
Optimization of In Vitro Th17 Polarization for Adoptive Cell Therapy in Chronic Lymphocytic Leukemia

 ,
,
Abstract

1. Introduction
2. Results
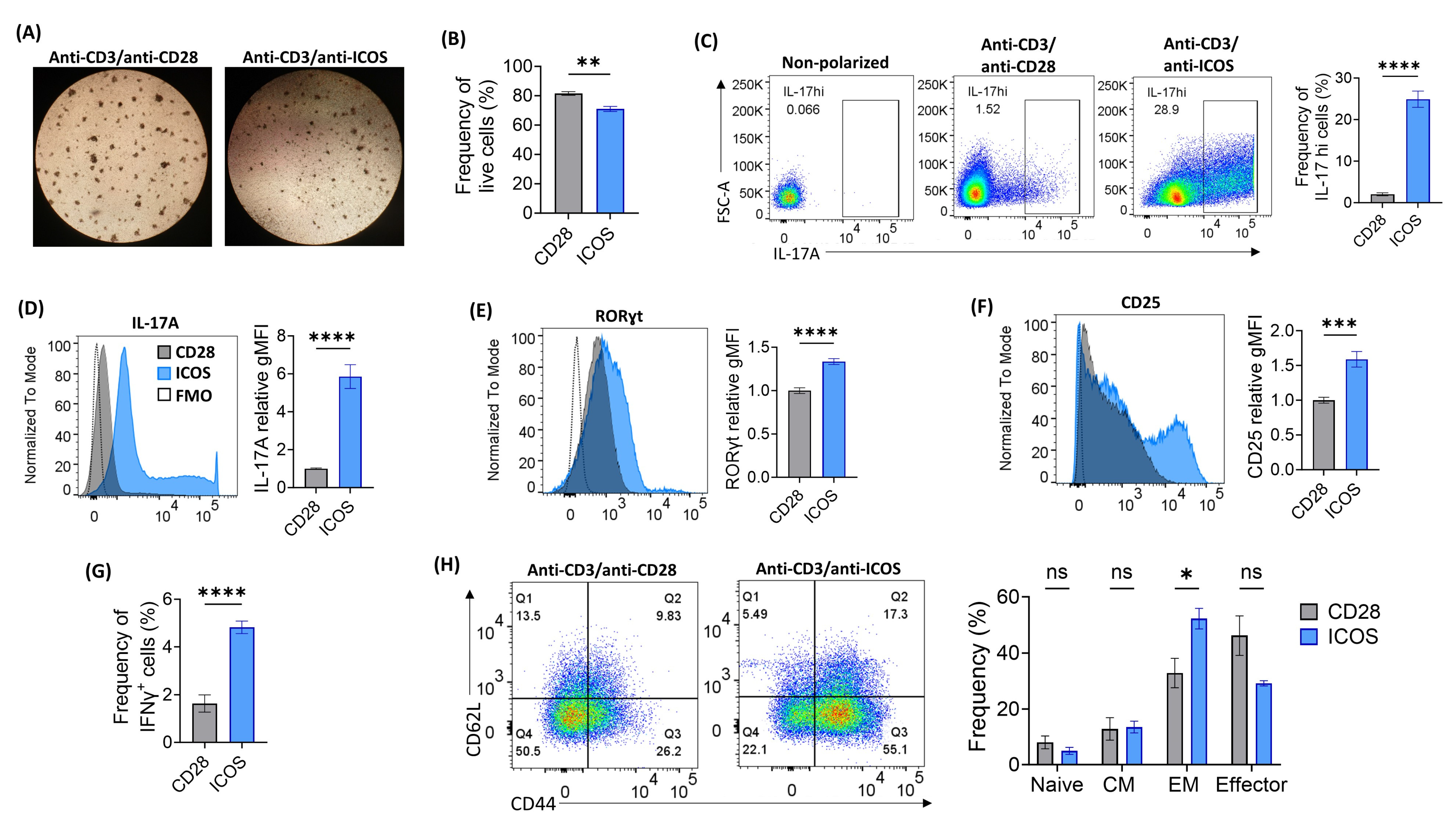
2.1. Optimization of ICOS-Based Co-Stimulation for In Vitro Th17 Polarization
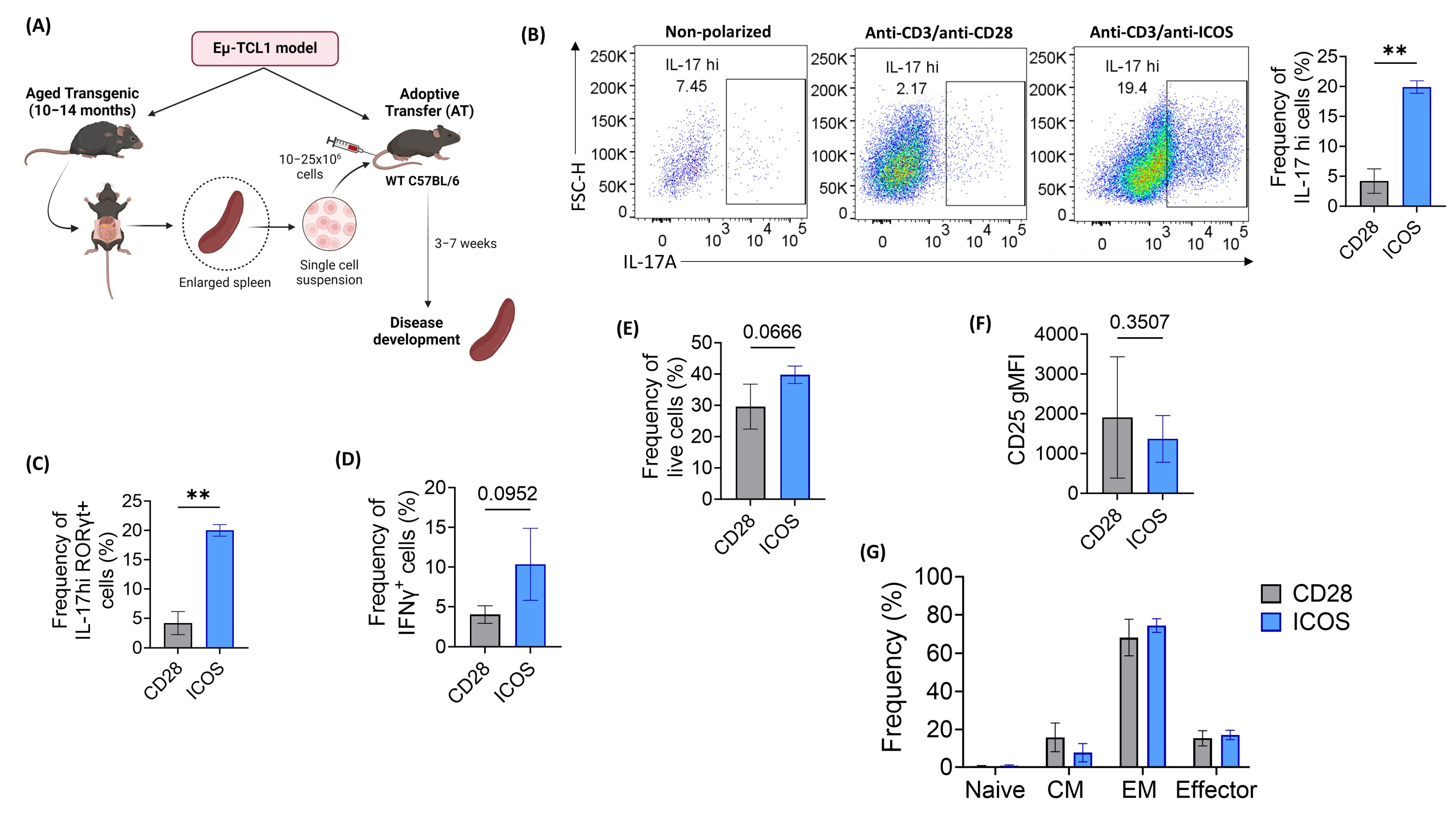
2.2. ICOS Co-Stimulation Effectively Polarizes Eµ-TCL1 CD4+ T Cells into Th17 Phenotype
2.3. Eµ-TCL1 In Vitro-Polarized Th17 Cells Show Signs of Enhanced Memory Formation and In Vivo Persistence
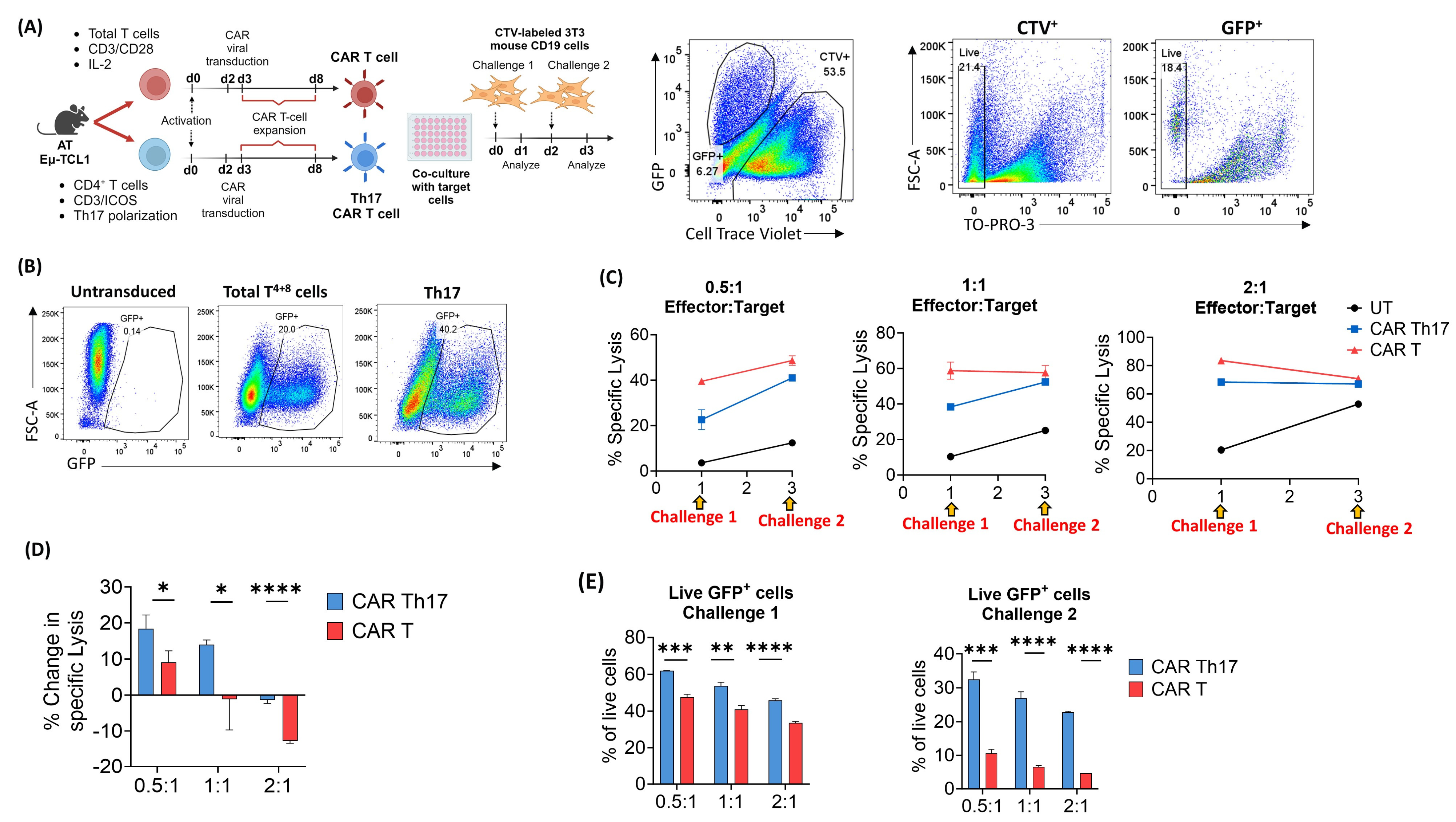
2.4. Eµ-TCL1 CAR Th17 Cells Maintain In Vitro Cytotoxic Potential with Improved Persistence
3. Discussion
4. Materials and Methods
4.1. Adoptive Transfer (AT) Eμ-TCL1 Murine Model
4.2. Th17 Cell Polarization and Culture
4.3. ACT Using Polarized Th17 Cells
4.4. Flow Cytometry Staining
4.5. Cell Trace Violet (CTV) Proliferation Assay
4.6. Flow Cytometry Analysis
4.7. T-Cell Viral Transduction
4.8. In Vitro Cytotoxicity Analysis
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Harrington, L.E.; Hatton, R.D.; Mangan, P.R.; Turner, H.; Murphy, T.L.; Murphy, K.M.; Weaver, C.T. Interleukin 17–producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat. Immunol. 2005, 6, 1123–1132. [Google Scholar] [CrossRef]
- Park, H.; Li, Z.; Yang, X.O.; Chang, S.H.; Nurieva, R.; Wang, Y.-H.; Wang, Y.; Hood, L.; Zhu, Z.; Tian, Q. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat. Immunol. 2005, 6, 1133–1141. [Google Scholar] [CrossRef]
- Steinman, L. A brief history of TH17, the first major revision in the TH1/TH2 hypothesis of T cell–mediated tissue damage. Nat. Med. 2007, 13, 139–145. [Google Scholar] [CrossRef]
- Ouyang, W.; Kolls, J.K.; Zheng, Y. The biological functions of T helper 17 cell effector cytokines in inflammation. Immunity 2008, 28, 454–467. [Google Scholar] [CrossRef]
- Sutton, C.; Brereton, C.; Keogh, B.; Mills, K.H.; Lavelle, E.C. A crucial role for interleukin (IL)-1 in the induction of IL-17–producing T cells that mediate autoimmune encephalo-myelitis. J. Exp. Med. 2006, 203, 1685–1691. [Google Scholar] [CrossRef]
- Krebs, C.F.; Paust, H.-J.; Krohn, S.; Koyro, T.; Brix, S.R.; Riedel, J.-H.; Bartsch, P.; Wiech, T.; Meyer-Schwesinger, C.; Huang, J.; et al. Autoimmune renal disease is exacerbated by S1P-receptor-1-dependent intestinal Th17 cell migration to the kidney. Immunity 2016, 45, 1078–1092. [Google Scholar] [CrossRef]
