
Structural Insights into the Mechanisms Underlying Polyaminopathies

Abstract
1. Introduction
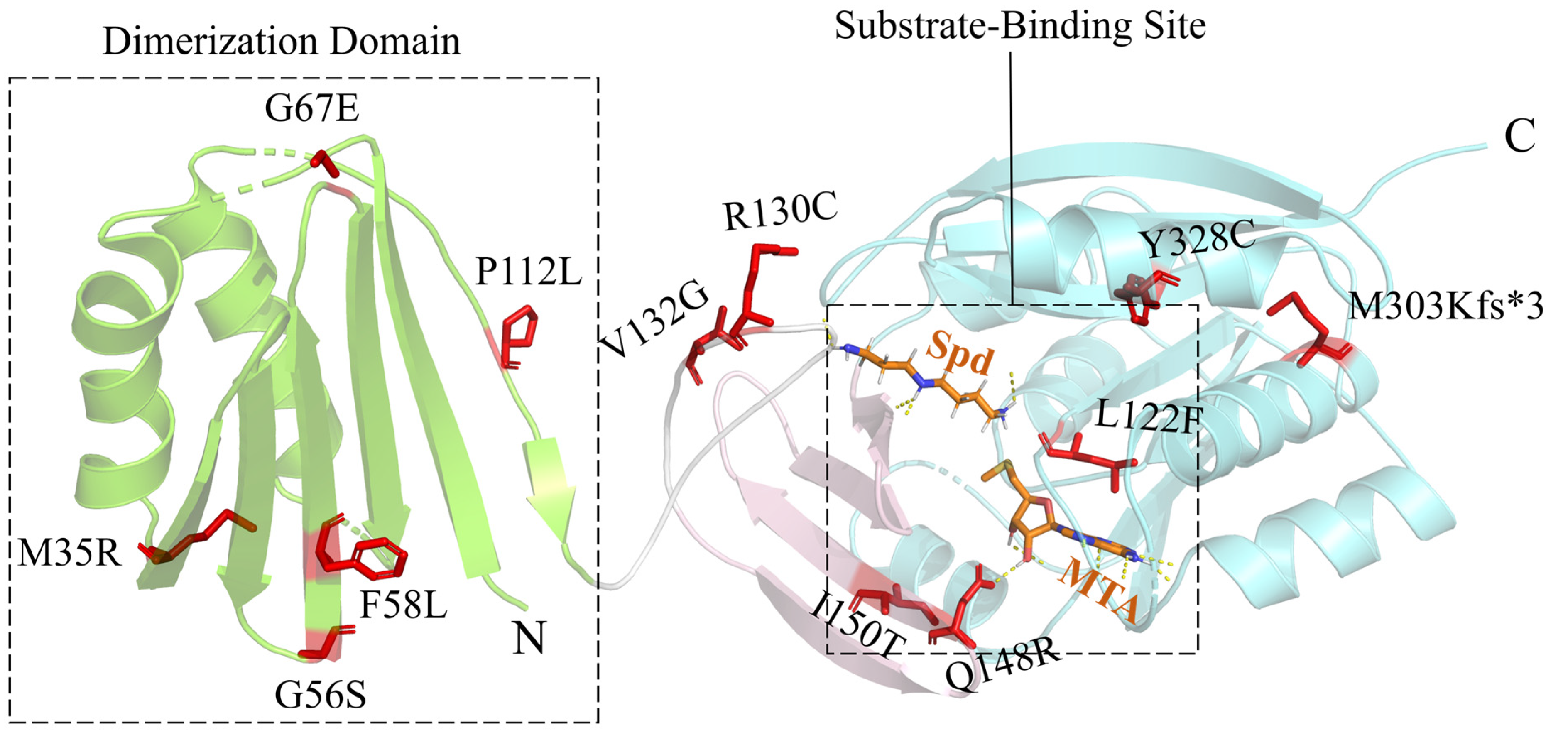
2. Snyder–Robinson Syndrome
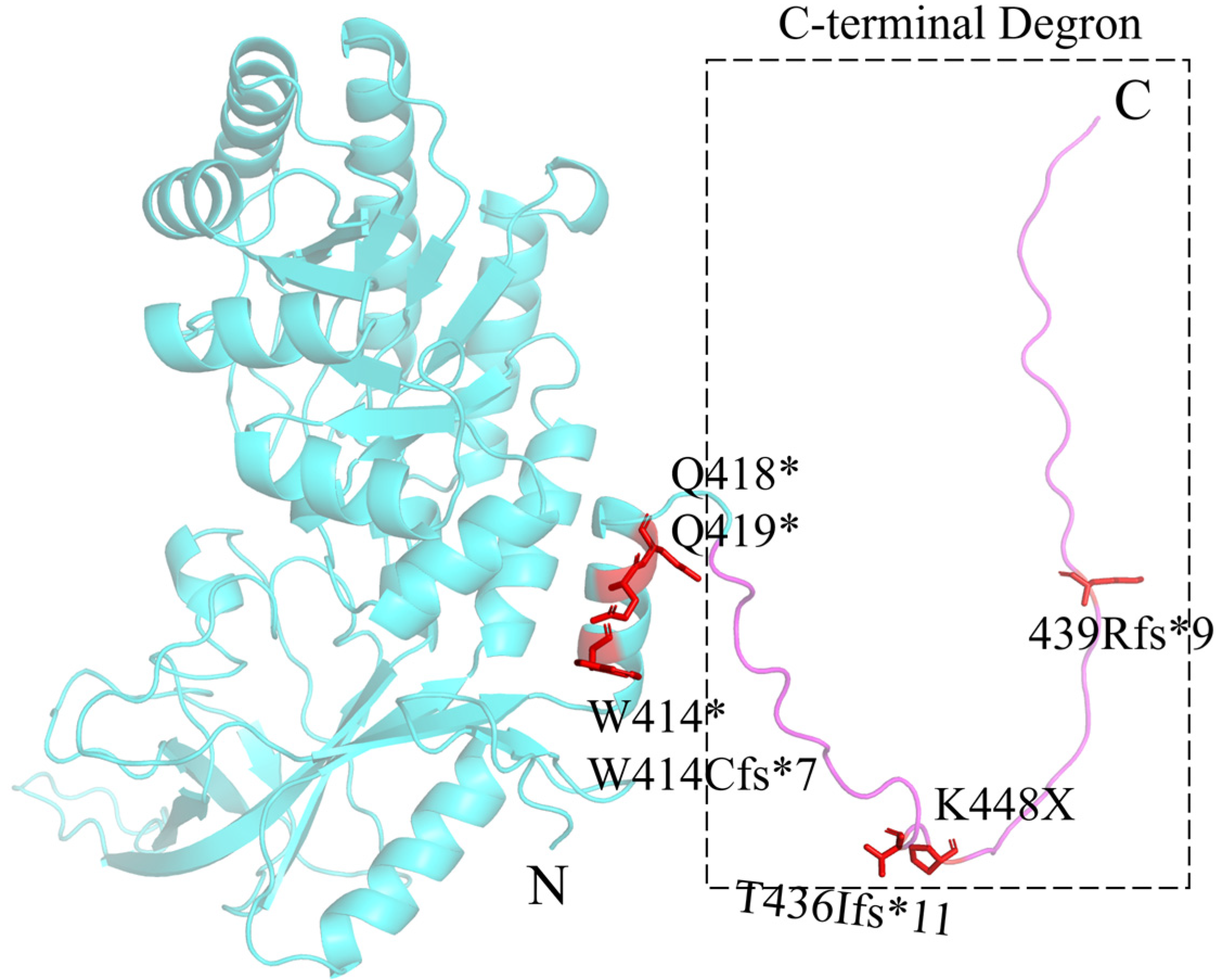
3. Bachmann–Bupp Syndrome
4. Deoxyhypusine Synthase Disorder
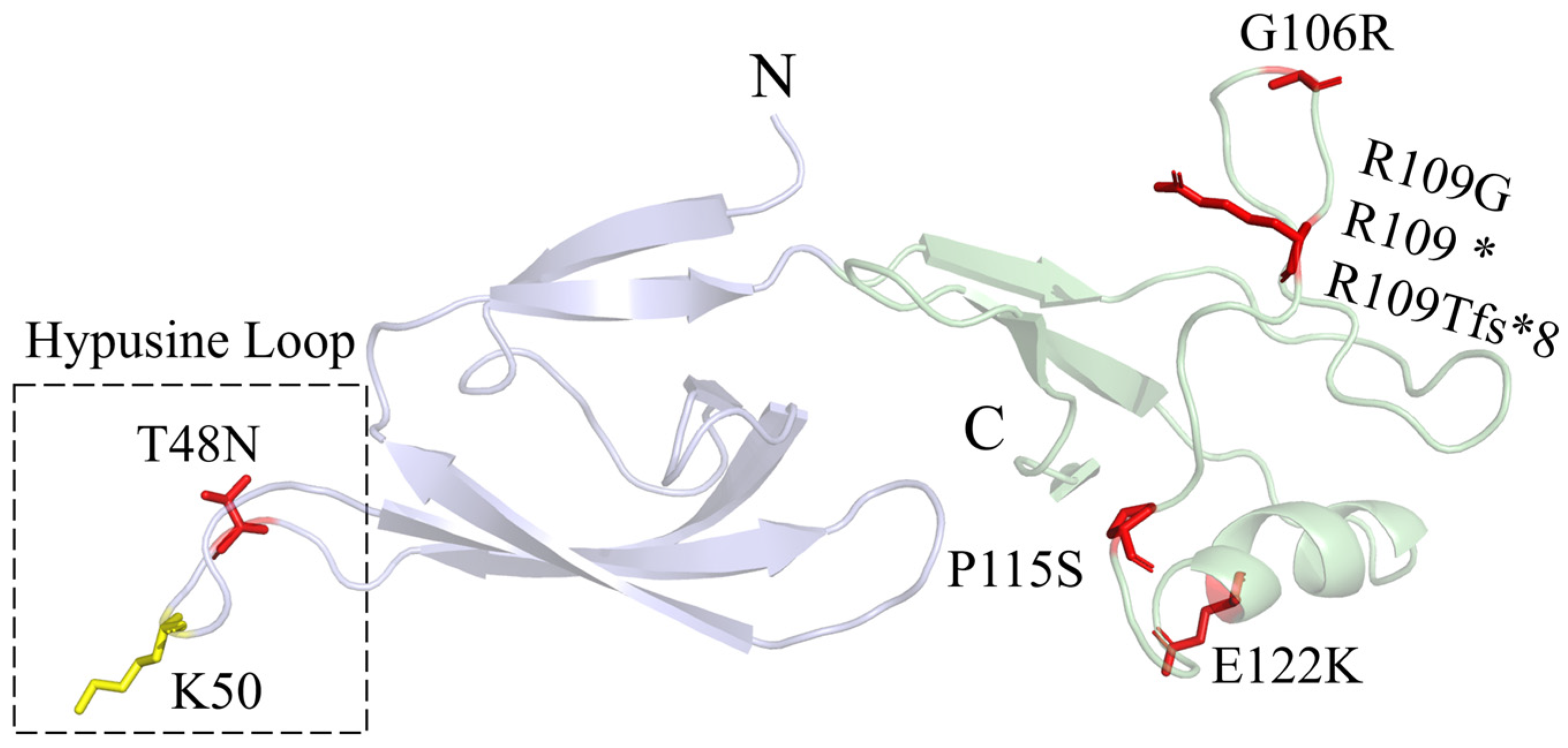
5. Faundes–Banka Syndrome
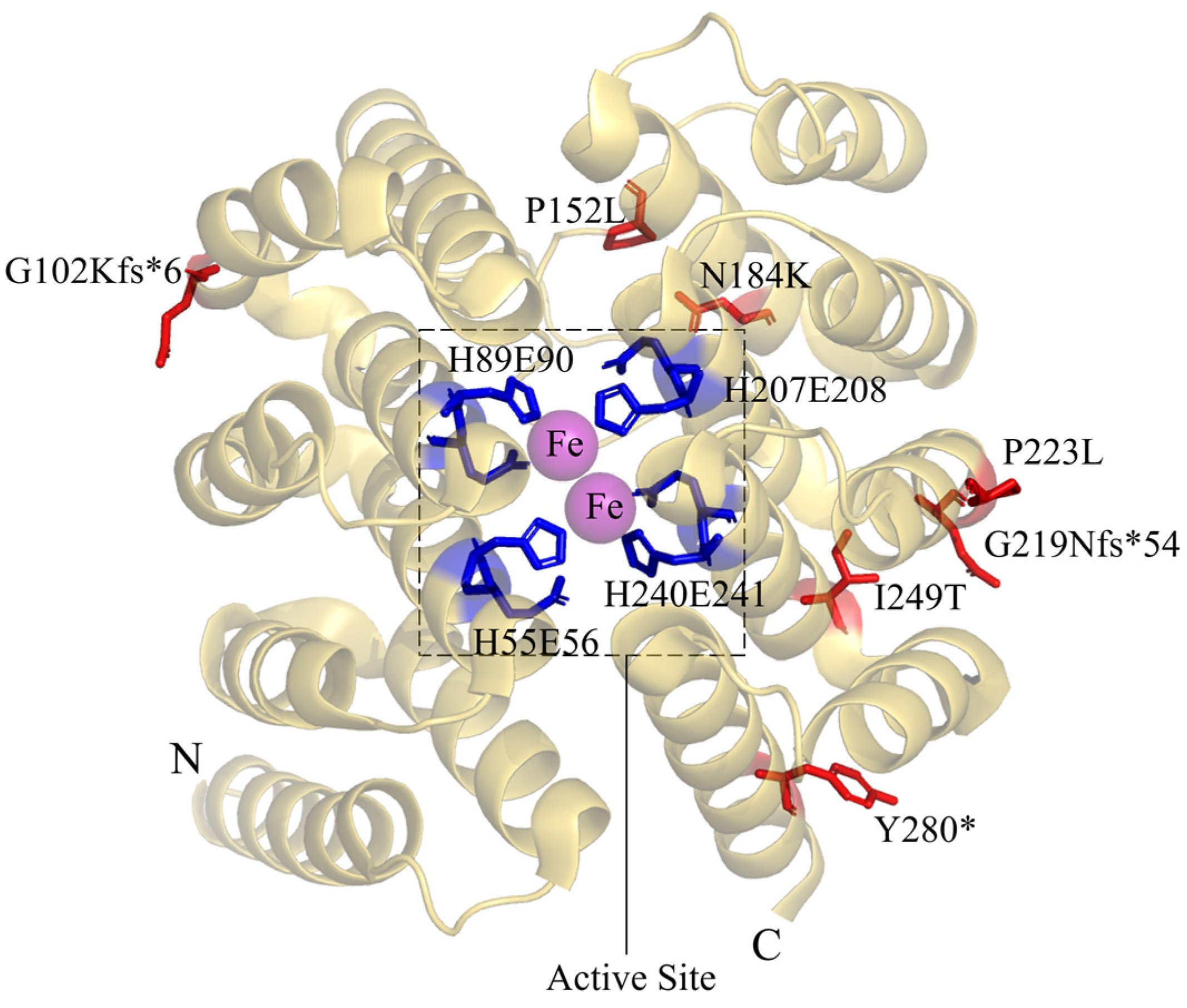
6. Deoxyhypusine Hydroxylase Disorder
7. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Xuan, M.; Gu, X.; Li, J.; Huang, D.; Xue, C.; He, Y. Polyamines: Their Significance for Maintaining Health and Contributing to Diseases. Cell Commun. Signal. 2023, 21, 348. [Google Scholar] [CrossRef] [PubMed]
- Uemura, T.; Yerushalmi, H.F.; Tsaprailis, G.; Stringer, D.E.; Pastorian, K.E.; Hawel, L.; Byus, C.V.; Gerner, E.W. Identification and Characterization of a Diamine Exporter in Colon Epithelial Cells. J. Biol. Chem. 2008, 283, 26428–26435. [Google Scholar] [CrossRef] [PubMed]
- Sekhar, V.; Andl, T.; Phanstiel, O. ATP13A3 Facilitates Polyamine Transport in Human Pancreatic Cancer Cells. Sci. Rep. 2022, 12, 4045. [Google Scholar] [CrossRef] [PubMed]
- Barba-Aliaga, M.; Alepuz, P. Role of eIF5A in Mitochondrial Function. Int. J. Mol. Sci. 2022, 23, 1284. [Google Scholar] [CrossRef] [PubMed]
- Tauc, M.; Cougnon, M.; Carcy, R.; Melis, N.; Hauet, T.; Pellerin, L.; Blondeau, N.; Pisani, D.F. The Eukaryotic Initiation Factor 5A (eIF5A1), the Molecule, Mechanisms and Recent Insights into the Pathophysiological Roles. Cell Biosci. 2021, 11, 219. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Wu, B.-K.; Kanchwala, M.; Cai, J.; Wang, L.; Xing, C.; Zheng, Y.; Pan, D. YAP/TAZ Drives Cell Proliferation and Tumour Growth via a Polyamine–eIF5A Hypusination–LSD1 Axis. Nat. Cell Biol. 2022, 24, 373–383. [Google Scholar] [CrossRef] [PubMed]
- Kang, B.; Wang, X.; An, X.; Ji, C.; Ling, W.; Qi, Y.; Li, S.; Jiang, D. Polyamines in Ovarian Aging and Disease. Int. J. Mol. Sci. 2023, 24, 15330. [Google Scholar] [CrossRef] [PubMed]
- Luo, D.; Lu, X.; Li, Y.; Xu, Y.; Zhou, Y.; Mao, H. Metabolism of Polyamines and Kidney Disease: A Promising Therapeutic Target. Kidney Dis. 2023, 9, 469–484. [Google Scholar] [CrossRef] [PubMed]
- Vrijsen, S.; Houdou, M.; Cascalho, A.; Eggermont, J.; Vangheluwe, P. Polyamines in Parkinson’s Disease: Balancing between Neurotoxicity and Neuroprotection. Annu. Rev. Biochem. 2023, 92, 435–464. [Google Scholar] [CrossRef]
- Holbert, C.E.; Cullen, M.T.; Casero, R.A.; Stewart, T.M. Polyamines in Cancer: Integrating Organismal Metabolism and Antitumour Immunity. Nat. Rev. Cancer 2022, 22, 467–480. [Google Scholar] [CrossRef]
