
Multiplexing LAMP Assays: A Methodological Review and Diagnostic Application


Abstract
1. Introduction
2. Multiplexing LAMP Assays
2.1. Methods Using Modified Primers
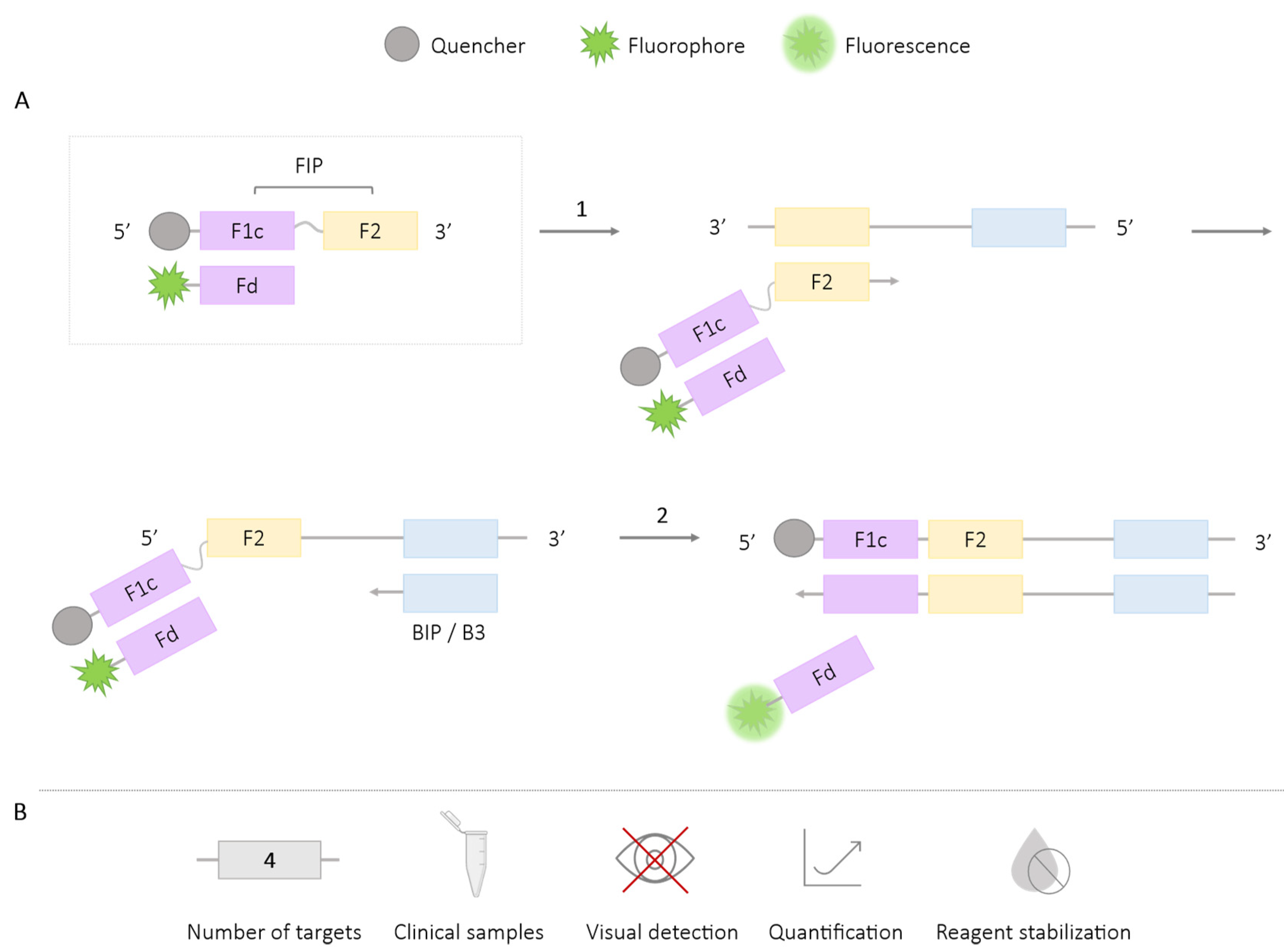
2.1.1. DARQ
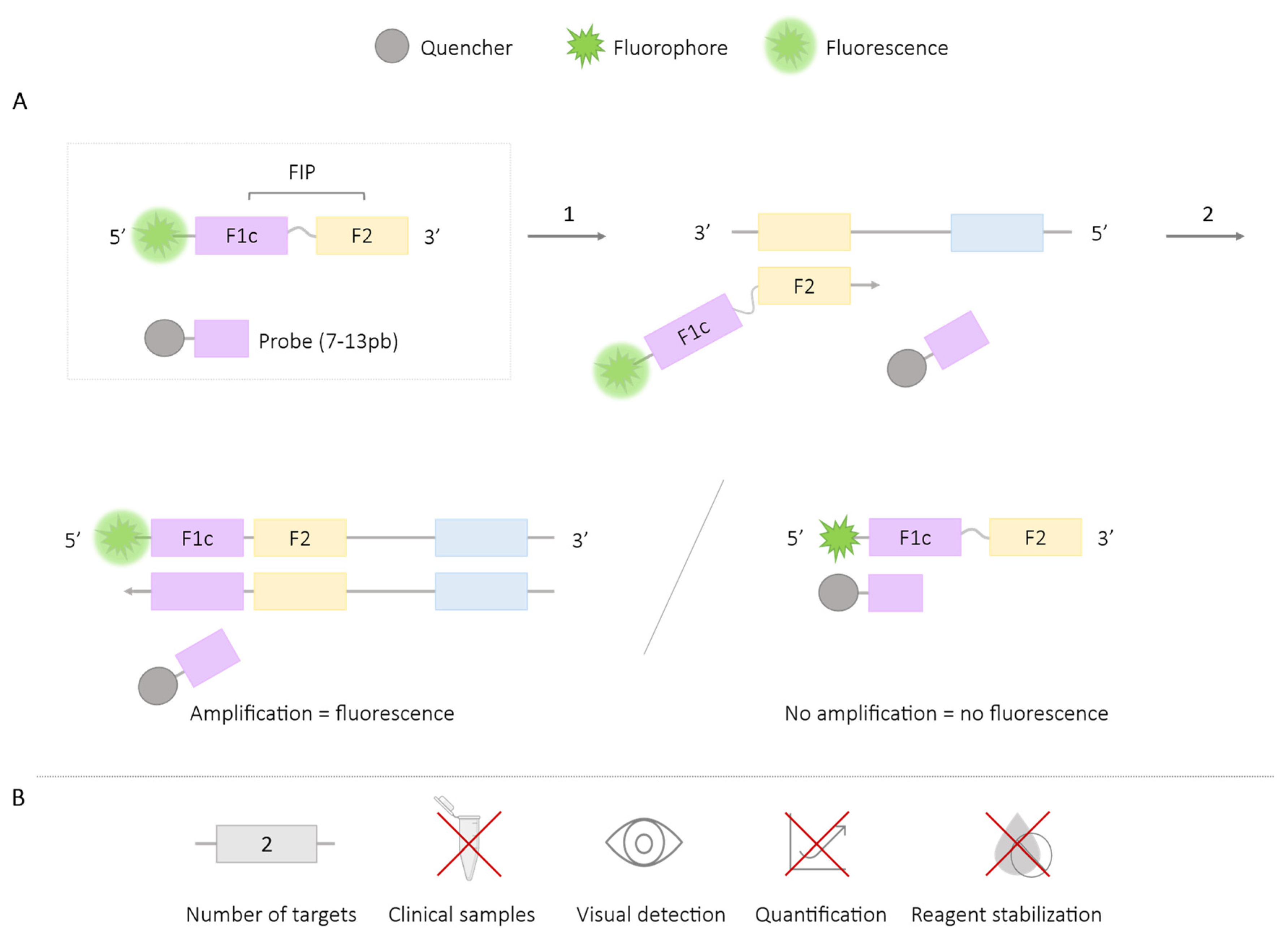
2.1.2. QUASR
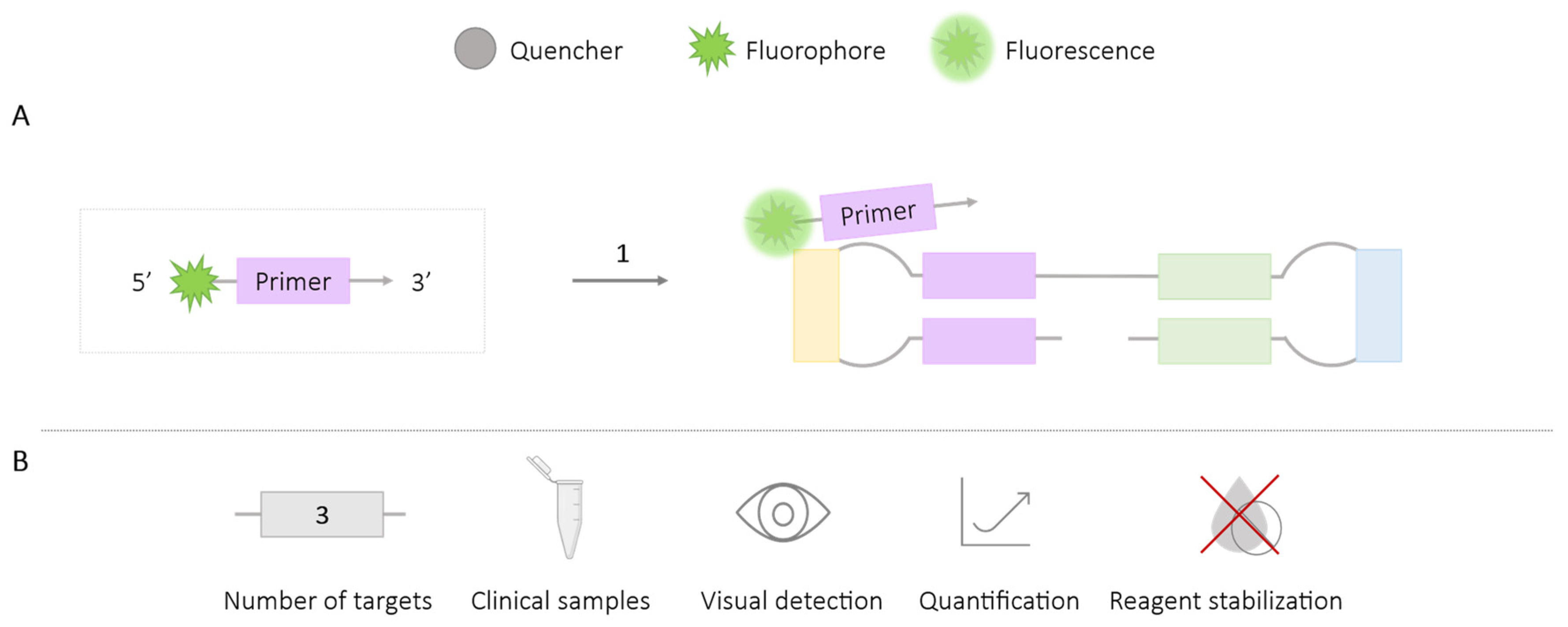
2.1.3. FLOS
2.1.4. Guanine Quenching
2.1.5. Assimilation Probes or Primer Fluorescence Probes
2.2. Methods Using Universal Probes
2.2.1. One-Step Strand Displacement (OSD) Probes
2.2.2. Molecular Beacon
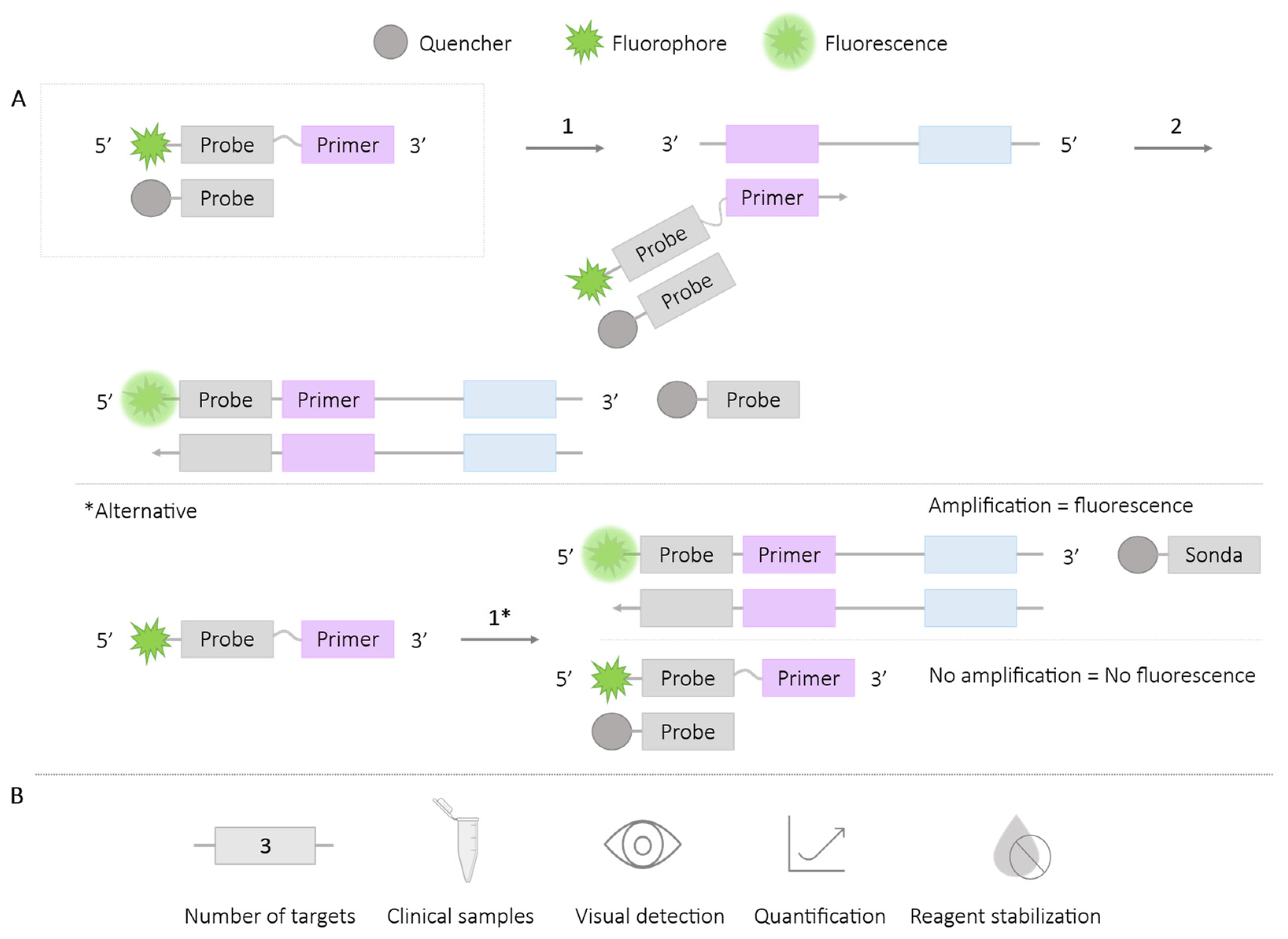
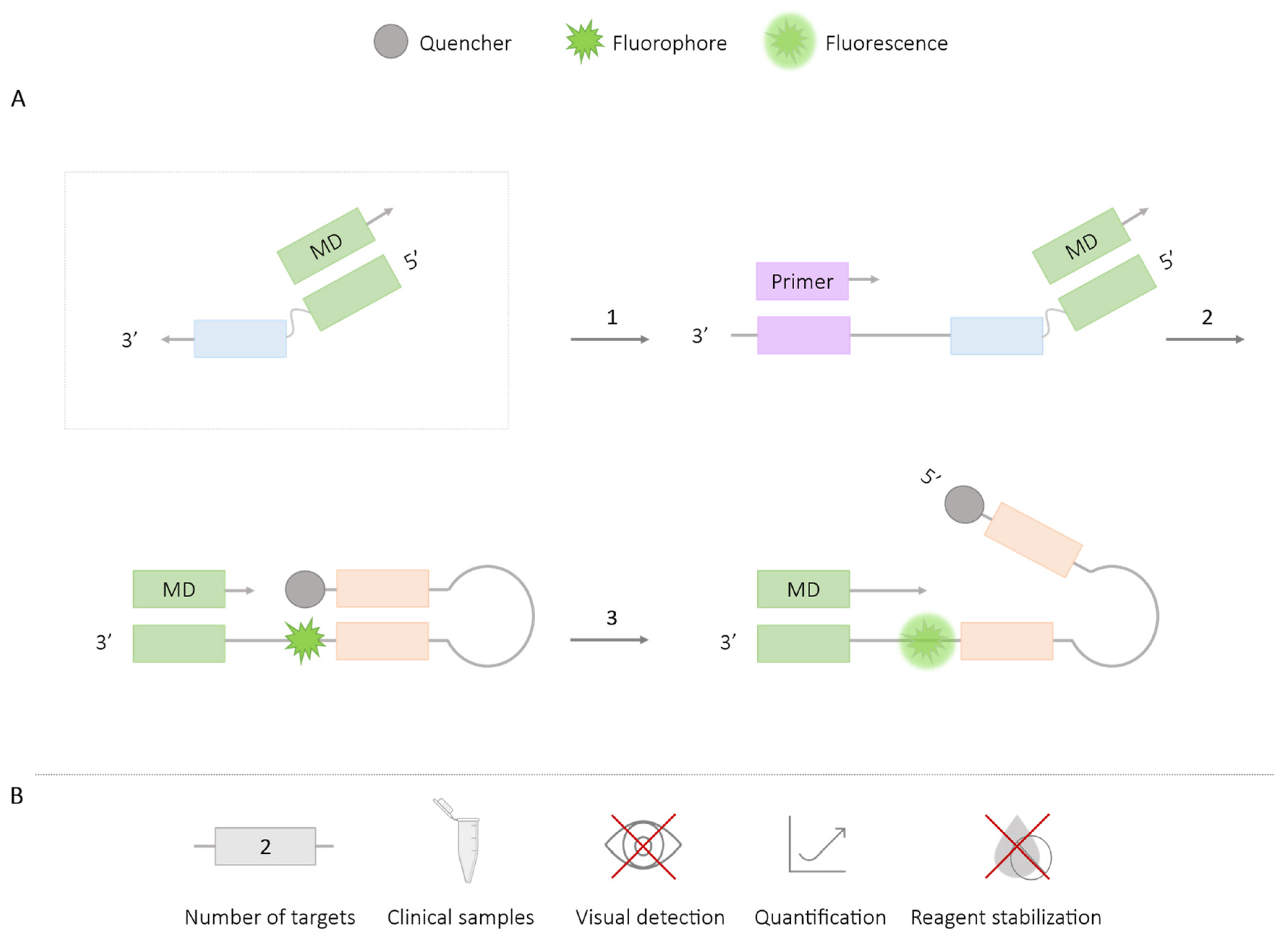
2.2.3. MD Probe
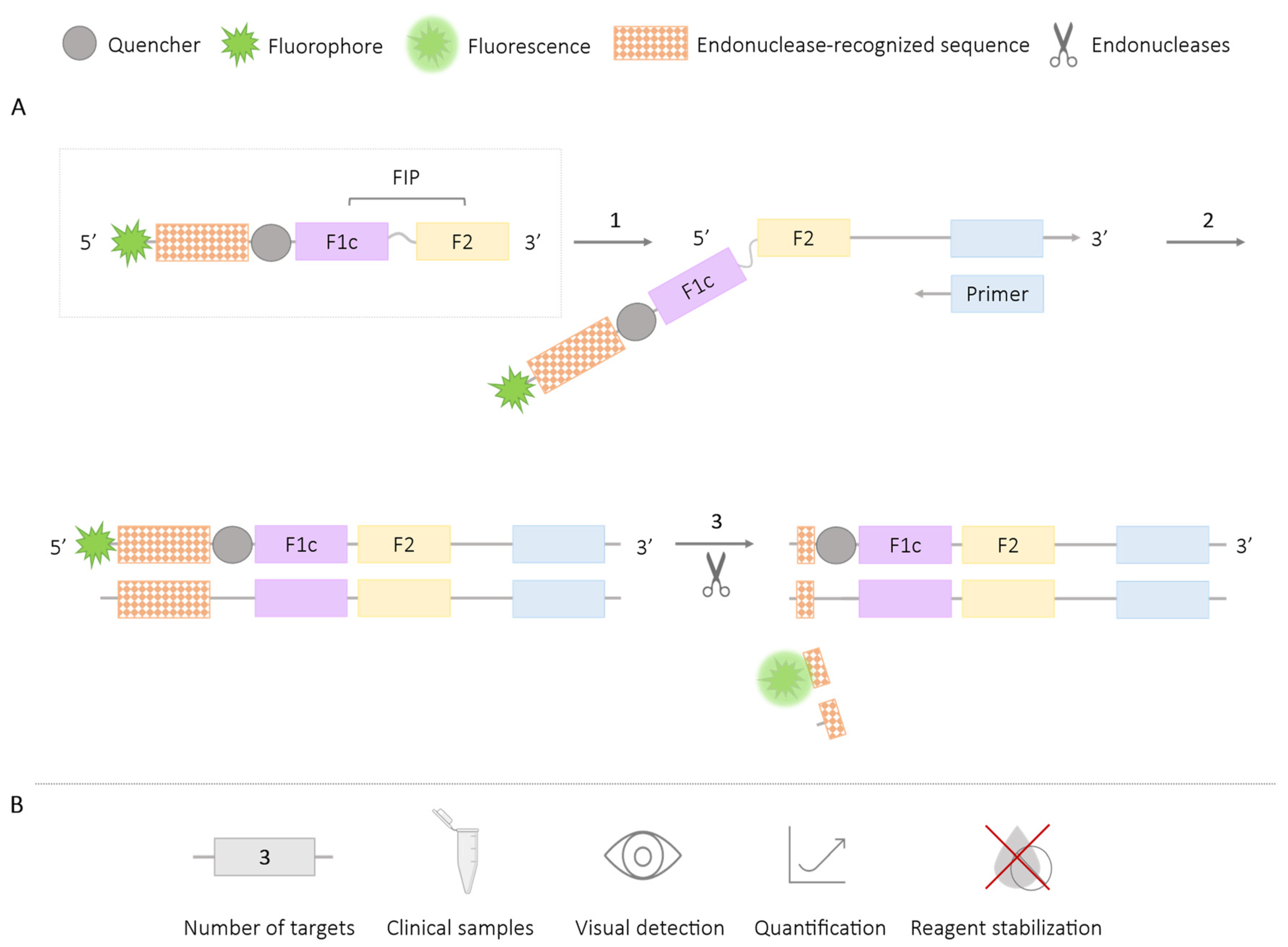
2.3. Methods Using Restriction Enzymes or Endonucleases
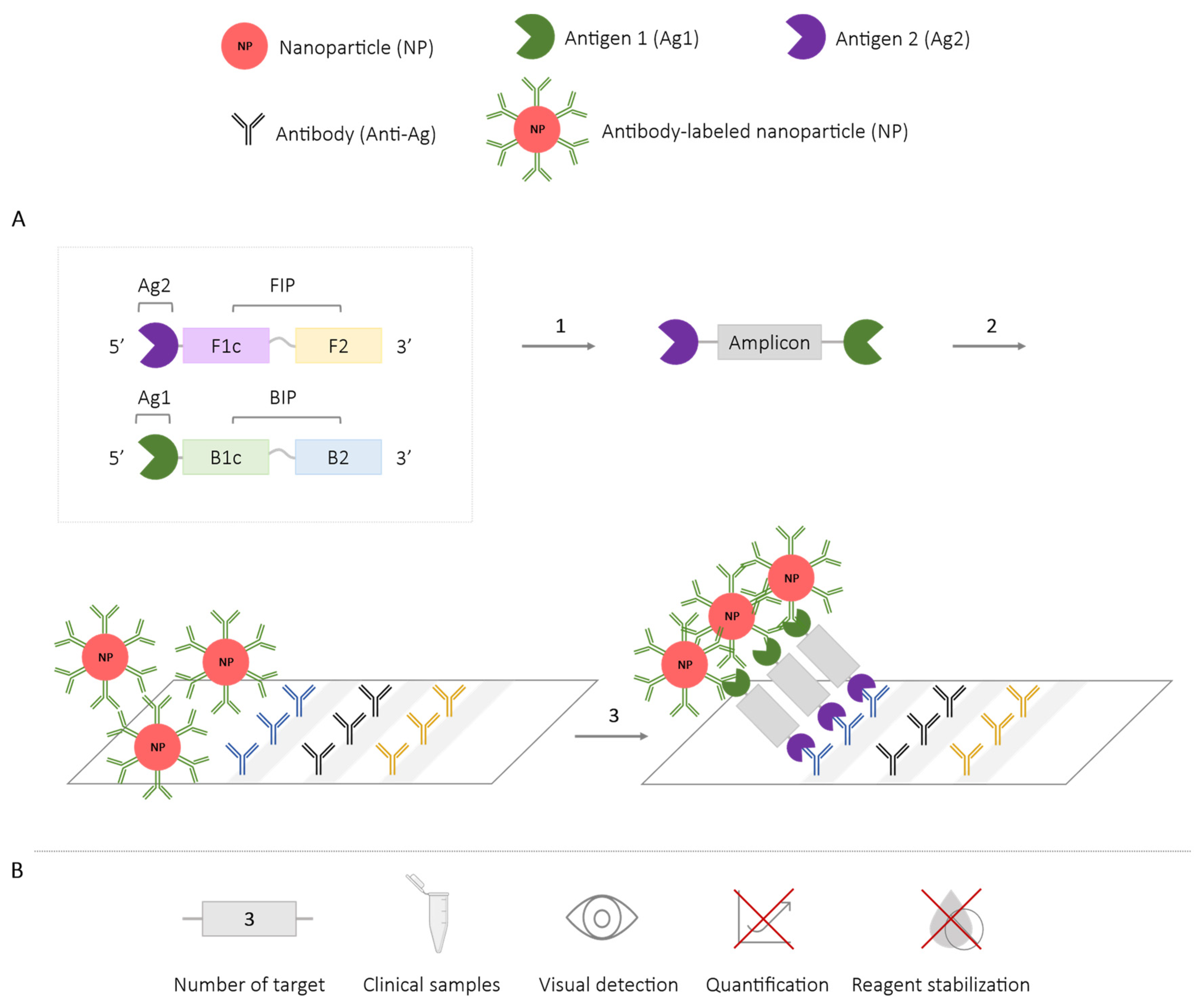
2.4. Methods Using Nanoparticles
2.5. Methods Using a Combination of Various Techniques
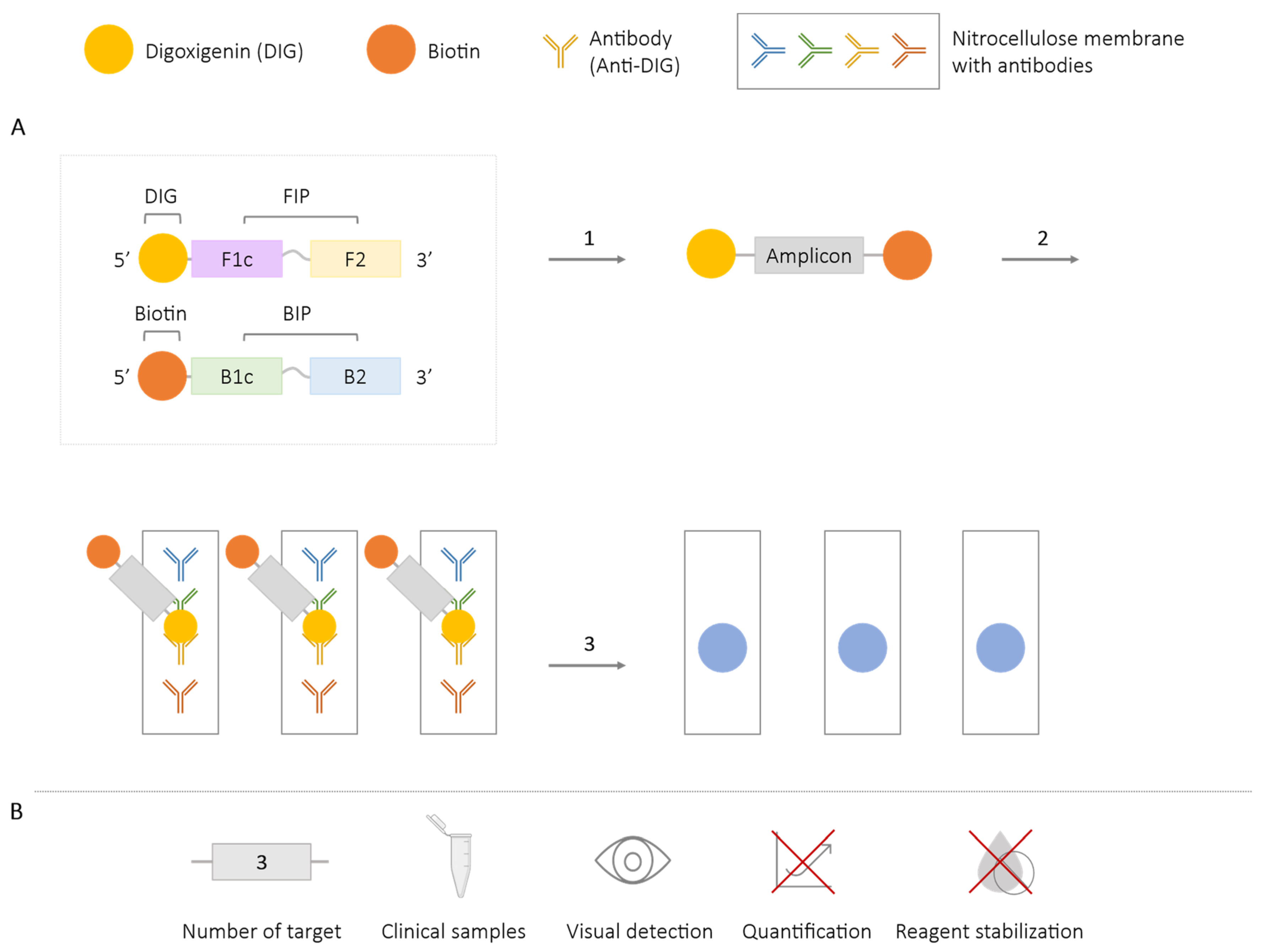
2.5.1. LAMP-ELISA
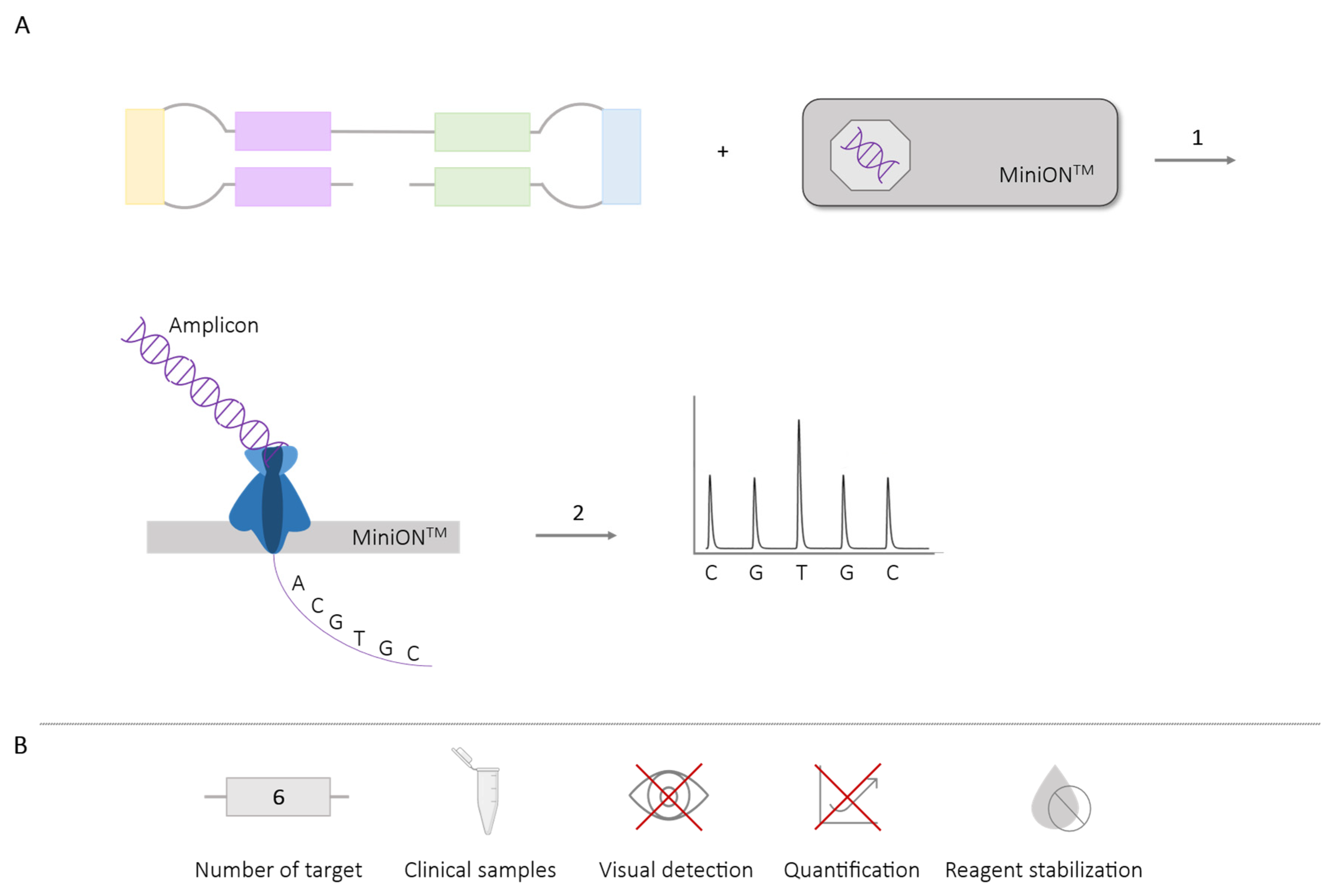
2.5.2. LAMP-Sequencing
2.6. Other Methods
3. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| Ag | Antigen |
| BRAF oncogene | B-Raf proto-oncogene serine/threonine-protein kinase |
| CRISPR-Cas | Clustered Regularly Interspaced Short Palindromic Repeats and associated protein Cas |
| CVB | Chrysanthemum B virus |
| DARQ | Detection of Amplification by Release of Quenching |
| DIG | Digoxigenin |
| ELISA | Enzyme-Linked Immunosorbent Assay |
| FITC | Fluorescein isothiocyanate |
| FLOS | Fluorescence of Loop primer upon Self-dequenching |
| hBRCA1 | gene human breast cancer 1 gene |
| HBV | Hepatitis B Virus |
| HCV | Hepatitis C Virus |
| HEV | Hepatitis E Virus |
| HIV | Human immunodeficiency virus |
| HPV | Human Papillomavirus |
| HTLV-1 | Human T-lymphotropic virus 1 |
| iNAATs | Isothermal nucleic acid amplification techniques |
| IV | Influenza virus |
| LAMP | loop-mediated isothermal amplification |
| LED light | Light-emitting diode light |
| LFA | lateral flow assays |
| MD | Mediaton displacement |
| MERS-CoV | Middle East Respiratory Syndrome Coronavirus |
| MERT-LAMP | Multiple Endonuclease Restriction Real-Time LAMP technology |
| mLAMP | multiplex LAMP |
| NPs | Nanoparticles |
| NTDs | Neglected Tropical Diseases |
| PEI polymer | Polyethylenimine or polyaziridine polymer |
| POC | point-of-care |
| qPCR | Quantitative polymerase chain reaction |
| QPD | Quencher Probe Duplex |
| QUASR | Quenching of Unincorporated Amplification Signal Reporters |
| RSV | Respiratory Syncytial Virus |
| SARS-CoV-2 | Severe Acute Respiratory Syndrome Coronavirus 2 |
| TAMRA | Tetramethylrhodamine |
| UV light | Ultraviolet light |
References
- Shen, C.-H. Diagnostic Molecular Biology; Academic Press: Cambridge, MA, USA, 2019. [Google Scholar]
- World Health Organization (WHO). Second Who Model List of Essential In Vitro Diagnostics; WHO/MVP/EMP/2019.05; World Health Organization: Geneva, Switzerland, 2019. [Google Scholar]
- Yu, A.C.H.; Vatcher, G.; Yue, X.; Dong, Y.; Li, M.H.; Tam, P.H.K.; Tsang, P.Y.L.; Wong, A.K.Y.; Hui, M.H.K.; Yang, B.; et al. Nucleic acid-based diagnostics for infectious diseases in public health affairs. Front. Med. China 2012, 6, 173–186. [Google Scholar] [CrossRef] [PubMed]
- Ballard, Z.; Ozcan, A. Nucleic acid quantification in the field. Nat. Biomed. Eng. 2018, 2, 629–630. [Google Scholar] [CrossRef] [PubMed]
- Glökler, J.; Lim, T.S.; Ida, J.; Frohme, M. Isothermal amplifications—A comprehensive review on current methods. Crit. Rev. Biochem. Mol. Biol. 2021, 56, 543–586. [Google Scholar] [CrossRef]
- Zhao, Y.; Chen, F.; Li, Q.; Wang, L.; Fan, C. Isothermal Amplification of Nucleic Acids. Chem. Rev. 2015, 115, 12491–12545. [Google Scholar] [CrossRef] [PubMed]
- Becherer, L.; Borst, N.; Bakheit, M.; Frischmann, S.; Zengerle, R.; von Stetten, F. Loop-mediated isothermal amplification (LAMP)—Review and classification of methods for sequence-specific detection. Anal. Methods 2020, 12, 717–746. [Google Scholar] [CrossRef]
- Notomi, T.; Okayama, H.; Masubuchi, H.; Yonekawa, T.; Watanabe, K.; Amino, N.; Hase, T. Loop-mediated isothermal amplification of DNA. Nucleic Acids Res. 2000, 28, e63. [Google Scholar] [CrossRef] [PubMed]