- Kumar, P.; Monin, L.; Castillo, P.; Elsegeiny, W.; Horne, W.; Eddens, T.; Vikram, A.V.A.; Good, M.; Schoenborn, A.A.; Bibby, K.; et al. Intestinal interleukin-17 receptor signaling mediates reciprocal control of the gut microbiota and autoimmune inflam-mation. Immunity 2016, 44, 659–671. [Google Scholar] [CrossRef]
- Zhang, J.-P.; Yan, J.; Xu, J.; Pang, X.-H.; Chen, M.-S.; Li, L.; Wu, C.; Li, S.-P.; Zheng, L. Increased intratumoral IL-17-producing cells correlate with poor survival in hepatocellular carcinoma patients. J. Hepatol. 2009, 50, 980–989. [Google Scholar] [CrossRef]
- Gu, K.; Li, M.-M.; Shen, J.; Liu, F.; Cao, J.-Y.; Jin, S.; Yu, Y. Interleukin-17-induced EMT promotes lung cancer cell migration and invasion via NF-κB/ZEB1 signal pathway. Am. J. Cancer Res. 2015, 5, 1169. [Google Scholar]
- Lin, Y.; Xu, J.; Su, H.; Zhong, W.; Yuan, Y.; Yu, Z.; Fang, Y.; Zhou, H.; Li, C.; Huang, K. Interleukin-17 is a favorable prognostic marker for colorectal cancer. Clin. Transl. Oncol. 2015, 17, 50–56. [Google Scholar] [CrossRef]
- Guéry, L.; Hugues, S. Th17 cell plasticity and functions in cancer immunity. BioMed Res. Int. 2015, 2015, 314620. [Google Scholar] [CrossRef]
- Muranski, P.; Borman, Z.; Kerkar, S.; Klebanoff, C.; Ji, Y.; Sanchez-Perez, L.; Sukumar, M.; Reger, R.; Yu, Z.; Kern, S. Th17 cells are long lived and retain a stem cell-like molecular signature. Immunity 2011, 35, 972–985. [Google Scholar] [CrossRef]
- Schnell, A.; Huang, L.; Singer, M.; Singaraju, A.; Barilla, R.M.; Regan, B.M.; Bollhagen, A.; Thakore, P.I.; Dionne, D.; Delorey, T.M. Stem-like intestinal Th17 cells give rise to pathogenic effector T cells during autoimmunity. Cell 2021, 184, 6281–6298.e23. [Google Scholar] [CrossRef]
- Kryczek, I.; Zhao, E.; Liu, Y.; Wang, Y.; Vatan, L.; Szeliga, W.; Moyer, J.; Klimczak, A.; Lange, A.; Zou, W. Human TH17 cells are long-lived effector memory cells. Sci. Transl. Med. 2011, 3, 104ra100. [Google Scholar] [CrossRef]
- Bowers, J.S.; Nelson, M.H.; Majchrzak, K.; Bailey, S.R.; Rohrer, B.; Kaiser, A.D.; Atkinson, C.; Gattinoni, L.; Paulos, C.M. Th17 cells are refractory to senescence and retain robust antitumor activity after long-term ex vivo expansion. JCI Insight 2017, 2, e90772. [Google Scholar] [CrossRef]
- Majchrzak, K.; Nelson, M.H.; Bowers, J.S.; Bailey, S.R.; Wyatt, M.M.; Wrangle, J.M.; Rubinstein, M.P.; Varela, J.C.; Li, Z.; Himes, R.A. β-catenin and PI3Kδ inhibition expands precursor Th17 cells with heightened stemness and antitumor activity. JCI Insight 2017, 2, e90547. [Google Scholar] [CrossRef]