- Milovic, V.; Turchanowa, L. Polyamines and Colon Cancer. Biochem. Soc. Trans. 2003, 31, 381–383. [Google Scholar] [CrossRef]
- Kulkarni, A.; Anderson, C.M.; Mirmira, R.G.; Tersey, S.A. Role of Polyamines and Hypusine in β Cells and Diabetes Pathogenesis. Metabolites 2022, 12, 344. [Google Scholar] [CrossRef]
- Fiches, G.N.; Wu, Z.; Zhou, D.; Biswas, A.; Li, T.-W.; Kong, W.; Jean, M.; Santoso, N.G.; Zhu, J. Polyamine Biosynthesis and eIF5A Hypusination Are Modulated by the DNA Tumor Virus KSHV and Promote KSHV Viral Infection. PLoS Pathog. 2022, 18, e1010503. [Google Scholar] [CrossRef]
- Smeltzer, S.; Quadri, Z.; Miller, A.; Zamudio, F.; Hunter, J.; Stewart, N.J.F.; Saji, S.; Lee, D.C.; Chaput, D.; Selenica, M.-L.B. Hypusination of Eif5a Regulates Cytoplasmic TDP-43 Aggregation and Accumulation in a Stress-Induced Cellular Model. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2021, 1867, 165939. [Google Scholar] [CrossRef] [PubMed]
- Sfakianos, A.P.; Raven, R.M.; Willis, A.E. The Pleiotropic Roles of eIF5A in Cellular Life and Its Therapeutic Potential in Cancer. Biochem. Soc. Trans. 2022, 50, 1885–1895. [Google Scholar] [CrossRef] [PubMed]
- Pendeville, H.; Carpino, N.; Marine, J.-C.; Takahashi, Y.; Muller, M.; Martial, J.A.; Cleveland, J.L. The Ornithine Decarboxylase Gene Is Essential for Cell Survival during Early Murine Development. Mol. Cell. Biol. 2001, 21, 6549–6558. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, K.; Nakatsu, F.; Kashiwagi, K.; Ohno, H.; Saito, T.; Igarashi, K. Essential Role of S-Adenosylmethionine Decarboxylase in Mouse Embryonic Development. Genes Cells Devoted Mol. Cell. Mech. 2002, 7, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, K.; Lee, S.B.; Park, J.H.; Park, M.H. Essential Role of eIF5A-1 and Deoxyhypusine Synthase in Mouse Embryonic Development. Amino Acids 2012, 42, 703–710. [Google Scholar] [CrossRef] [PubMed]
- Sievert, H.; Pällmann, N.; Miller, K.K.; Hermans-Borgmeyer, I.; Venz, S.; Sendoel, A.; Preukschas, M.; Schweizer, M.; Boettcher, S.; Janiesch, P.C.; et al. A Novel Mouse Model for Inhibition of DOHH-Mediated Hypusine Modification Reveals a Crucial Function in Embryonic Development, Proliferation and Oncogenic Transformation. Dis. Model. Mech. 2014, 7, 963–976. [Google Scholar] [CrossRef]
- Cason, A.L.; Ikeguchi, Y.; Skinner, C.; Wood, T.C.; Holden, K.R.; Lubs, H.A.; Martinez, F.; Simensen, R.J.; Stevenson, R.E.; Pegg, A.E.; et al. X-Linked Spermine Synthase Gene (SMS) Defect: The First Polyamine Deficiency Syndrome. Eur. J. Hum. Genet. 2003, 11, 937–944. [Google Scholar] [CrossRef]
