyping. Am. J. Med. Genet. A 2019, 179, 2214–2227. [Google Scholar] [CrossRef] [PubMed]
- Colclough, K.; Bellanne-Chantelot, C.; Saint-Martin, C.; Flanagan, S.E.; Ellard, S. Mutations in the genes encoding the transcription factors hepatocyte nuclear factor 1 alpha and 4 alpha in maturity-onset diabetes of the young and hyperinsulinemic hypoglycemia. Hum. Mutat. 2013, 34, 669–685. [Google Scholar] [CrossRef] [PubMed]
- Xu, A.; Lin, Y.; Sheng, H.; Cheng, J.; Mei, H.; Ting, T.H.; Zeng, C.; Liang, C.; Zhang, W.; Li, C.; et al. Molecular diagnosis of maturity-onset diabetes of the young in a cohort of Chinese children. Pediatr. Diabetes 2020, 21, 431–440. [Google Scholar] [CrossRef] [PubMed]
- Genome Aggregation Database. Available online: https://gnomad.broadinstitute.org/ (accessed on 1 April 2024).
- Masson, E.; Zou, W.B.; Génin, E.; Cooper, D.N.; Le Gac, G.; Fichou, Y.; Pu, N.; Rebours, V.; Férec, C.; Liao, Z.; et al. Expanding ACMG variant classification guidelines into a general framework. Hum. Genom. 2022, 16, 31. [Google Scholar] [CrossRef]
- ClinVar Database. Available online: https://www.ncbi.nlm.nih.gov/clinvar/ (accessed on 1 April 2024).
- Human Gene Mutation Database (HGMD) Database. Available online: https://digitalinsights.qiagen.com/products-overview/clinical-insights-portfolio/human-gene-mutation-database/ (accessed on 27 April 2024).
- dbSNP Database. Available online: https://www.ncbi.nlm.nih.gov/snp/ (accessed on 27 April 2024).
- Passanisi, S.; Salzano, G.; Bombaci, B.; Lombardo, F. Clinical and genetic features of maturity-onset diabetes of the young in pediatric patients: A 12-year monocentric experience. Diabetol. Metab. Syndr. 2021, 13, 96. [Google Scholar] [CrossRef]
- Alvelos, M.I.; Gonçalves, C.I.; Coutinho, E.; Almeida, J.T.; Bastos, M.; Sampaio, M.L.; Melo, M.; Martins, S.; Dinis, I.; Mirante, A.; et al. Maturity-Onset Diabetes of the Young (MODY) in Portugal: Novel GCK, HNFA1 and HNFA4 Mutations. J. Clin. Med. 2020, 9, 288. [Google Scholar] [CrossRef]
- Elashi, A.A.; Toor, S.M.; Diboun, I.; Al-Sarraj, Y.; Taheri, S.; Suhre, K.; Abou-Samra, A.B.; Albagha, O.M.E. The Genetic Spectrum of Maturity-Onset Diabetes of the Young (MODY) in Qatar, a Population-Based Study. Int. J. Mol. Sci. 2022, 24, 130. [Google Scholar] [CrossRef] [PubMed]
- Ben Khelifa, S.; Martinez, R.; Dandana, A.; Khochtali, I.; Ferchichi, S.; Castaño, L. Maturity Onset Diabetes of the Young (MODY) in Tunisia: Low frequencies of GCK and HNF1A mutations. Gene 2018, 651, 44–48. [Google Scholar] [CrossRef] [PubMed]
- Barrio, R.; Bellanné-Chantelot, C.; Moreno, J.C.; Morel, V.; Calle, H.; Alonso, M.; Mustieles, C. Nine novel mutations in maturity-onset diabetes of the young (MODY) candidate genes in 22 Spanish families. J. Clin. Endocrinol. Metab. 2002, 87, 2532–2539. [Google Scholar] [CrossRef] [PubMed]
- Firdous, P.; Nissar, K.; Ali, S.; Ganai, B.A.; Shabir, U.; Hassan, T.; Masoodi, S.R. Genetic Testing of Maturity-Onset Diabetes of the Young Current Status and Future Perspectives. Front. Endocrinol. 2018, 9, 253. [Google Scholar] [CrossRef] [PubMed]
- Skała-Zamorowska, E.; Deja, G.; Borowiec, M.; Fendler, W.; Małachowska, B.; Kamińska, H.; Młynarski, W.; Jarosz-Chobot, P. Genetic counseling in monogenic diabetes GCK MODY. Pediatr. Endocrinol. Diabetes Metab. 2016, 22, 54–59. [Google Scholar] [CrossRef] [PubMed]