- Paulos, C.M.; Carpenito, C.; Plesa, G.; Suhoski, M.M.; Varela-Rohena, A.; Golovina, T.N.; Carroll, R.G.; Riley, J.L.; June, C.H. The inducible costimulator (ICOS) is critical for the development of human TH17 cells. Sci. Transl. Med. 2010, 2, 55ra78. [Google Scholar] [CrossRef]
- Guedan, S.; Chen, X.; Madar, A.; Carpenito, C.; McGettigan, S.E.; Frigault, M.J.; Lee, J.; Posey, A.D., Jr.; Scholler, J.; Scholler, N.; et al. ICOS-based chimeric antigen receptors program bipolar TH17/TH1 cells. Blood 2014, 124, 1070–1080. [Google Scholar] [CrossRef]
- Guedan, S.; Posey, A.D., Jr.; Shaw, C.; Wing, A.; Da, T.; Patel, P.R.; McGettigan, S.E.; Casado-Medrano, V.; Kawalekar, O.U.; Uribe-Herranz, M.; et al. Enhancing CAR T cell persistence through ICOS and 4-1BB costimulation. JCI Insight 2018, 3, e96976. [Google Scholar] [CrossRef]
- Moreno-Cortes, E.; Franco-Fuquen, P.; Garcia-Robledo, J.E.; Forero, J.; Booth, N.; Castro, J.E. ICOS and OX40 tandem co-stimulation enhances CAR T-cell cytotoxicity and promotes T-cell persistence phenotype. Front. Oncol. 2023, 13, 1200914. [Google Scholar] [CrossRef]
- Hallek, M.; Al-Sawaf, O. Chronic lymphocytic leukemia: 2022 update on diagnostic and therapeutic procedures. Am. J. Hematol. 2021, 96, 1679–1705. [Google Scholar] [CrossRef]
- Herman, S.E.M.; Gordon, A.L.; Hertlein, E.; Ramanunni, A.; Zhang, X.; Jaglowski, S.; Flynn, J.; Jones, J.; Blum, K.A.; Buggy, J.J.; et al. Bruton tyrosine kinase represents a promising therapeutic target for treatment of chronic lymphocytic leukemia and is effectively targeted by PCI-32765. Blood 2011, 117, 6287–6296. [Google Scholar] [CrossRef]
- Souers, A.J.; Leverson, J.D.; Boghaert, E.R.; Ackler, S.L.; Catron, N.D.; Chen, J.; Dayton, B.D.; Ding, H.; Enschede, S.H.; Fairbrother, W.J.; et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 2013, 19, 202–208. [Google Scholar] [CrossRef]
- Khan, A.N.; Asija, S.; Pendhari, J.; Purwar, R. CAR-T cell therapy in hematological malignancies: Where are we now and where are we heading for? Eur. J. Haematol. 2024, 112, 6–18. [Google Scholar] [CrossRef]
- Siddiqi, T.; Maloney, D.G.; Kenderian, S.S.; Brander, D.M.; Dorritie, K.; Soumerai, J.; Riedell, P.A.; Shah, N.N.; Nath, R.; Fakhri, B.; et al. Lisocabtagene Maraleucel (liso-cel) in R/R CLL/SLL: 24-Month Median Follow-up of TRANSCEND CLL 004. Blood 2023, 142, 330. [Google Scholar] [CrossRef]