- Bupp, C.P.; Schultz, C.R.; Uhl, K.L.; Rajasekaran, S.; Bachmann, A.S. Novel de Novo Pathogenic Variant in the ODC1 Gene in a Girl with Developmental Delay, Alopecia, and Dysmorphic Features. Am. J. Med. Genet. Part A 2018, 176, 2548–2553. [Google Scholar] [CrossRef] [PubMed]
- Ganapathi, M.; Padgett, L.R.; Yamada, K.; Devinsky, O.; Willaert, R.; Person, R.; Au, P.-Y.B.; Tagoe, J.; McDonald, M.; Karlowicz, D.; et al. Recessive Rare Variants in Deoxyhypusine Synthase, an Enzyme Involved in the Synthesis of Hypusine, Are Associated with a Neurodevelopmental Disorder. Am. J. Hum. Genet. 2019, 104, 287–298. [Google Scholar] [CrossRef]
- Faundes, V.; Jennings, M.D.; Crilly, S.; Legraie, S.; Withers, S.E.; Cuvertino, S.; Davies, S.J.; Douglas, A.G.L.; Fry, A.E.; Harrison, V.; et al. Impaired eIF5A Function Causes a Mendelian Disorder That Is Partially Rescued in Model Systems by Spermidine. Nat. Commun. 2021, 12, 833. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, A.; Steindl, K.; Hanner, A.S.; Kumar Kar, R.; Prouteau, C.; Boland, A.; Deleuze, J.F.; Coubes, C.; Bézieau, S.; Küry, S.; et al. Bi-Allelic Variants in DOHH, Catalyzing the Last Step of Hypusine Biosynthesis, Are Associated with a Neurodevelopmental Disorder. Am. J. Hum. Genet. 2022, 109, 1549–1558. [Google Scholar] [CrossRef] [PubMed]
- Bachmann, A.S.; VanSickle, E.A.; Michael, J.; Vipond, M.; Bupp, C.P. Bachmann-Bupp Syndrome and Treatment. Dev. Med. Child Neurol. 2024, 66, 445–455. [Google Scholar] [CrossRef] [PubMed]
- Snyder, R.D.; Robinson, A. Recessive Sex-Linked Mental Retardation in the Absence of Other Recognizable Abnormalities: Report of a Family. Clin. Pediatr. 1969, 8, 669–674. [Google Scholar] [CrossRef]
- Wu, H.; Min, J.; Zeng, H.; McCloskey, D.E.; Ikeguchi, Y.; Loppnau, P.; Michael, A.J.; Pegg, A.E.; Plotnikov, A.N. Crystal Structure of Human Spermine Synthase: Implications of Substrate Binding and Catalytic Mechanism. J. Biol. Chem. 2008, 283, 16135–16146. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Norris, J.; Schwartz, C.; Alexov, E. Revealing the Effects of Missense Mutations Causing Snyder-Robinson Syndrome on the Stability and Dimerization of Spermine Synthase. Int. J. Mol. Sci. 2016, 17, 77. [Google Scholar] [CrossRef] [PubMed]
- de Alencastro, G.; McCloskey, D.E.; Kliemann, S.E.; Maranduba, C.M.C.; Pegg, A.E.; Wang, X.; Bertola, D.R.; Schwartz, C.E.; Passos-Bueno, M.R.; Sertié, A.L. New SMS Mutation Leads to a Striking Reduction in Spermine Synthase Protein Function and a Severe Form of Snyder-Robinson X-Linked Recessive Mental Retardation Syndrome. J. Med. Genet. 2008, 45, 539–543. [Google Scholar] [CrossRef]