- Wong, Y.P.; Othman, S.; Lau, Y.L.; Radu, S.; Chee, H.Y. Loop-mediated isothermal amplification (LAMP): A versatile technique for detection of micro-organisms. J. Appl. Microbiol. 2018, 124, 626–643. [Google Scholar] [CrossRef] [PubMed]
- Goto, M.; Honda, E.; Ogura, A.; Nomoto, A.; Hanaki, K.I. Colorimetric detection of loop-mediated isothermal amplification reaction by using hydroxy naphthol blue. Biotechniques 2009, 46, 167–172. [Google Scholar] [CrossRef] [PubMed]
- Mori, Y.; Kitao, M.; Tomita, N.; Notomi, T. Real-time turbidimetry of LAMP reaction for quantifying template DNA. J. Biochem. Biophys. Methods 2004, 59, 145–157. [Google Scholar] [CrossRef]
- Oscorbin, I.P.; Belousova, E.A.; Zakabunin, A.I.; Boyarskikh, U.A.; Filipenko, M.L. Comparison of fluorescent intercalating dyes for quantitative loop-mediated isothermal amplification (qLAMP). Biotechniques 2016, 61, 20–25. [Google Scholar] [CrossRef]
- Agarwal, S.; Warmt, C.; Henkel, J.; Schrick, L.; Nitsche, A.; Bier, F.F. Lateral flow–based nucleic acid detection of SARS-CoV-2 using enzymatic incorporation of biotin-labeled dUTP for POCT use. Anal. Bioanal. Chem. 2022, 414, 3177–3186. [Google Scholar] [CrossRef] [PubMed]
- Selvam, K.; Najid, M.A.; Khalid, M.; Mohamad, S.; Palaz, F.; Ozsoz, M.; Aziah, I. RT-LAMP CRISPR-Cas12/13-Based SARS-CoV-2 Detection Methods. Diagnostics 2021, 11, 1646. [Google Scholar] [CrossRef] [PubMed]
- Carter, C.; Akrami, K.; Hall, D.; Smith, D.; Aronoff-Spencer, E. Lyophilized visually readable loop-mediated isothermal reverse transcriptase nucleic acid amplification test for detection Ebola Zaire RNA. J. Virol. Methods 2017, 244, 32–38. [Google Scholar] [CrossRef] [PubMed]
- García-Bernalt Diego, J.; Fernández-Soto, P.; Crego-Vicente, B.; Alonso-Castrillejo, S.; Febrer-Sendra, B.; Gómez-Sánchez, A.; Vicente, B.; López-Abán, J.; Muro, A. Progress in loop-mediated isothermal amplification assay for detection of Schistosoma mansoni DNA: Towards a ready-to-use test. Sci. Rep. 2019, 9, 14744. [Google Scholar] [CrossRef] [PubMed]
- García-Bernalt Diego, J.; Fernández-Soto, P.; Muro, A. Lamp in neglected tropical diseases: A focus on parasites. Diagnostics 2021, 11, 521. [Google Scholar] [CrossRef] [PubMed]
- Francois, P.; Tangomo, M.; Hibbs, J.; Bonetti, E.J.; Boehme, C.C.; Notomi, T.; Perkins, M.D.; Schrenzel, J. Robustness of a loop-mediated isothermal amplification reaction for diagnostic applications. FEMS Immunol. Med. Microbiol. 2011, 62, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Garg, N.; Ahmad, F.J.; Kar, S. Recent advances in loop-mediated isothermal amplification (LAMP) for rapid and efficient detection of pathogens. Curr. Res. Microb. Sci. 2022, 3, 100120. [Google Scholar] [CrossRef]
- Petney, T.N.; Andrews, R.H. Multiparasite communities in animals and humans: Frequency, structure and pathogenic significance. Int. J. Parasitol. 1998, 28, 377–393. [Google Scholar] [CrossRef] [PubMed]
- Vaumourin, E.; Vourc’h, G.; Gasqui, P.; Vayssier-Taussat, M. The importance of multiparasitism: Examining the consequences of co-infections for human and animal health. Parasites Vectors 2015, 8, 545. [Google Scholar] [CrossRef]
- Becherer, L.; Bakheit, M.; Frischmann, S.; Stinco, S.; Borst, N.; Zengerle, R.; Von Stetten, F. Simplified Real-Time Multiplex Detection of Loop-Mediated Isothermal Amplification Using Novel Mediator Displacement Probes with Universal Reporters. Anal. Chem. 2018, 90, 4741–4748. [Google Scholar] [CrossRef]
- Biorad. Fluorophore Reference Guide Spectra. Group. 350:400–50. Available online: http://www.bio-rad.com/webroot/web/pdf/lsr/literature/Bulletin_2421.pdf (accessed on 7 February 2024).
- Tanner, N.A.; Zhang, Y.; Evans, T.C. Simultaneous multiple target detection in real-time loop-mediated isothermal amplification. Biotechniques 2012, 53, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Tanner, N.A.; Evans, T.C. Loop-mediated isothermal amplification for detection of nucleic acids. Curr. Protoc. Mol. Biol. 2014, 105, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Nanayakkara, I.A.; White, I.M. Demonstration of a quantitative triplex LAMP assay with an improved probe-based readout for the detection of MRSA. Analyst 2019, 144, 3878–3885. [Google Scholar] [CrossRef] [PubMed]