- Riches, J.C.; Davies, J.K.; McClanahan, F.; Fatah, R.; Iqbal, S.; Agrawal, S.; Ramsay, A.G.; Gribben, J.G. T cells from CLL patients exhibit features of T-cell exhaustion but retain capacity for cytokine production. Blood J. Am. Soc. Hematol. 2013, 121, 1612–1621. [Google Scholar] [CrossRef]
- Görgün, G.; Holderried, T.A.; Zahrieh, D.; Neuberg, D.; Gribben, J.G. Chronic lymphocytic leukemia cells induce changes in gene expression of CD4 and CD8 T cells. J. Clin. Investig. 2005, 115, 1797–1805. [Google Scholar] [CrossRef]
- Ramsay, A.G.; Johnson, A.J.; Lee, A.M.; Gorgün, G.; Le Dieu, R.; Blum, W.; Byrd, J.C.; Gribben, J.G. Chronic lymphocytic leukemia T cells show impaired immunological synapse formation that can be reversed with an immunomodulating drug. J. Clin. Investig. 2008, 118, 2427–2437. [Google Scholar] [CrossRef]
- Solomon, B.M.; Rabe, K.G.; Slager, S.L.; Brewer, J.D.; Cerhan, J.R.; Shanafelt, T.D. Overall and cancer-specific survival of patients with breast, colon, kidney, and lung cancers with and without chronic lymphocytic leukemia: A SEER population-based study. J. Clin. Oncol. 2013, 31, 930–937. [Google Scholar] [CrossRef]
- Teh, B.W.; Tam, C.S.; Handunnetti, S.; Worth, L.J.; Slavin, M.A. Infections in patients with chronic lymphocytic leukaemia: Mitigating risk in the era of targeted therapies. Blood Rev. 2018, 32, 499–507. [Google Scholar] [CrossRef]
- Hus, I.; Bojarska-Junak, A.; Chocholska, S.; Tomczak, W.; Woś, J.; Dmoszyńska, A.; Roliński, J. Th17/IL-17A might play a protective role in chronic lymphocytic leukemia immunity. PLoS ONE 2013, 8, e78091. [Google Scholar] [CrossRef]
- Jadidi-Niaragh, F.; Ghalamfarsa, G.; Memarian, A.; Asgarian-Omran, H.; Razavi, S.M.; Sarrafnejad, A.; Shokri, F. Downregulation of IL-17-producing T cells is associated with regulatory T cell expansion and disease progression in chronic lymphocytic leukemia. Tumor Biol. 2013, 34, 929–940. [Google Scholar] [CrossRef]
- Gamal, W.; Sahakian, E.; Pinilla-Ibarz, J. The role of Th17 cells in chronic lymphocytic leukemia: Friend or foe? Blood Adv. 2023, 7, 2401–2417. [Google Scholar] [CrossRef]
- Fraietta, J.A.; Lacey, S.F.; Orlando, E.J.; Pruteanu-Malinici, I.; Gohil, M.; Lundh, S.; Boesteanu, A.C.; Wang, Y.; O’Connor, R.S.; Hwang, W.-T. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat. Med. 2018, 24, 563–571. [Google Scholar] [CrossRef]
- Buggins, A.; Patten, P.; Richards, J.; Thomas, N.; Mufti, G.; Devereux, S. Tumor-derived IL-6 may contribute to the immunological defect in CLL. Leukemia 2008, 22, 1084–1087. [Google Scholar] [CrossRef]
