
Streamlined Full-Length Total RNA Sequencing of Paraformaldehyde-Fixed Brain Tissues
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
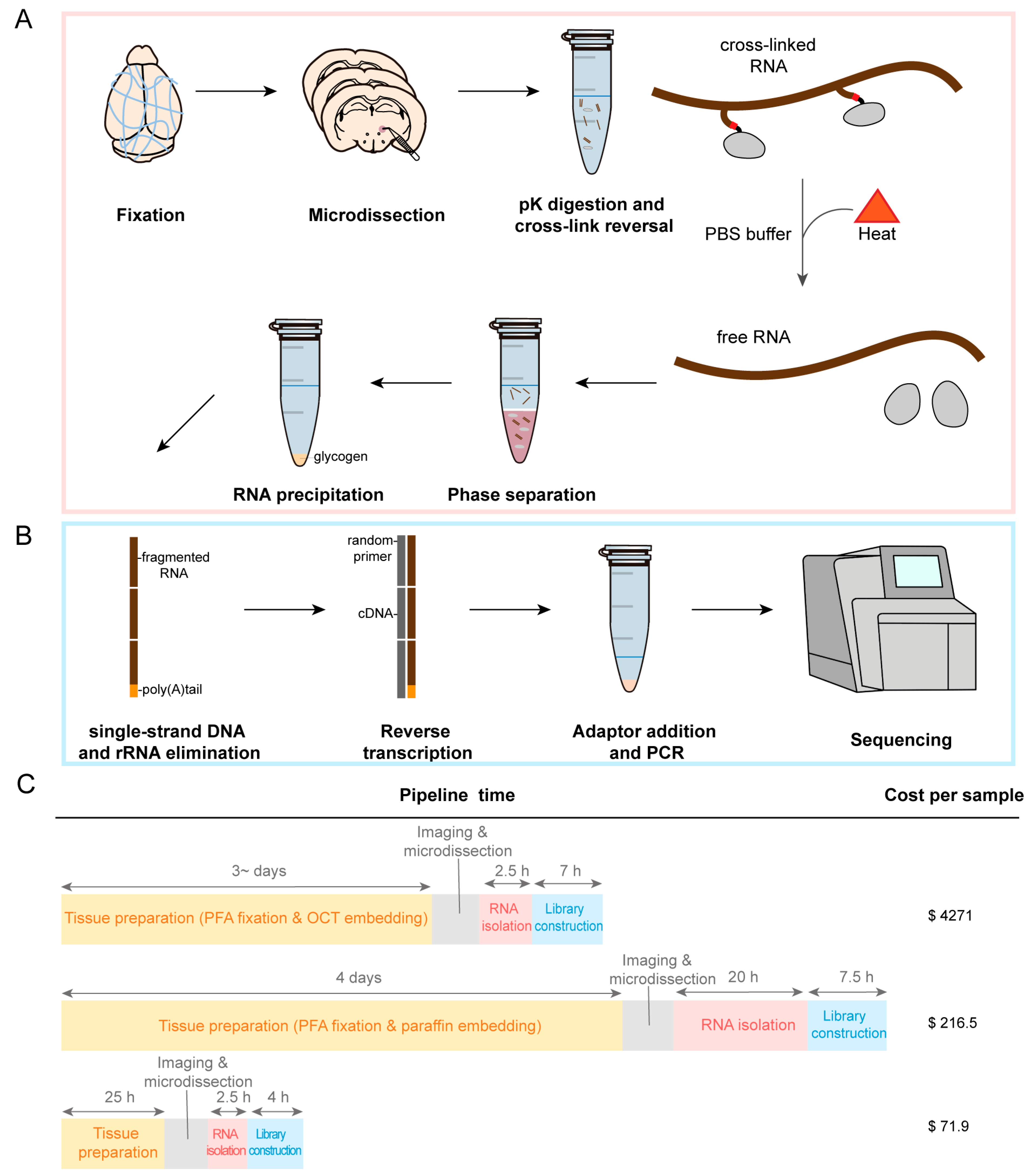
2.1. Development of an RNA Sequencing Method in PFA-Fixed Samples
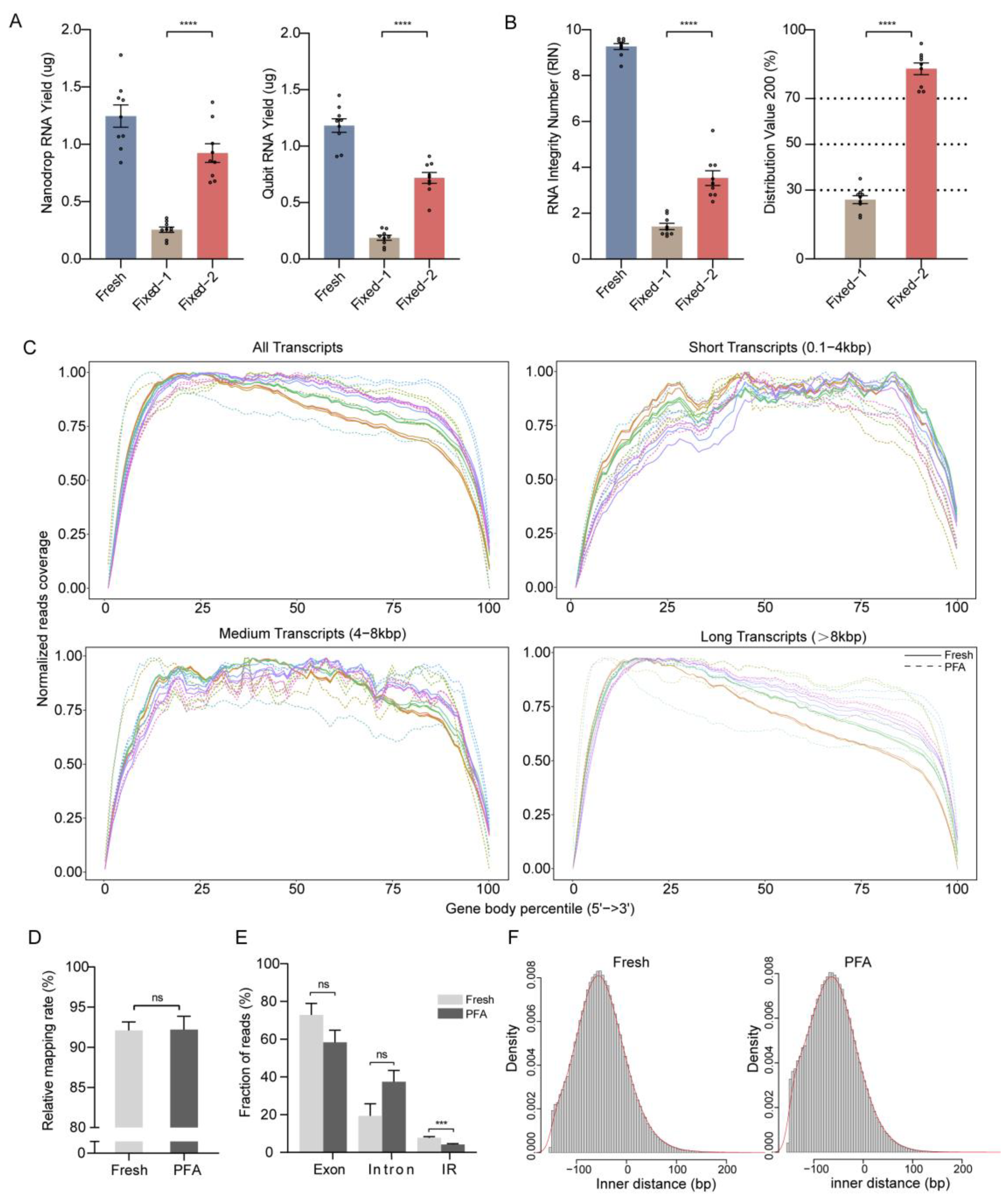
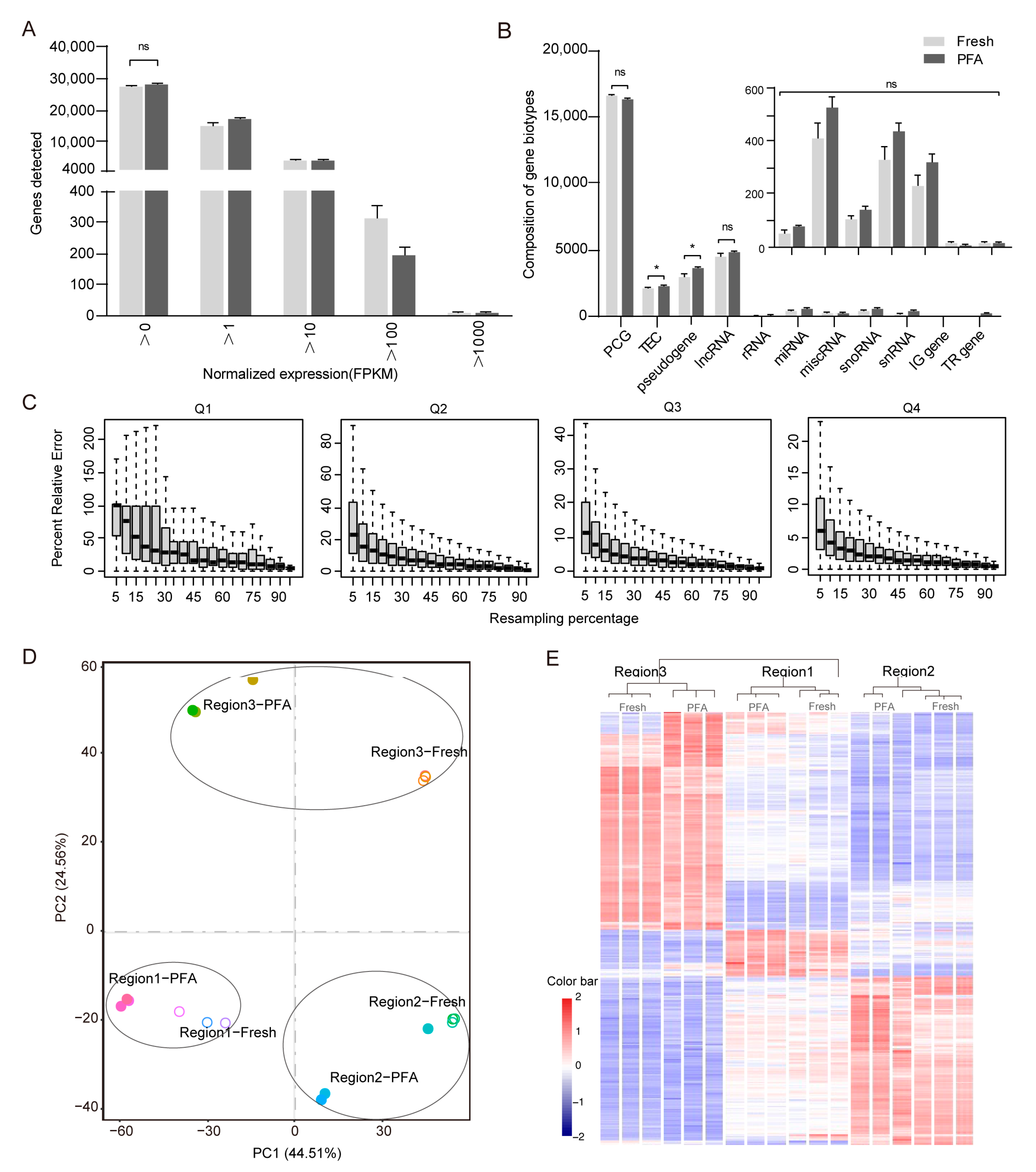
2.2. Validation of Method Compared to Reference Fresh Samples
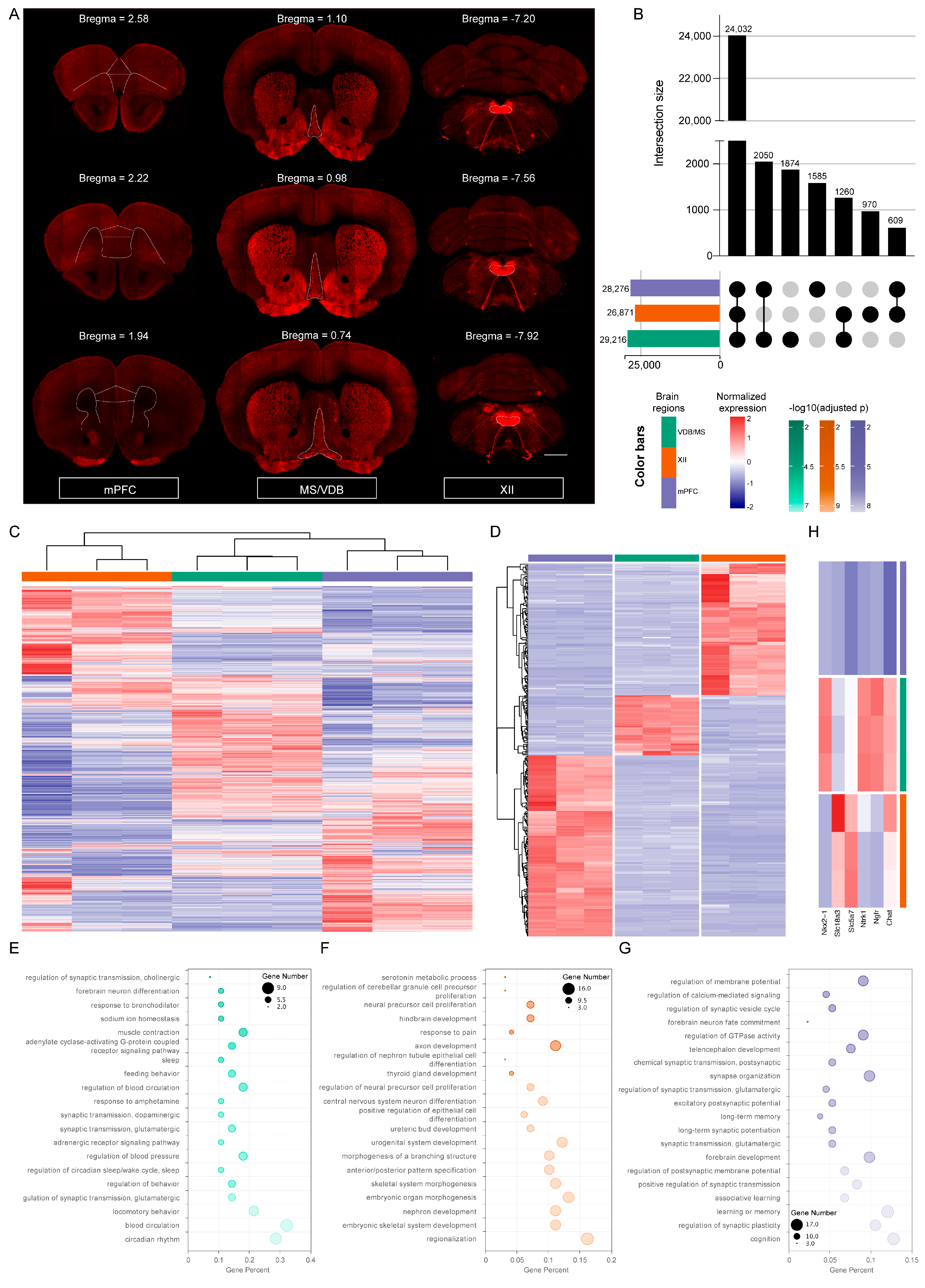
2.3. The Diversity of Gene Expression Profiling in Different Regions of the Mouse Brain
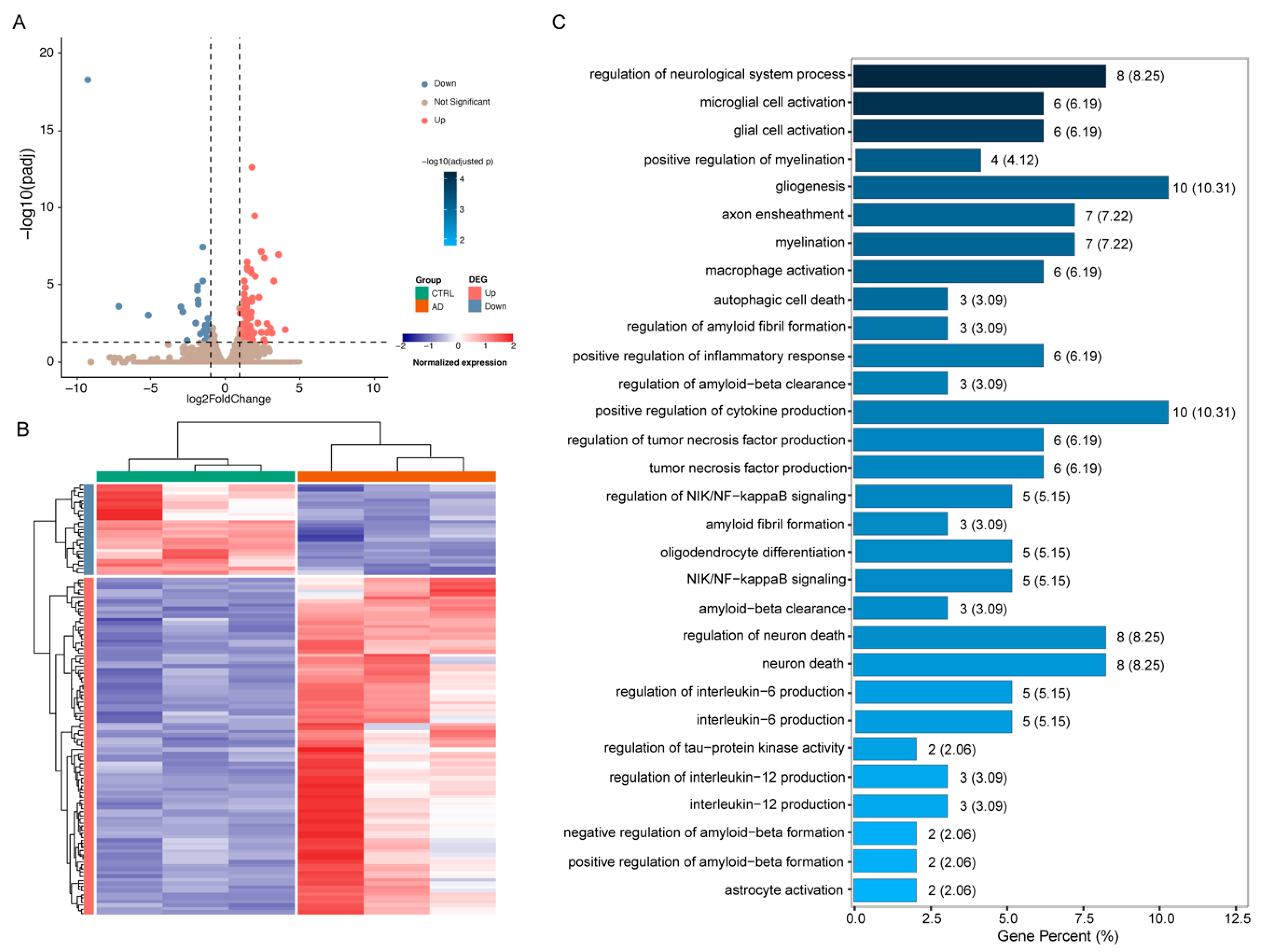
2.4. The Molecular Signatures Associated with Pathological Features in the AD Mouse Model
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Perfusion Fixation
4.3. Microdissection
4.4. RNA Isolation
4.5. Library Construction and Sequencing
4.6. Read Alignment
4.7. Gene Expression Quantification