- Tosur, M.; Philipson, L.H. Precision diabetes: Lessons learned from maturity-onset diabetes of the young (MODY). J. Diabetes Investig. 2022, 13, 1465–1471. [Google Scholar] [CrossRef] [PubMed]
- Clarke, A.J.; Wallgren-Pettersson, C. Ethics in genetic counselling. J. Community Genet. 2019, 1, 3–33. [Google Scholar] [CrossRef] [PubMed]
- Gloyn, A.L. The use of precision diagnostics for monogenic diabetes: A systematic review and expert opinion. Commun. Med. 2023, 3, 136. [Google Scholar]
- van der Zwaag, A.M.; Weinreich, S.S.; Bosma, A.R.; Rigter, T.; Losekoot, M.; Henneman, L.; Cornel, M.C. Current and best practices of genetic testing for maturity onset diabetes of the young: Views of professional experts. Public Health Genom. 2015, 18, 52–59. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| MODY Type | Gene Function | OMIM | Locus | Pathophysiology | Other Features | Treatment |
|---|---|---|---|---|---|---|
| HNF4A-MODY 1 (5–10%) | TF | 600281 | 20q13.12 | β-CD | Hyperinsulinism during infancy, low TG level | Sulfonylureas |
| GCK-MODY 2 (30–50%) | Glucokinase (Hexokinase 4) | 138079 | 7p13 | β-CD | Mild fasting hyperglycemia | Diet |
| HNF1A-MODY 3 (30–65%) | TF | 142410 | 12q24.31 | β-CD | Glycosuria | Sulfonylureas |
| PDX1-MODY 4 (1%) | TF | 600733 | 13q12.2 | β-CD | Pancreatic agenesis in homozygote/compound heterozygote | Diet or OAD or insulin |
| HNF1B-MODY 5 (<5%) | TF | 189907 | 17q12 | β-CD | Renal/genital anomalies, pancreatic hypoplasia | Insulin |
| NEUROD1-MODY 6 (<1%) | TF | 601724 | 2q31.3 | β-CD | Susceptibility for T2DM in heterozygous; neonatal diabetes; neurological abnormalities in homozygous mice | OAD or insulin |
| KLF11-MODY 7 (<1%) | TF | 603301 | 2p25.1 | β-CD | Early-onset T2DM | OAD or insulin |
| CEL-MODY 8 (<1%) | Controls pancreatic functions; plays essential role in digestion and intestinal absorption of dietary lipids | 114840 | 9q34.13 | Pancreas endocrine and exocrine dysfunction | Lipomatosis | OAD or insulin |
| PAX4-MODY 9 (<1%) | TF | 167413 | 7q32.1 | β-CD | Ketoacidosis | OAD, diet, or insulin |
| INS-MODY 10 (<1%) | Encode the proinsulin precursor | 176730 | 11p15.5 | Severe insulin deficiency | PND | OAD, diet, or insulin |
| BLK-MODY 11 (<1%) | Encodes B-lymphoid TK | 191305 | 8p23.1 | Abnormal IS | Overweight | OAD, diet, or insulin |
| ABCC8-MODY 12 (<1%) | Regulation of IS | 600509 | 11p15.1 | KATP dysfunction | PND, TND, HHF1 | Sulfonylurea |
| KCNJ11-MODY 13 (<1%) | Regulation of IS | 600937 | 11p15.1 | KATP dysfunction | HHF2, PND2, TND3 | OAD or insulin |
| APPL1-MODY 14 (<1%) | Insulin signaling pathways | 604299 | 3p14.3 | Insulin IS defect | Higher adiponectin levels among T2DM patients | Diet, OAD, or inulin |
| Patient ID | Mutation | Exon | Genotype | Effect of Mutation/ Pathogenicity | Protein | GnomAD | dbSNP Database | ClinVar | Previous Studies |
|---|---|---|---|---|---|---|---|---|---|
| P01 (I.C) | GCK c.106C>T | exon 2 | Hz | Missense/Pathogen | p.Arg36Trp | gnomAD (0.008%) | rs762263694 | Variation ID 431973 | [16,17,18,19,20,21,22,23] |
| P02 (C.I) | GCK c.106C>T | exon 2 | Hz | Missense/Pathogen | p.Arg36Trp | gnomAD (0.008%) | rs762263694 | Variation ID:431973 | [16,17,18,19,20,21,22,23] |
| P03 (D.P) | GCK c.106C>T | exon 2 | Hz | Missense/Pathogen | pp.Arg36Trp | gnomAD (0.008%) | rs762263694 | Variation ID:431973 | [16,17,18,19,20,21,22,23] |
| P04 (S.L.) | GCK c.1072C>T | exon 9 | Hz | Nonsense/Pathogen | p.Arg358* | gnomAD (0.00043%) | rs780716926 | [24,25,26,27] | |