- Podhorecka, M.; Dmoszynska, A.; Rolinski, J.; Wasik, E. T type 1/type 2 subsets balance in B-cell chronic lymphocytic leukemia—The three-color flow cytometry analysis. Leuk. Res. 2002, 26, 657–660. [Google Scholar] [CrossRef]
- Rossmann, E.D.; Lewin, N.; Jeddi-Tehrani, M.; Österborg, A.; Mellstedt, H. Intracellular T cell cytokines in patients with B cell chronic lymphocytic leukaemia (B-CLL). Eur. J. Haematol. 2002, 68, 299–306. [Google Scholar] [CrossRef]
- McClanahan, F.; Riches, J.C.; Miller, S.; Day, W.P.; Kotsiou, E.; Neuberg, D.; Croce, C.M.; Capasso, M.; Gribben, J.G. Mechanisms of PD-L1/PD-1–mediated CD8 T-cell dysfunction in the context of aging-related immune defects in the Eµ-TCL1 CLL mouse model. Blood J. Am. Soc. Hematol. 2015, 126, 212–221. [Google Scholar] [CrossRef]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 2014, 371, 1507–1517. [Google Scholar] [CrossRef]
- Roschewski, M.; Longo, D.L.; Wilson, W.H. CAR T-cell therapy for large B-cell lymphoma—Who, when, and how? N. Engl. J. Med. 2022, 386, 692–696. [Google Scholar] [CrossRef]
- Boulch, M.; Cazaux, M.; Cuffel, A.; Guerin, M.V.; Garcia, Z.; Alonso, R.; Lemaître, F.; Beer, A.; Corre, B.; Menger, L.; et al. Tumor-intrinsic sensitivity to the pro-apoptotic effects of IFN-γ is a major determinant of CD4+ CAR T-cell antitumor activity. Nat. Cancer 2023, 4, 968–983. [Google Scholar] [CrossRef]
- Yang, Y.; Kohler, M.E.; Chien, C.D.; Sauter, C.T.; Jacoby, E.; Yan, C.; Hu, Y.; Wanhainen, K.; Qin, H.; Fry, T.J. TCR engagement negatively affects CD8 but not CD4 CAR T cell expansion and leukemic clearance. Sci. Transl. Med. 2017, 9, eaag1209. [Google Scholar] [CrossRef]
- Kruse, B.; Buzzai, A.C.; Shridhar, N.; Braun, A.D.; Gellert, S.; Knauth, K.; Pozniak, J.; Peters, J.; Dittmann, P.; Mengoni, M.; et al. CD4+ T cell-induced inflammatory cell death controls immune-evasive tumours. Nature 2023, 618, 1033–1040. [Google Scholar] [CrossRef]
- Hunder, N.N.; Wallen, H.; Cao, J.; Hendricks, D.W.; Reilly, J.Z.; Rodmyre, R.; Jungbluth, A.; Gnjatic, S.; Thompson, J.A.; Yee, C. Treatment of Metastatic Melanoma with Autologous CD4+ T Cells against NY-ESO-1. N. Engl. J. Med. 2008, 358, 2698–2703. [Google Scholar] [CrossRef]