- Mashooq, M.; Kumar, D.; Niranjan, A.K.; Agarwal, R.K.; Rathore, R. Development and evaluation of probe based real time loop mediated isothermal amplification for Salmonella: A new tool for DNA quantification. J. Microbiol. Methods 2016, 126, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Crego-Vicente, B.; Fernández-Soto, P.; García-Bernalt Diego, J.; Febrer-Sendra, B.; Muro, A. Development of a Duplex LAMP Assay with Probe-Based Readout for Simultaneous Real-Time Detection of Schistosoma mansoni and Strongyloides spp. -A Laboratory Approach to Point-Of-Care. Int. J. Mol. Sci. 2023, 24, 893. [Google Scholar] [CrossRef] [PubMed]
- Jang, W.S.; Lim, D.H.; Choe, Y.; Jee, H.; Moon, K.C.; Kim, C.; Choi, M.; Park, I.S.; Lim, C.S. Development of a multiplex loop-mediated isothermal amplification assay for diagnosis of plasmodium spp., plasmodium falciparum and plasmodium vivax. Diagnostics 2021, 11, 1950. [Google Scholar] [CrossRef] [PubMed]
- Ball, C.S.; Light, Y.K.; Koh, C.Y.; Wheeler, S.S.; Coffey, L.L.; Meagher, R.J. Quenching of Unincorporated Amplification Signal Reporters in Reverse-Transcription Loop-Mediated Isothermal Amplification Enabling Bright, Single-Step, Closed-Tube, and Multiplexed Detection of RNA Viruses. Anal. Chem. 2016, 88, 3562–3568. [Google Scholar] [CrossRef] [PubMed]
- Priye, A.; Bird, S.W.; Light, Y.K.; Ball, C.S.; Negrete, O.A.; Meagher, R.J. A smartphone-based diagnostic platform for rapid detection of Zika, chikungunya, and dengue viruses. Sci. Rep. 2017, 7, 44778. [Google Scholar] [CrossRef] [PubMed]
- Priye, A.; Ball, C.S.; Meagher, R.J. Colorimetric-Luminance Readout for Quantitative Analysis of Fluorescence Signals with a Smartphone CMOS Sensor. Anal. Chem. 2018, 90, 12385–12389. [Google Scholar] [CrossRef]
- Meagher, R.J.; Priye, A.; Light, Y.K.; Huang, C.; Wang, E. Impact of primer dimers and self-amplifying hairpins on reverse transcription loop-mediated isothermal amplification detection of viral RNA. Analyst 2018, 143, 1924–1933. [Google Scholar] [CrossRef]
- Gadkar, V.J.; Goldfarb, D.M.; Gantt, S.; Tilley, P.A.G. Real-time Detection and Monitoring of Loop Mediated Amplification (LAMP) Reaction Using Self-quenching and De-quenching Fluorogenic Probes. Sci. Rep. 2018, 8, 5548. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, R.; Tanaka, H.; Suzuki, K.; Fukuta, S.; Kato, S.; Ohno, T. Multiplex loop-mediated isothermal amplification assay for the identification of three major whitefly species in the greenhouse. J. Appl. Entomol. 2018, 142, 745–754. [Google Scholar] [CrossRef]
- Fukuta, S.; Nagai, H.; Suzuki, R.; Matsumoto, Y.; Kato, S.; Saka, N.; Horikawa, H.; Kato, S.; Miyake, N. Detection of Fomitiporia torreyae and Fulviformes umbrinellus by multiplex loop-mediated isothermal amplification (mLAMP) for diagnosis of Japanese pear dwarf. Ann. Appl. Biol. 2016, 170, 170–178. [Google Scholar] [CrossRef]
- Takayama, I.; Nakauchi, M.; Takahashi, H.; Oba, K.; Semba, S.; Kaida, A.; Kubo, H.; Saito, S.; Nagata, S.; Odagiri, T.; et al. Development of real-time fluorescent reverse transcription loop-mediated isothermal amplification assay with quenching primer for influenza virus and respiratory syncytial virus. J. Virol. Methods 2019, 267, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Kurata, S.; Kanagawa, T.; Yamada, K.; Torimura, M.; Yokomaku, T.; Kamagata, Y.; Kurane, R. Fluorescent quenching-based quantitative detection of specific DNA/RNA using a BODIPY® FL-labeled probe or primer. Nucleic Acids Res. 2001, 29, e34. [Google Scholar] [CrossRef] [PubMed]
- Tani, H.; Teramura, T.; Adachi, K.; Tsuneda, S.; Kurata, S.; Nakamura, K.; Kanagawa, T.; Noda, N. Technique for quantitative detection of specific DNA sequences using alternately binding quenching probe competitive assay combined with loop-mediated isothermal amplification. Anal. Chem. 2007, 79, 5608–5613. [Google Scholar] [CrossRef] [PubMed]