- Lemke, J.R.; Riesch, E.; Scheurenbrand, T.; Schubach, M.; Wilhelm, C.; Steiner, I.; Hansen, J.; Courage, C.; Gallati, S.; Bürki, S.; et al. Targeted next Generation Sequencing as a Diagnostic Tool in Epileptic Disorders. Epilepsia 2012, 53, 1387–1398. [Google Scholar] [CrossRef]
- Peron, A.; Spaccini, L.; Norris, J.; Bova, S.M.; Selicorni, A.; Weber, G.; Wood, T.; Schwartz, C.E.; Mastrangelo, M. Snyder-Robinson Syndrome: A Novel Nonsense Mutation in Spermine Synthase and Expansion of the Phenotype. Am. J. Med. Genet. Part A 2013, 161A, 2316–2320. [Google Scholar] [CrossRef]
- Abela, L.; Simmons, L.; Steindl, K.; Schmitt, B.; Mastrangelo, M.; Joset, P.; Papuc, M.; Sticht, H.; Baumer, A.; Crowther, L.M.; et al. N(8)-Acetylspermidine as a Potential Plasma Biomarker for Snyder-Robinson Syndrome Identified by Clinical Metabolomics. J. Inherit. Metab. Dis. 2016, 39, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Becerra-Solano, L.E.; Butler, J.; Castañeda-Cisneros, G.; McCloskey, D.E.; Wang, X.; Pegg, A.E.; Schwartz, C.E.; Sánchez-Corona, J.; García-Ortiz, J.E. A Missense Mutation, p.V132G, in the X-Linked Spermine Synthase Gene (SMS) Causes Snyder-Robinson Syndrome. Am. J. Med. Genet. Part A 2009, 149A, 328–335. [Google Scholar] [CrossRef]
- Albert, J.S.; Bhattacharyya, N.; Wolfe, L.A.; Bone, W.P.; Maduro, V.; Accardi, J.; Adams, D.R.; Schwartz, C.E.; Norris, J.; Wood, T.; et al. Impaired Osteoblast and Osteoclast Function Characterize the Osteoporosis of Snyder-Robinson Syndrome. Orphanet J. Rare Dis. 2015, 10, 27. [Google Scholar] [CrossRef]
- Zhang, Z.; Teng, S.; Wang, L.; Schwartz, C.E.; Alexov, E. Computational Analysis of Missense Mutations Causing Snyder-Robinson Syndrome. Hum. Mutat. 2010, 31, 1043–1049. [Google Scholar] [CrossRef] [PubMed]
- Starks, R.; Kirby, P.; Ciliberto, M.; Hefti, M. Snyder-Robinson Syndrome. Autops. Case Rep. 2018, 8, e2018031. [Google Scholar] [CrossRef]
- Larcher, L.; Norris, J.W.; Lejeune, E.; Buratti, J.; Mignot, C.; Garel, C.; Keren, B.; Schwartz, C.E.; Whalen, S. The Complete Loss of Function of the SMS Gene Results in a Severe Form of Snyder-Robinson Syndrome. Eur. J. Med. Genet. 2020, 63, 103777. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Norris, J.; Kalscheuer, V.; Wood, T.; Wang, L.; Schwartz, C.; Alexov, E.; Van Esch, H. A Y328C Missense Mutation in Spermine Synthase Causes a Mild Form of Snyder-Robinson Syndrome. Hum. Mol. Genet. 2013, 22, 3789–3797. [Google Scholar] [CrossRef]