- Lu, Y.-C.; Parker, L.L.; Lu, T.; Zheng, Z.; Toomey, M.A.; White, D.E.; Yao, X.; Li, Y.F.; Robbins, P.F.; Feldman, S.A.; et al. Treatment of Patients With Metastatic Cancer Using a Major Histocompatibility Complex Class II–Restricted T-Cell Receptor Targeting the Cancer Germline Antigen MAGE-A3. J. Clin. Oncol. 2017, 35, 3322–3329. [Google Scholar] [CrossRef]
- Tran, E.; Turcotte, S.; Gros, A.; Robbins, P.F.; Lu, Y.-C.; Dudley, M.E.; Wunderlich, J.R.; Somerville, R.P.; Hogan, K.; Hinrichs, C.S.; et al. Cancer Immunotherapy Based on Mutation-Specific CD4+ T Cells in a Patient with Epithelial Cancer. Science 2014, 344, 641–645. [Google Scholar] [CrossRef]
- Sommermeyer, D.; Hudecek, M.; Kosasih, P.L.; Gogishvili, T.; Maloney, D.G.; Turtle, C.J.; Riddell, S.R. Chimeric antigen receptor-modified T cells derived from defined CD8+ and CD4+ subsets confer superior antitumor reactivity in vivo. Leukemia 2016, 30, 492–500. [Google Scholar] [CrossRef]
- Turtle, C.J.; Hanafi, L.-A.; Berger, C.; Hudecek, M.; Pender, B.; Robinson, E.; Hawkins, R.; Chaney, C.; Cherian, S.; Chen, X.; et al. Immunotherapy of non-Hodgkin’s lymphoma with a defined ratio of CD8+ and CD4+ CD19-specific chimeric antigen receptor-modified T cells. Sci. Transl. Med. 2016, 8, 355ra116. [Google Scholar] [CrossRef]
- Turtle, C.J.; Hanafi, L.-A.; Berger, C.; Gooley, T.A.; Cherian, S.; Hudecek, M.; Sommermeyer, D.; Melville, K.; Pender, B.; Budiarto, T.M.; et al. CD19 CAR–T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J. Clin. Investig. 2016, 126, 2123–2138. [Google Scholar] [CrossRef]
- Wang, D.; Aguilar, B.; Starr, R.; Alizadeh, D.; Brito, A.; Sarkissian, A.; Ostberg, J.R.; Forman, S.J.; Brown, C.E. Glioblastoma-targeted CD4+ CAR T cells mediate superior antitumor activity. JCI Insight 2018, 3, e99048. [Google Scholar] [CrossRef]
- Melenhorst, J.J.; Chen, G.M.; Wang, M.; Porter, D.L.; Chen, C.; Collins, M.A.; Gao, P.; Bandyopadhyay, S.; Sun, H.; Zhao, Z.; et al. Decade-long leukaemia remissions with persistence of CD4+ CAR T cells. Nature 2022, 602, 503–509. [Google Scholar] [CrossRef]
- Muranski, P.; Boni, A.; Antony, P.A.; Cassard, L.; Irvine, K.R.; Kaiser, A.; Paulos, C.M.; Palmer, D.C.; Touloukian, C.E.; Ptak, K. Tumor-specific Th17-polarized cells eradicate large established melanoma. Blood J. Am. Soc. Hematol. 2008, 112, 362–373. [Google Scholar] [CrossRef]
- Nelson, M.H.; Knochelmann, H.M.; Bailey, S.R.; Huff, L.W.; Bowers, J.S.; Majchrzak-Kuligowska, K.; Wyatt, M.M.; Rubinstein, M.P.; Mehrotra, S.; Nishimura, M.I.; et al. Identification of human CD4+ T cell populations with distinct antitumor activity. Sci. Adv. 2020, 6, eaba7443. [Google Scholar] [CrossRef]