- Shirato, K.; Semba, S.; El-Kafrawy, S.A.; Hassan, A.M.; Tolah, A.M.; Takayama, I.; Kageyama, T.; Notomi, T.; Kamitani, W.; Matsuyama, S.; et al. Development of fluorescent reverse transcription loop-mediated isothermal amplification (RT-LAMP) using quenching probes for the detection of the Middle East respiratory syndrome coronavirus. J. Virol. Methods 2018, 258, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Kubota, R.; Alvarez, A.M.; Su, W.W.; Jenkins, D.M. FRET-Based Assimilating Probe for Sequence-Specific Real-Time Monitoring of Loop-Mediated Isothermal Amplification (LAMP). Biol. Eng. Trans. 2011, 4, 81–100. [Google Scholar] [CrossRef]
- Kubota, R.; Jenkins, D.M. Real-time duplex applications of loop-mediated AMPlification (LAMP) by assimilating probes. Int. J. Mol. Sci. 2015, 16, 4786–4799. [Google Scholar] [CrossRef]
- Yaren, O.; Alto, B.W.; Gangodkar, P.V.; Ranade, S.R.; Patil, K.N.; Bradley, K.M.; Yang, Z.; Phadke, N.; Benner, S.A. Point of sampling detection of Zika virus within a multiplexed kit capable of detecting dengue and chikungunya. BMC Infect. Dis. 2017, 17, 1–13. [Google Scholar] [CrossRef]
- Kim, M.J.; Kim, H.Y. Direct duplex real-time loop mediated isothermal amplification assay for the simultaneous detection of cow and goat species origin of milk and yogurt products for field use. Food Chem. 2018, 246, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Yaren, O.; Alto, B.W.; Bradley, K.M.; Moussatche, P.; Glushakova, L.; Benner, S.A. Multiplexed isothermal amplification based diagnostic platform to detect zika, chikungunya, and dengue 1. J. Vis. Exp. 2018, 2018, e57051. [Google Scholar] [CrossRef] [PubMed]
- Yaren, O.; McCarter, J.; Phadke, N.; Bradley, K.M.; Overton, B.; Yang, Z.; Ranade, S.; Patil, K.; Bangale, R.; Benner, S.A. Ultra-rapid detection of SARS-CoV-2 in public workspace environments. PLoS ONE 2021, 16, e0240524. [Google Scholar] [CrossRef] [PubMed]
- Khamlor, T.; Pongpiachan, P.; Parnpai, R.; Punyawai, K.; Sangsritavong, S.; Chokesajjawatee, N. Bovine embryo sex determination by multiplex loop-mediated isothermal amplification. Theriogenology 2015, 83, 891–896. [Google Scholar] [CrossRef] [PubMed]
- Curtis, K.A.; Rudolph, D.L.; Owen, S.M. Sequence-specific detection method for reverse transcription, loop-mediated isothermal amplification of HIV-1. J. Med. Virol. 2009, 81, 966–972. [Google Scholar] [CrossRef] [PubMed]
- Curtis, K.A.; Rudolph, D.L.; Morrison, D.; Guelig, D.; Diesburg, S.; McAdams, D.; Burton, R.A.; LaBarre, P.; Owen, M. Single-use, electricity-free amplification device for detection of HIV-1. J. Virol. Methods 2016, 237, 132–137. [Google Scholar] [CrossRef] [PubMed]
- Curtis, K.A.; Morrison, D.; Rudolph, D.L.; Shankar, A.; Bloomfield, L.S.P.; Switzer, W.M.; Owen, S.M. A multiplexed RT-LAMP assay for detection of group M HIV-1 in plasma or whole blood. J. Virol. Methods 2018, 255, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.S.; Bhadra, S.; Li, B.; Wu, Y.R.; Milligan, J.N.; Ellington, A.D. Robust Strand Exchange Reactions for the Sequence-Specific, Real-Time Detection of Nucleic Acid Amplicons. Anal. Chem. 2015, 87, 3314–3320. [Google Scholar] [CrossRef]
- Jiang, Y.S.; Riedel, T.E.; Popoola, J.A.; Morrow, B.R.; Cai, S.; Ellington, A.D.; Bhadra, S. Portable platform for rapid in-field identification of human fecal pollution in water. Water Res. 2018, 131, 186–195. [Google Scholar] [CrossRef]
- Tang, Y.; Lu, B.; Zhu, Z.; Li, B. Establishment of a universal and rational gene detection strategy through three-way junction-based remote transduction. Chem. Sci. 2018, 9, 760–769. [Google Scholar] [CrossRef]
- Bhadra, S.; Riedel, T.E.; Saldaña, M.A.; Hegde, S.; Pederson, N.; Hughes, G.L.; Ellington, A.D. Direct nucleic acid analysis of mosquitoes for high fidelity species identification and detection of Wolbachia using a cellphone. PLoS Negl. Trop. Dis. 2018, 12, e0006671. [Google Scholar] [CrossRef] [PubMed]
- Bhadra, S.; Jiang, Y.S.; Kumar, M.R.; Johnson, R.F.; Hensley, L.E.; Ellington, A.D. Real-time sequence-validated loop-mediated isothermal amplification assays for detection of Middle East respiratory syndrome coronavirus (MERS-CoV). PLoS ONE 2015, 10, e0123126. [Google Scholar] [CrossRef] [PubMed]