- Wang, X.; Levic, S.; Gratton, M.A.; Doyle, K.J.; Yamoah, E.N.; Pegg, A.E. Spermine Synthase Deficiency Leads to Deafness and a Profound Sensitivity to Alpha-Difluoromethylornithine. J. Biol. Chem. 2009, 284, 930–937. [Google Scholar] [CrossRef]
- Tantak, M.P.; Sekhar, V.; Tao, X.; Zhai, R.G.; Phanstiel, O. Development of a Redox-Sensitive Spermine Prodrug for the Potential Treatment of Snyder Robinson Syndrome. J. Med. Chem. 2021, 64, 15593–15607. [Google Scholar] [CrossRef]
- Tao, X.; Zhu, Y.; Diaz-Perez, Z.; Yu, S.-H.; Foley, J.R.; Stewart, T.M.; Casero, R.A.; Steet, R.; Zhai, R.G. Phenylbutyrate Modulates Polyamine Acetylase and Ameliorates Snyder-Robinson Syndrome in a Drosophila Model and Patient Cells. JCI Insight 2022, 7, e158457. [Google Scholar] [CrossRef] [PubMed]
- Stewart, T.M.; Khomutov, M.; Foley, J.R.; Guo, X.; Holbert, C.E.; Dunston, T.T.; Schwartz, C.E.; Gabrielson, K.; Khomutov, A.; Casero, R.A. (R,R)-1,12-Dimethylspermine Can Mitigate Abnormal Spermidine Accumulation in Snyder-Robinson Syndrome. J. Biol. Chem. 2020, 295, 3247–3256. [Google Scholar] [CrossRef] [PubMed]
- Stewart, T.M.; Foley, J.R.; Holbert, C.E.; Khomutov, M.; Rastkari, N.; Tao, X.; Khomutov, A.R.; Zhai, R.G.; Casero, R.A. Difluoromethylornithine Rebalances Aberrant Polyamine Ratios in SNYDER–ROBINSON Syndrome. EMBO Mol. Med. 2023, 15, e17833. [Google Scholar] [CrossRef]
- Rodan, L.H.; Anyane-Yeboa, K.; Chong, K.; Klein Wassink-Ruiter, J.S.; Wilson, A.; Smith, L.; Kothare, S.V.; Rajabi, F.; Blaser, S.; Ni, M.; et al. Gain-of-Function Variants in the ODC1 Gene Cause a Syndromic Neurodevelopmental Disorder Associated with Macrocephaly, Alopecia, Dysmorphic Features, and Neuroimaging Abnormalities. Am. J. Med. Genet. Part A 2018, 176, 2554–2560. [Google Scholar] [CrossRef]
- VanSickle, E.A.; Michael, J.; Bachmann, A.S.; Rajasekaran, S.; Prokop, J.W.; Kuzniecky, R.; Hofstede, F.C.; Steindl, K.; Rauch, A.; Lipson, M.H.; et al. Expanding the Phenotype: Four New Cases and Hope for Treatment in Bachmann-Bupp Syndrome. Am. J. Med. Genet. Part A 2021, 185, 3485–3493. [Google Scholar] [CrossRef]
- Michael, J.; VanSickle, E.; Vipond, M.; Dalman, A.; Prokop, J.; Schwartz, C.E.; Rajasekaran, S.; Bachmann, A.S.; Barth, M.; Prouteau, C.; et al. Two New Cases of Bachmann-Bupp Syndrome Identified through the International Center for Polyamine Disorders. Med. Sci. 2023, 11, 29. [Google Scholar] [CrossRef] [PubMed]
- Kahana, C. The Antizyme Family for Regulating Polyamines. J. Biol. Chem. 2018, 293, 18730–18735. [Google Scholar] [CrossRef] [PubMed]