- Bürgler, S.; Gimeno, A.; Parente-Ribes, A.; Wang, D.; Os, A.; Devereux, S.; Jebsen, P.; Bogen, B.; Tjønnfjord, G.E.; Munthe, L.A. Chronic Lymphocytic Leukemia Cells Express CD38 in Response to Th1 Cell–Derived IFN-γ by a T-bet–Dependent Mechanism. J. Immunol. 2015, 194, 827–835. [Google Scholar] [CrossRef]
- Os, A.; Bürgler, S.; Ribes, A.P.; Funderud, A.; Wang, D.; Thompson, K.M.; Tjønnfjord, G.E.; Bogen, B.; Munthe, L.A. Chronic lymphocytic leukemia cells are activated and proliferate in response to specific T helper cells. Cell Rep. 2013, 4, 566–577. [Google Scholar] [CrossRef]
- Roessner, P.M.; Hanna, B.S.; Öztürk, S.; Schulz, R.; Llaó Cid, L.; Yazdanparast, H.; Scheffold, A.; Colomer, D.; Stilgenbauer, S.; Lichter, P. TBET-expressing Th1 CD4+ T cells accumulate in chronic lymphocytic leukaemia without affecting disease progression in Eµ-TCL1 mice. Br. J. Haematol. 2020, 189, 133–145. [Google Scholar] [CrossRef]
- Lad, D.P.; Varma, S.; Varma, N.; Sachdeva, M.U.S.; Bose, P.; Malhotra, P. Regulatory T-cells in B-cell chronic lymphocytic leukemia: Their role in disease progression and autoimmune cytopenias. Leuk. Lymphoma 2013, 54, 1012–1019. [Google Scholar] [CrossRef]
- Giannopoulos, K.; Schmitt, M.; Własiuk, P.; Chen, J.; Bojarska-Junak, A.; Kowal, M.; Rolinski, J.; Dmoszynska, A. The high frequency of T regulatory cells in patients with B-cell chronic lymphocytic leukemia is diminished through treatment with thalidomide. Leukemia 2008, 22, 222–224. [Google Scholar] [CrossRef]
- Beyer, M.; Kochanek, M.; Darabi, K.; Popov, A.; Jensen, M.; Endl, E.; Knolle, P.A.; Thomas, R.K.; von Bergwelt-Baildon, M.; Debey, S. Reduced frequencies and suppressive function of CD4+ CD25hi regulatory T cells in patients with chronic lymphocytic leukemia after therapy with fludarabine. Blood 2005, 106, 2018–2025. [Google Scholar] [CrossRef]
- D’Arena, G.; Laurenti, L.; Minervini, M.M.; Deaglio, S.; Bonello, L.; De Martino, L.; De Padua, L.; Savino, L.; Tarnani, M.; De Feo, V. Regulatory T-cell number is increased in chronic lymphocytic leukemia patients and correlates with progressive disease. Leuk. Res. 2011, 35, 363–368. [Google Scholar] [CrossRef]
- Weiss, L.; Melchardt, T.; Egle, A.; Grabmer, C.; Greil, R.; Tinhofer, I. Regulatory T cells predict the time to initial treatment in early stage chronic lymphocytic leukemia. Cancer 2011, 117, 2163–2169. [Google Scholar] [CrossRef]