- Bhadra, S.; Saldaña, M.A.; Han, H.G.; Hughes, G.L.; Ellington, A.D. Simultaneous detection of different zika virus lineages via molecular computation in a point-of-care assay. Viruses 2018, 10, 714. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Huang, S.; Liu, N.; Dong, D.; Yang, Z.; Tang, Y.; Ma, W.; He, X.; Ao, D.; Xu, Y.; et al. Establishment of an accurate and fast detection method using molecular beacons in loop-mediated isothermal amplification assay. Sci. Rep. 2017, 7, 40125. [Google Scholar] [CrossRef]
- Lee, J.E.; Mun, H.; Kim, S.R.; Kim, M.G.; Chang, J.Y.; Shim, W.B. A colorimetric Loop-mediated isothermal amplification (LAMP) assay based on HRP-mimicking molecular beacon for the rapid detection of Vibrio parahaemolyticus. Biosens. Bioelectron. 2020, 151, 111968. [Google Scholar] [CrossRef]
- Tan, Y.L.; Huang, A.Q.; Tang, L.J.; Jiang, J.H. Multiplexed droplet loop-mediated isothermal amplification with scorpion-shaped probes and fluorescence microscopic counting for digital quantification of virus RNAs. Chem. Sci. 2021, 12, 8445–8451. [Google Scholar] [CrossRef]
- Wan, L.; Chen, T.; Gao, J.; Dong, C.; Wong, A.H.H.; Jia, Y.; Mak, P.I.; Deng, C.X.; Martins, R.P. A digital microfluidic system for loop-mediated isothermal amplification and sequence specific pathogen detection. Sci. Rep. 2017, 7, 14586. [Google Scholar] [CrossRef] [PubMed]
- Nyan, D.C.; Swinson, K.L. A novel multiplex isothermal amplification method for rapid detection and identification of viruses. Sci. Rep. 2015, 5, 17925. [Google Scholar] [CrossRef]
- Becherer, L.; Knauf, S.; Marks, M.; Lueert, S.; Frischmann, S.; Borst, N.; Von Stetten, F.; Bieb, S.; Adu-Sarkodie, Y.; Asiedu, K.; et al. Multiplex mediator displacement loop-mediated isothermal amplification for detection of treponema pallidum and haemophilus ducreyi. Emerg. Infect. Dis. 2020, 26, 282–288. [Google Scholar] [CrossRef]
- Zhang, D.Y.; Seelig, G. Dynamic DNA nanotechnology using strand-displacement reactions. Nat. Chem. 2011, 3, 103–113. [Google Scholar] [CrossRef]
- Wang, Y.; Li, D.; Wang, Y.; Li, K.; Ye, C. Rapid and sensitive detection of vibrio parahaemolyticus and vibrio vulnificus by multiple endonuclease restriction real-Time loop-Mediated isothermal amplification technique. Molecules 2016, 21, 111. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, Y.; Lan, R.; Xu, H.; Ma, A.; Li, D.; Dai, H.; Yuan, X.; Xu, J.; Ye, C. Multiple endonuclease restriction real-time loop-mediated isothermal amplification: A novel analytically rapid, sensitive, multiplex loop-mediated isothermal amplification detection technique. J. Mol. Diagn. 2015, 17, 392–401. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, Y.; Luo, L.; Liu, D.; Luo, X.; Xu, Y.; Hu, S.; Niu, L.; Xu, J.; Ye, C. Rapid and sensitive detection of Shigella spp. and Salmonella spp. by multiple endonuclease restriction real-time loop-mediated isothermal amplification technique. Front. Microbiol. 2015, 6, 1400. [Google Scholar] [CrossRef] [PubMed]
- Iseki, H.; Alhassan, A.; Ohta, N.; Thekisoe, O.M.M.; Yokoyama, N.; Inoue, N.; Nambota, A.; Yasuda, J.; Igarashi, I. Development of a multiplex loop-mediated isothermal amplification (mLAMP) method for the simultaneous detection of bovine Babesia parasites. J. Microbiol. Methods 2007, 71, 281–287. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Zhuang, L.; Zhang, D.; Zhang, P.; Dou, X.; Wang, C. Establishment of a Multiplex Loop-Mediated Isothermal Amplification Method for Rapid Detection of Sulfonamide Resistance Genes (sul1, sul2, sul3) in Clinical Enterobacteriaceae Isolates from Poultry. Foodborne Pathog. Dis. 2018, 15, 413–419. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.L.; Zhao, X.T.; Muhammad, I.; Ge, B.B.; Hong, B. Multiplex reverse transcription loop-mediated isothermal amplification for the simultaneous detection of CVB and CSVd in chrysanthemum. J. Virol. Methods 2014, 210, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.; Chu, Y.; Cheng, S.; Wu, H.; Kajiyama, T.; Kambara, H.; Zhou, G. Multiplex loop-mediated isothermal amplification detection by sequence-based barcodes coupled with nicking endonuclease-mediated pyrosequencing. Anal. Chem. 2012, 84, 3758–3763. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Xiao, J.; Ma, X.; Wang, Q.; Zhang, Y. An effective established biosensor of bifunctional probes-labeled AuNPs combined with LAMP for detection of fish pathogen Streptococcus iniae. Appl. Microbiol. Biotechnol. 2018, 102, 5299–5308. [Google Scholar] [CrossRef] [PubMed]