- Rajasekaran, S.; Bupp, C.P.; Leimanis-Laurens, M.; Shukla, A.; Russell, C.; Junewick, J.; Gleason, E.; VanSickle, E.A.; Edgerly, Y.; Wittmann, B.M.; et al. Repurposing Eflornithine to Treat a Patient with a Rare ODC1 Gain-of-Function Variant Disease. eLife 2021, 10, e67097. [Google Scholar] [CrossRef] [PubMed]
- Wątor, E.; Wilk, P.; Grudnik, P. Half Way to Hypusine—Structural Basis for Substrate Recognition by Human Deoxyhypusine Synthase. Biomolecules 2020, 10, 522. [Google Scholar] [CrossRef]
- Liao, D.I.; Wolff, E.C.; Park, M.H.; Davies, D.R. Crystal Structure of the NAD Complex of Human Deoxyhypusine Synthase: An Enzyme with a Ball-and-Chain Mechanism for Blocking the Active Site. Structure 1998, 6, 23–32. [Google Scholar] [CrossRef]
- Wątor, E.; Wilk, P.; Biela, A.; Rawski, M.; Zak, K.M.; Steinchen, W.; Bange, G.; Glatt, S.; Grudnik, P. Cryo-EM Structure of Human eIF5A-DHS Complex Reveals the Molecular Basis of Hypusination-Associated Neurodegenerative Disorders. Nat. Commun. 2023, 14, 1698. [Google Scholar] [CrossRef] [PubMed]
- Umland, T.C.; Wolff, E.C.; Park, M.H.; Davies, D.R. A New Crystal Structure of Deoxyhypusine Synthase Reveals the Configuration of the Active Enzyme and of an Enzyme.NAD.Inhibitor Ternary Complex. J. Biol. Chem. 2004, 279, 28697–28705. [Google Scholar] [CrossRef] [PubMed]
- Tong, Y.; Park, I.; Hong, B.; Nedyalkova, L.; Tempel, W.; Park, H. Crystal Structure of Human eIF5A1: Insight into Functional Similarity of Human eIF5A1 and eIF5A2. Proteins 2009, 75, 1040–1045. [Google Scholar] [CrossRef]
- Cano, V.S.P.; Jeon, G.A.; Johansson, H.E.; Henderson, C.A.; Park, J.-H.; Valentini, S.R.; Hershey, J.W.B.; Park, M.H. Mutational Analyses of Human eIF5A-1--Identification of Amino Acid Residues Critical for eIF5A Activity and Hypusine Modification. FEBS J. 2008, 275, 44–58. [Google Scholar] [CrossRef] [PubMed]
- Zanelli, C.F.; Valentini, S.R. Is There a Role for eIF5A in Translation? Amino Acids 2007, 33, 351–358. [Google Scholar] [CrossRef]
- Park, M.H.; Kar, R.K.; Banka, S.; Ziegler, A.; Chung, W.K. Post-Translational Formation of Hypusine in eIF5A: Implications in Human Neurodevelopment. Amino Acids 2022, 54, 485–499. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.; Sakai, N.; Böttger, L.H.; Klinke, S.; Hauber, J.; Trautwein, A.X.; Hilgenfeld, R. Crystal Structure of the Peroxo-Diiron(III) Intermediate of Deoxyhypusine Hydroxylase, an Oxygenase Involved in Hypusination. Structure 2015, 23, 882–892. [Google Scholar] [CrossRef]