- Davila, M.L.; Kloss, C.C.; Gunset, G.; Sadelain, M. CD19 CAR-Targeted T Cells Induce Long-Term Remission and B Cell Aplasia in an Immunocompetent Mouse Model of B Cell Acute Lymphoblastic Leukemia. PLoS ONE 2013, 8, e61338. [Google Scholar] [CrossRef]
- Boucher, J.C.; Li, G.; Kotani, H.; Cabral, M.L.; Morrissey, D.; Lee, S.B.; Spitler, K.; Beatty, N.J.; Cervantes, E.V.; Shrestha, B.; et al. CD28 Costimulatory Domain-Targeted Mutations Enhance Chimeric Antigen Receptor T-cell Function. Cancer Immunol. Res. 2021, 9, 62–74. [Google Scholar] [CrossRef]
- Li, G.; Park, K.; Davila, M.L. Gammaretroviral Production and T Cell Transduction to Genetically Retarget Primary T Cells Against Cancer. Methods Mol. Biol. 2017, 1514, 111–118. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Activation | Activation signal | Beads:cells ratio |
| CD3/ICOS (M-450 Tosylactivated dynabeads loaded with 50% anti-CD3 and 50% anti-ICOS purified antibodies) | 2:1 | |
| Cytokine/Ab mixture | Cytokine/Ab | Final concentration |
| IL-6 | 30 ng/mL | |
| IL-21 | 100 ng/mL | |
| IL-1β | 10 ng/mL | |
| TGF-β | 3 ng/mL | |
| Anti-IL-4 | 10 µg/mL | |
| Anti-IFNγ | 10 µg/mL | |
| IL-23 (Starting day 3 of culture) | 10 ng/mL | |
| IL-2 (Starting day 3 of culture) | 100 IU/mL |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gamal, W.; Mediavilla-Varela, M.; Uriepero-Palma, A.; Pinilla-Ibarz, J.; Sahakian, E. Optimization of In Vitro Th17 Polarization for Adoptive Cell Therapy in Chronic Lymphocytic Leukemia. Int. J. Mol. Sci. 2024, 25, 6324. https://doi.org/10.3390/ijms25126324
Gamal W, Mediavilla-Varela M, Uriepero-Palma A, Pinilla-Ibarz J, Sahakian E. Optimization of In Vitro Th17 Polarization for Adoptive Cell Therapy in Chronic Lymphocytic Leukemia. International Journal of Molecular Sciences. 2024; 25(12):6324. https://doi.org/10.3390/ijms25126324
Chicago/Turabian StyleGamal, Wael, Melanie Mediavilla-Varela, Angimar Uriepero-Palma, Javier Pinilla-Ibarz, and Eva Sahakian. 2024. "Optimization of In Vitro Th17 Polarization for Adoptive Cell Therapy in Chronic Lymphocytic Leukemia" International Journal of Molecular Sciences 25, no. 12: 6324. https://doi.org/10.3390/ijms25126324
APA StyleGamal, W., Mediavilla-Varela, M., Uriepero-Palma, A., Pinilla-Ibarz, J., & Sahakian, E. (2024). Optimization of In Vitro Th17 Polarization for Adoptive Cell Therapy in Chronic Lymphocytic Leukemia. International Journal of Molecular Sciences, 25(12), 6324. https://doi.org/10.3390/ijms25126324