4.8. Analysis of Gene Expression Differences
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lu, K.; Ye, W.; Zhou, L.; Collins, L.B.; Chen, X.; Gold, A.; Ball, L.M.; Swenberg, J.A. Structural Characterization of Formaldehyde-Induced Cross-Links Between Amino Acids and Deoxynucleosides and Their Oligomers. J. Am. Chem. Soc. 2010, 132, 3388–3399. [Google Scholar] [CrossRef] [PubMed]
- Qidwai, K.; Afkhami, M.; Day, C.E. The Pathologist’s Guide to Fixatives; Springer: New York, NY, USA, 2014; pp. 21–30. [Google Scholar]
- Tainaka, K.; Kuno, A.; Kubota, S.I.; Murakami, T.; Ueda, H.R. Chemical Principles in Tissue Clearing and Staining Protocols for Whole-Body Cell Profiling. Annu. Rev. Cell Dev. Biol. 2016, 32, 713–741. [Google Scholar] [CrossRef] [PubMed]
- Suvarna, K.S.; Layton, C.; Bancroft, J.D. Bancroft’s Theory and Practice of Histological Techniques; Elsevier: Amsterdam, The Netherlands, 2018. [Google Scholar]
- Thavarajah, R.; Mudimbaimannar, V.K.; Elizabeth, J.; Rao, U.K.; Ranganathan, K. Chemical and physical basics of routine formaldehyde fixation. J. Oral Maxillofac. Pathol. 2012, 16, 400. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.W.; Guan, W.; Chung, K. Basic principles of hydrogel-based tissue transformation technologies and their applications. Cell 2021, 184, 4115–4136. [Google Scholar] [CrossRef] [PubMed]
- Tomer, R.; Ye, L.; Hsueh, B.; Deisseroth, K. Advanced CLARITY for rapid and high-resolution imaging of intact tissues. Nat. Protoc. 2014, 9, 1682–1697. [Google Scholar] [CrossRef] [PubMed]
- Chung, K.; Wallace, J.; Kim, S.Y.; Kalyanasundaram, S.; Andalman, A.S.; Davidson, T.J.; Mirzabekov, J.J.; Zalocusky, K.A.; Mattis, J.; Denisin, A.K.; et