- Park, J.-H.; Aravind, L.; Wolff, E.C.; Kaevel, J.; Kim, Y.S.; Park, M.H. Molecular Cloning, Expression, and Structural Prediction of Deoxyhypusine Hydroxylase: A HEAT-Repeat-Containing Metalloenzyme. Proc. Natl. Acad. Sci. USA 2006, 103, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Karamanou, M.; Poulakou-Rebelakou, E.; Tzetis, M.; Androutsos, G. Anton van Leeuwenhoek (1632–1723): Father of Micromorphology and Discoverer of Spermatozoa. Rev. Argent. Microbiol. 2010, 42, 311–314. [Google Scholar]
- Hofer, S.J.; Liang, Y.; Zimmermann, A.; Schroeder, S.; Dengjel, J.; Kroemer, G.; Eisenberg, T.; Sigrist, S.J.; Madeo, F. Spermidine-Induced Hypusination Preserves Mitochondrial and Cognitive Function during Aging. Autophagy 2021, 17, 2037–2039. [Google Scholar] [CrossRef]
- Eisenberg, T.; Knauer, H.; Schauer, A.; Büttner, S.; Ruckenstuhl, C.; Carmona-Gutierrez, D.; Ring, J.; Schroeder, S.; Magnes, C.; Antonacci, L.; et al. Induction of Autophagy by Spermidine Promotes Longevity. Nat. Cell Biol. 2009, 11, 1305–1314. [Google Scholar] [CrossRef] [PubMed]
- Madeo, F.; Eisenberg, T.; Pietrocola, F.; Kroemer, G. Spermidine in Health and Disease. Science 2018, 359, eaan2788. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.B.; Park, J.H.; Folk, J.E.; Deck, J.A.; Pegg, A.E.; Sokabe, M.; Fraser, C.S.; Park, M.H. Inactivation of Eukaryotic Initiation Factor 5A (eIF5A) by Specific Acetylation of Its Hypusine Residue by Spermidine/Spermine Acetyltransferase 1 (SSAT1). Biochem. J. 2011, 433, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Alayoubi, A.M.; Iqbal, M.; Aman, H.; Hashmi, J.A.; Alayadhi, L.; Al-Regaiey, K.; Basit, S. Loss-of-Function Variant in Spermidine/Spermine N1-Acetyl Transferase like 1 (SATL1) Gene as an Underlying Cause of Autism Spectrum Disorder. Sci. Rep. 2024, 14, 5765. [Google Scholar] [CrossRef]
- Schwarz, C.; Stekovic, S.; Wirth, M.; Benson, G.; Royer, P.; Sigrist, S.J.; Pieber, T.; Dammbrueck, C.; Magnes, C.; Eisenberg, T.; et al. Safety and Tolerability of Spermidine Supplementation in Mice and Older Adults with Subjective Cognitive Decline. Aging 2018, 10, 19–33. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polyaminopathies | Name | Mutant Gene | Genetic Mode | Mutant Form | First Case | Cases | Deaths | Clinical Symptoms |
|---|---|---|---|---|---|---|---|---|
| Polyamine biosynthesis-related diseases | SRS | SMS | X-linked recessive | Monogenic mutations | 2003 | 24 males | 2 males | Developmental delay, intellectual disability, hypotonia, seizures, osteoporosis, kyphosis, genital abnormalities, facial dysmorphism |
| BABS | ODC | autosomal dominant | Monogenic mutations | 2018 | 6 males 5 females | 1 male (labor induction) | Developmental delay, intellectual disability, hypotonia, non-congenital alopecia, abnormal brain MRI, non-specific dysmorphic features, macrocephaly | |