- Kumvongpin, R.; Jearanaikool, P.; Wilailuckana, C.; Sae-ung, N.; Prasongdee, P.; Daduang, S.; Wongsena, M.; Boonsiri, P.; Kiatpathomchai, W.; Swangvaree, S.S.; et al. High sensitivity, loop-mediated isothermal amplification combined with colorimetric gold-nanoparticle probes for visual detection of high risk human papillomavirus genotypes 16 and 18. J. Virol. Methods 2016, 234, 90–95. [Google Scholar] [CrossRef]
- Heo, J.H.; Yi, G.S.; Lee, B.S.; Cho, H.H.; Lee, J.W.; Lee, J.H. A significant enhancement of color transition from an on-off type achromatic colorimetric nanosensor for highly sensitive multi-analyte detection with the naked eye. Nanoscale 2016, 8, 18341–18351. [Google Scholar] [CrossRef]
- Chen, Y.; Cheng, N.; Xu, Y.; Huang, K.; Luo, Y.; Xu, W. Point-of-care and visual detection of P. aeruginosa and its toxin genes by multiple LAMP and lateral flow nucleic acid biosensor. Biosens. Bioelectron. 2016, 81, 317–323. [Google Scholar] [CrossRef]
- Wang, Y.; Li, H.; Wang, Y.; Zhang, L.; Xu, J.; Ye, C. Loop-mediated isothermal amplification label-based gold nanoparticles lateral flow biosensor for detection of Enterococcus faecalis and Staphylococcus aureus. Front. Microbiol. 2017, 8, 248861. [Google Scholar] [CrossRef]
- Zhu, X.; Wang, X.; Han, L.; Chen, T.; Wang, L.; Li, H.; Li, S.; He, L.; Fu, X.; Chen, S.; et al. Multiplex reverse transcription loop-mediated isothermal amplification combined with nanoparticle-based lateral flow biosensor for the diagnosis of COVID-19. Biosens. Bioelectron. 2020, 166, 112437. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.H.; Oh, S.J.; Kim, Y.T.; Kim, S.Y.; Kim, W.; Jung, J.; Seo, T.S. Combination of multiplex reverse-transcription loop-mediated isothermal amplification with an immunochromatographic strip for subtyping influenza A virus. Anal. Chim. Acta 2015, 853, 541–547. [Google Scholar] [CrossRef] [PubMed]
- Nurul Najian, A.B.; Engku Nur Syafirah, E.A.R.; Ismail, N.; Mohamed, M.; Yean, C.Y. Development of multiplex loop mediated isothermal amplification (m-LAMP) label-based gold nanoparticles lateral flow dipstick biosensor for detection of pathogenic Leptospira. Anal. Chim. Acta 2016, 903, 142–148. [Google Scholar] [CrossRef]
- Lee, M.F.; Chen, Y.H.; Peng, C.F. Evaluation of reverse transcription loop-mediated isothermal amplification in conjunction with ELISA-hybridization assay for molecular detection of Mycobacterium tuberculosis. J. Microbiol. Methods 2009, 76, 174–180. [Google Scholar] [CrossRef]
- Ravan, H.; Yazdanparast, R. Development and evaluation of a loop-mediated isothermal amplification method in conjunction with an enzyme-linked immunosorbent assay for specific detection of Salmonella serogroup D. Anal. Chim. Acta 2012, 733, 64–70. [Google Scholar] [CrossRef]
- Ravan, H.; Yazdanparast, R. Loop region-specific oligonucleotide probes for loop-mediated isothermal amplification-enzyme-linked immunosorbent assay truly minimize the instrument needed for detection process. Anal. Biochem. 2013, 439, 102–108. [Google Scholar] [CrossRef] [PubMed]
- Pappas, M.G.; Hajkowski, R.; Hockmeyer, W.T. Dot enzyme-linked immunosorbent assay (Dot-ELISA): A micro technique for the rapid diagnosis of visceral leishmaniasis. J. Immunol. Methods 1983, 64, 205–214. [Google Scholar] [CrossRef]
- Pappas, M.G. Recent applications of the Dot-ELISA in immunoparasitology. Vet. Parasitol. 1988, 29, 105–129. [Google Scholar] [CrossRef]
- Nkouawa, A.; Sako, Y.; Okamoto, M.; Ito, A. Simple identification of human taenia species by multiplex loop-mediated isothermal amplification in combination with dot enzyme-linked immunosorbent assay. Am. J. Trop. Med. Hyg. 2016, 94, 1318–1323. [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, J.; Runtuwene, L.R.; Hayashida, K.; Mongan, A.E.; Thi, L.A.N.; Thuy, L.N.; Nhat, C.N.; Limkittikul, K.; Sirivichayakul, C.; Sathirapongsasuti, N.; et al. Serotyping dengue virus with isothermal amplification and a portable sequencer. Sci. Rep. 2017, 7, 3510. [Google Scholar] [CrossRef] [PubMed]