| P05 (I.L) | GCK c.617C>T | exon 6 | Hz | Missense/Pathogen | p.Thr206Met | gnomAD (0.000004) | rs1441649062 | Variation ID:1191898 | [28,29,30,31,32,33] |
| P06 (G.B) | GCK c.1019+1G>A | Intron 8 (splice donor) | Hz | Splice_donor_variant | gnomAD (0/0) | Variation ID 2664360 | [34,35,36,37,38,39,40,41,42,43,44] | ||
| P07 (F.L.Z) | KCNJ11 c.616C>T | exon 1 | Hz | Missense/Pathogen | p.Arg206Cys | gnomAD (0.01%) | rs775204908 | Variation ID:1338615 | [45,46,47,48] |
| P08 (E.T) | HNF4A c.733C>T | exon 7 | Hz | Missense/Pathogen | p.Arg245Cys | gnomAD (0.00001) | rs1290868034 | Variation ID: 804917 | [46,49,50] |
| Criteria | P01 (C.I) | P02 (C.I.) | P03 (D.P.) | P04 (S.L) | P05 (I.L.) | P06 (G.B) | P07 (F.L.Z) | P08 (E.T.) |
|---|---|---|---|---|---|---|---|---|
| Age of presentation (years) | 2.5 y | 7.6 y | 4y | 9.2 y | 3.4 y | 10 y | 15.9 y | 12 y |
| Follow-up months (m)/years (y) | 7 m | 3 m | 11 y 4 m | - | 3 m | 2 y | 2 y | 2 y 3 m |
| Sex | M | M | M | M | M | M | M | F |
| Clinical symptoms | - | - | - | - | + | + | + (DKA) | - |
| Higher hyperglycemia level (mg/dL) | 150 | 150 | 140 | 200 | 148 | 160 | 248 | 189 |
| Obesity | - | - | - | - | - | + | + | + |
| Initial HbA1c | 5.9% | 6.3% | 6.2% | 6.5% | 6.8% | 6.3% | 11.5% | 7.4% |
| Type of autoantibodies tested/results | ||||||||
| GAD65 | - | - | - | - | - | - | - | - |
| ICA | - | - | - | - | - | - | - | - |
| IA-2 | - | - | - | |||||
| C-peptide (ng/mL) | normal | normal | low | normal | normal | normal | normal | normal |
| Family history of diabetes | + | + | + | + | + | + | + | - |
| Pharmacological Treatment | - | - | + | - | - | - | + | + |
| Type of treatment | diet | diet | insulin | diet | diet | diet | insulin + sulfonylurea | sulfonylurea |
| Last HbA1c | 6% | 6% | 6.7% | 6.1% | 6.8% | 6.3% | 11.2% | 5.8% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Butnariu, L.I.; Bizim, D.A.; Oltean, C.; Rusu, C.; Pânzaru, M.C.; Păduraru, G.; Gimiga, N.; Ghiga, G.; Moisă, Ș.M.; Țarcă, E.; et al. The Importance of Molecular Genetic Testing for Precision Diagnostics, Management, and Genetic Counseling in MODY Patients. Int. J. Mol. Sci. 2024, 25, 6318. https://doi.org/10.3390/ijms25126318
Butnariu LI, Bizim DA, Oltean C, Rusu C, Pânzaru MC, Păduraru G, Gimiga N, Ghiga G, Moisă ȘM, Țarcă E, et al. The Importance of Molecular Genetic Testing for Precision Diagnostics, Management, and Genetic Counseling in MODY Patients. International Journal of Molecular Sciences. 2024; 25(12):6318. https://doi.org/10.3390/ijms25126318
Chicago/Turabian StyleButnariu, Lăcrămioara Ionela, Delia Andreia Bizim, Carmen Oltean, Cristina Rusu, Monica Cristina Pânzaru, Gabriela Păduraru, Nicoleta Gimiga, Gabriela Ghiga, Ștefana Maria Moisă, Elena Țarcă, and et al. 2024. "The Importance of Molecular Genetic Testing for Precision Diagnostics, Management, and Genetic Counseling in MODY Patients" International Journal of Molecular Sciences 25, no. 12: 6318. https://doi.org/10.3390/ijms25126318
APA StyleButnariu, L. I., Bizim, D. A., Oltean, C., Rusu, C., Pânzaru, M. C., Păduraru, G., Gimiga, N., Ghiga, G., Moisă, Ș. M., Țarcă, E., Starcea, I. M., Popa, S., & Trandafir, L. M. (2024). The Importance of Molecular Genetic Testing for Precision Diagnostics, Management, and Genetic Counseling in MODY Patients. International Journal of Molecular Sciences, 25(12), 6318. https://doi.org/10.3390/ijms25126318