| Polyamine utilization-related diseases | DHPS disorder | DHPS | autosomal recessive | Biallelic mutations | 2019 | 1 male 4 females | none | Developmental delay, intellectual disability, seizures, dystonia, pregnancy problems |
| FABAS | EIF5A | autosomal dominant | Monogenic mutations | 2021 | 3 males 4 females | none | Developmental delay, intellectual disability, facial deformity, microcephaly | |
| DOHH disorder | DOHH | autosomal recessive | Biallelic mutations | 2022 | 3 males 2 females | 1 male 1 female | Developmental delay, intellectual disability, brain MRI abnormalities, microcephaly, congenital cardiac malformations |
| Polyaminopathies | Name | Mutant Gene | Variants (Gene) | Variants (Protein) | ||||
|---|---|---|---|---|---|---|---|---|
| Polyamine biosynthesis-related diseases | SRS | SMS | c.104T>G c.166G>A c.174T>A c.200G>A c.329+5G>A | c.335C>T c.388C>T c.395T>G c.443A>G c.449T>C | c.831G>T c.908_911del c.983A>G | p.M35R p.G56S p.F58L p.G67E p.? | p.P112L p.R130C p.V132G p.Q148R p.I150T | p.L277F p.M303Kfs*3 p.Y328C |
| BABS | ODC | c.1240_1241dupTG c.1241+1G>T c.1242_1263del22 | c.1242-2A>G c.1252C>T c.1255C>T | c.1307_1311delinsT c.1342A>T c.1313_1316delCTGT | p.W414Cfs*7 p.? p.W414* | p.? p.Q418* p.Q419* | p.T436Ifs*11 p.K448K p.438Rfs*9 | |
| Polyamine utilization-related diseases | DHPS disorder | DHPS | c.1A>G c.518A>G | c.912_917delTTACAT | c.1014+1G>A | p.Met1? p.N173S | p.Y305_I306del | p.? |
| FABAS | EIF5A | c.143C>A c.316G>A c.324dupA | c.325C>G c.325C>T c.343C>T | c.364G>A | p.T48N p.G106R p.R109Tfs*8 | p.R109G p.R109* p.P115S | p.E122K | |
| DOHH disorder | DOHH | c.304delG c.455C>T c.552C>A | c.654_655insAACC c.668C>T c.746T>C | c.840T>A | p.G102Kfs*6 p.P152L p.N184K | p.G219Nfs*54 p.P223L p.I249T | p.Y280* | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, B.; Liu, S. Structural Insights into the Mechanisms Underlying Polyaminopathies. Int. J. Mol. Sci. 2024, 25, 6340. https://doi.org/10.3390/ijms25126340
Wu B, Liu S. Structural Insights into the Mechanisms Underlying Polyaminopathies. International Journal of Molecular Sciences. 2024; 25(12):6340. https://doi.org/10.3390/ijms25126340
Chicago/Turabian StyleWu, Bing, and Sen Liu. 2024. "Structural Insights into the Mechanisms Underlying Polyaminopathies" International Journal of Molecular Sciences 25, no. 12: 6340. https://doi.org/10.3390/ijms25126340
APA StyleWu, B., & Liu, S. (2024). Structural Insights into the Mechanisms Underlying Polyaminopathies. International Journal of Molecular Sciences, 25(12), 6340. https://doi.org/10.3390/ijms25126340