- Hayashida, K.; Orba, Y.; Sequeira, P.C.; Sugimoto, C.; Hall, W.W.; Eshita, Y.; Suzuki, Y.; Runtuwene, L.; Brasil, P.; Calvet, G.; et al. Field diagnosis and genotyping of chikungunya virus using a dried reverse transcription loop-mediated isothermal amplification (LAMP) assay and MinION sequencing. PLoS Negl. Trop. Dis. 2019, 13, e0007480. [Google Scholar] [CrossRef] [PubMed]
- Check Hayden, E. Nanopore genome sequencer makes its debut. Nature News 2012, 1–2. [Google Scholar] [CrossRef]
- Imai, K.; Tarumoto, N.; Runtuwene, L.R.; Sakai, J.; Hayashida, K.; Eshita, Y.; Maeda, R.; Tuda, J.; Ohno, H.; Murakami, T.; et al. An innovative diagnostic technology for the codon mutation C580Y in kelch13 of Plasmodium falciparum with MinION nanopore sequencer. Malar. J. 2018, 17, 217. [Google Scholar] [CrossRef] [PubMed]
- Imai, K.; Tarumoto, N.; Misawa, K.; Runtuwene, L.R.; Sakai, J.; Hayashida, K.; Eshita, Y.; Maeda, R.; Tuda, J.; Murakami, T.; et al. A novel diagnostic method for malaria using loop-mediated isothermal amplification (LAMP) and MinIONTM nanopore sequencer. BMC Infect. Dis. 2017, 17, 621. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Zou, D.; Dong, D.; Yang, Z.; Ao, D.; Liu, W.; Huang, L. Development of a multiplex loop-mediated isothermal amplification method for the simultaneous detection of Salmonella spp. and Vibrio parahaemolyticus. Sci. Rep. 2017, 7, 45601. [Google Scholar] [CrossRef] [PubMed]
- Joshi, S.; Dixit, K.K.; Sharma, V.; Ramesh, V.; Singh, R.; Salotra, P. Rapid Multiplex Loop-Mediated Isothermal Amplification (m-LAMP) Assay for Differential Diagnosis of Leprosy and Post–Kala-Azar Dermal Leishmaniasis. Am. J. Trop. Med. Hyg. 2021, 104, 2085–2090. [Google Scholar] [CrossRef]
- Shao, Y.; Zhu, S.; Jin, C.; Chen, F. Development of multiplex loop-mediated isothermal amplification-RFLP (mLAMP-RFLP) to detect Salmonella spp. and Shigella spp. in milk. Int. J. Food Microbiol. 2011, 148, 75–79. [Google Scholar] [CrossRef]
- Aonuma, H.; Yoshimura, A.; Kobayashi, T.; Okado, K.; Badolo, A.; Nelson, B.; Kanuka, H.; Fukumoto, S. A single fluorescence-based LAMP reaction for identifying multiple parasites in mosquitoes. Exp. Parasitol. 2010, 125, 179–183. [Google Scholar] [CrossRef]
- Wang, H.; Ma, Z.; Qin, J.; Shen, Z.; Liu, Q.; Chen, X.; Wang, H.; An, Z.; Liu, W.; Li, M. A versatile loop-mediated isothermal amplification microchip platform for Streptococcus pneumoniae and Mycoplasma pneumoniae testing at the point of care. Biosens. Bioelectron. 2019, 126, 373–380. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Chen, Y.; Wang, Z.; Qiao, L.; Xie, R.; Zhang, J.; Bian, S.; Li, H.; Zhang, Y.; Chen, A. Multiple authentications of high-value milk by centrifugal microfluidic chip-based real-time fluorescent LAMP. Food Chem. 2021, 351, 129348. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Liu, L.; Ye, Z.; Gong, J.; Hao, P.; Ping, J.; Ying, Y. TriD-LAMP: A pump-free microfluidic chip for duplex droplet digital loop-mediated isothermal amplification analysis. Anal. Chim. Acta 2022, 1233, 340513. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Jia, Y.; Li, R.; Wen, Y.; Liang, Y.; Lao, G.; Liu, X.; Zhou, W.; Liu, H.; Xie, J.; et al. Rapid and simultaneous detection of multiple pathogens in the lower reproductive tract during pregnancy based on loop-mediated isothermal amplification-microfluidic chip. BMC Microbiol. 2022, 22, 260. [Google Scholar] [CrossRef]
- García-Bernalt, J.; Fernández-Soto, P.; Domínguez-Gil, M.; Belhassen-García, M.; Muñoz, J.L.; Muro, A. A Simple, Affordable, Rapid, Stabilized, Colorimetric, Versatile RT-LAMP Assay to Detect SARS-CoV-2. Diagnostics 2021, 11, 438. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Methodologies | Number of Targets | Species/Target Amplified | Application in Clinical Samples | Visual Detection | Quantification | Reagent Stabilization |
|---|---|---|---|---|---|---|
| DARQ | 4 | Escherichia coli, Caenorhabditis elegans, bacteriophage λ, hBRCA1 [24] Methicillin-resistant Staphyloccus aureus genes (femB, mecA, spa) [26] Salmonella spp. [27] Plasmodium spp., P. vivax, P. falciparum, Human actin [29] Schistosoma mansoni, Strongyloides spp. [28] | ✓ | ✗ | ✓ | ✓ |
| QUASR | 2 | West Nile virus, Chikungunya [30] Zika, Chikungunya [31] Yellow fever virus, Dengue [33] | ✓ | ✓ | ✗ | ✗ |
| FLOS | 3 | Varicella-zoster virus [34] Trialeurodes vaporariorum, Bemisia tabaco (MEAM1) and B. tabaci (MED) [35] Fulviformes umbrinellus and Fomitiporia torreyae [36] | ✓ | ✓ | ✓ | ✗ |
| Guanine quenching * | 1 | IV [37] RSV [37] Nitrosomonas europaea [39] MERS-CoV [40] | ✓ | ✗ | ✓ | ✗ |
| Assimilation probes or primer fluorescence probes | 3 | Zika, Dengue, Chikungunya [43,45] SARS CoV-2 and hARNasa P [46] Salmonella enterica and Enterobacteria phage λ [42] Ralstonia salanacearum and R. solanacearum R3B2 [41,42] Mitochondrial cytochrome b gene (cow and goat) [44] HIV-1 [48,49,50] Sex embryos [47] | ✓ | ✓ | ✓ | ✓ |
| OSD probes | 4 | HSV1 and P. falciparum [51] Polymorphic detection in BRAF gen [51] Human fecal contamination (Bacteroides HF183) [52] Wolbachia spp. [54] MERS-CoV [55] Zika (4 genotypes) [56] | ✗ | ✗ | ✓ | ✓ |
| Molecular beacon | 6 | Vibrio cholerae [57] HIV, HBV, HCV, HEV, Dengue and West Nile virus [61] HIV and HCV [59] Vibrio parahaemolyticus [58] | ✓ | ✓ | ✓ | ✗ |
| MD probe | 2 | HIV-1 and HTLV-1 [22] Haemophilus ducreyi and Treponema pallidum [22] T. pallidum and H. ducreyi [62] | ✓ | ✗ | ✓ | ✗ |
| Endonucleases | 3 | Sulfonamide resistance genes (sul1, sul2, sul3) [68] Babesia bigemina and B. bovis [67] Crysanthemum virus B (CVB) and Crysanthemum stunt viroid (CSVd) [69] Listeria monocytogenes and L. invanovii [65] V. parahaemolyticus and Vibrio vulnificus [64] Shigella spp. and Salmonella spp. [66] | ✓ | ✓ | ✓ | ✗ |
| Endonucleases + pyrosequencing | 4 | HBV, HCV, HIV and T. pallidum [70] | ✓ | ✗ | ✗ | ✗ |
| Nanoparticles * | 1 | HPV [72] | ✗ | ✓ | ✗ | ✗ |
| Nanoparticles + lateral flow assay | 3 | Streptococcus iniae [71] Pseudomonas aeruginosa (ecfx) and toxin ExoS and ExoU [74] Influenza A virus (2 subtypes) [77] Leptospira spp. [78] Enterococcus fecalis and S. aureus [75] SARS-CoV-2 (2 genes) [76] | ✓ | ✓ | ✗ | ✗ |
| LAMP-ELISA/dot-ELISA | 3 | Salmonella spp. [80,81] Mycobacterium tuberculosis [79] Taenia solium, T. saginata and T. asiatica [84] | ✓ | ✓ | ✗ | ✗ |
| LAMP- sequencing | 6 | Dengue (4 serotypes) [85] P. falciparum (artemisinin resistance mutation) [88] Plasmodium (6 spp.) [89] Chikungunya (genotypes) [86] | ✓ | ✗ | ✗ | ✓ |
| Melting curve | 2 | Salmonella spp. and V. parahaemolyticus [90] Leishmania donovani and Mycobacterium leprae [91] | ✓ | ✗ | ✓ | ✗ |
| Gel electrophoresis | 2 | Salmonella spp. and Shigella spp. [92] Plasmodium berghei and Dirofilaria immitis [93] | ✓ | ✓ | ✗ | ✗ |
| Microfluidic chips | 5 | Streptococcus pneumoniae and Mycoplasma pneumoniae [94] Specific genes cow, camel, goat, horse and yak [95] E. coli and bacteriophage λ [96] Streptococcus agalactiae, Enterococcus faecalis, Gardnerella vaginalis, Candida albicans and Chlamydia trachomatis [97] | ✓ | ✓ | ✓ | ✗ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Crego-Vicente, B.; del Olmo, M.D.; Muro, A.; Fernández-Soto, P. Multiplexing LAMP Assays: A Methodological Review and Diagnostic Application. Int. J. Mol. Sci. 2024, 25, 6374. https://doi.org/10.3390/ijms25126374
Crego-Vicente B, del Olmo MD, Muro A, Fernández-Soto P. Multiplexing LAMP Assays: A Methodological Review and Diagnostic Application. International Journal of Molecular Sciences. 2024; 25(12):6374. https://doi.org/10.3390/ijms25126374
Chicago/Turabian StyleCrego-Vicente, Beatriz, Manuel Diego del Olmo, Antonio Muro, and Pedro Fernández-Soto. 2024. "Multiplexing LAMP Assays: A Methodological Review and Diagnostic Application" International Journal of Molecular Sciences 25, no. 12: 6374. https://doi.org/10.3390/ijms25126374
APA StyleCrego-Vicente, B., del Olmo, M. D., Muro, A., & Fernández-Soto, P. (2024). Multiplexing LAMP Assays: A Methodological Review and Diagnostic Application. International Journal of Molecular Sciences, 25(12), 6374. https://doi.org/10.3390/ijms25126374