al. Structural and molecular interrogation of intact biological systems. Nature 2013, 497, 332–337. [Google Scholar] [CrossRef] [PubMed]
- Masuda, N.; Ohnishi, T.; Kawamoto, S.; Monden, M.; Okubo, K. Analysis of chemical modification of RNA from formalin-fixed samples and optimization of molecular biology applications for such samples. Nucleic Acids Res. 1999, 27, 4436–4443. [Google Scholar] [CrossRef]
- Evers, D.L.; Fowler, C.B.; Cunningham, B.R.; Mason, J.T.; O’Leary, T.J. The effect of formaldehyde fixation on RNA: Optimization of formaldehyde adduct removal. J. Mol. Diagn. 2011, 13, 282–288. [Google Scholar] [CrossRef] [PubMed]
- Zeka, F.; Vanderheyden, K.; De Smet, E.; Cuvelier, C.A.; Mestdagh, P.; Vandesompele, J. Straightforward and sensitive RT-qPCR based gene expression analysis of FFPE samples. Sci. Rep. 2016, 6, 21418. [Google Scholar] [CrossRef]
- Channathodiyil, P.; Houseley, J. Glyoxal fixation facilitates transcriptome analysis after antigen staining and cell sorting by flow cytometry. PLoS ONE 2021, 16, e0240769. [Google Scholar] [CrossRef]
- Salmén, F.; Ståhl, P.L.; Mollbrink, A.; Navarro, J.F.; Vickovic, S.; Frisén, J.; Lundeberg, J. Barcoded solid-phase RNA capture for Spatial Transcriptomics profiling in mammalian tissue sections. Nat. Protoc. 2018, 13, 2501–2534. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Lloyd, R.V. In situ hybridization: Methods and applications. J. Clin. Lab. Anal. 1997, 11, 2–9. [Google Scholar] [CrossRef]
- Chen, K.H.; Boettiger, A.N.; Moffitt, J.R.; Wang, S.Y.; Zhuang, X.W. Spatially resolved, highly multiplexed RNA profiling in single cells. Science 2015, 348, aaa6090. [Google Scholar] [CrossRef] [PubMed]
- Eng, C.L.; Lawson, M.; Zhu, Q.; Dries, R.; Koulena, N.; Takei, Y.; Yun, J.; Cronin, C.; Karp, C.; Yuan, G.C.; et al. Transcriptome-scale super-resolved imaging in tissues by RNA seqFISH. Nature 2019, 568, 235–239. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Yang, M.Y.; Deng, Y.X.; Su, G.; Enninful, A.; Guo, C.C.; Tebaldi, T.; Zhang, D.; Kim, D.; Bai, Z.L.; et al. High-Spatial-Resolution Multi-Omics Sequencing via Deterministic Barcoding in Tissue. Cell 2020, 183, 1665. [Google Scholar] [CrossRef] [PubMed]
- Su, G.; Qin, X.; Enninful, A.; Bai, Z.; Deng, Y.; Liu, Y.; Fan, R. Spatial multi-omics sequencing for fixed tissue via DBiT-seq. STAR Protoc. 2021, 2, 100532. [Google Scholar] [CrossRef] [PubMed]
- Phan, H.V.; Van Gent, M.; Drayman, N.; Basu, A.; Gack, M.U.; Tay, S. High-throughput RNA sequencing of paraformaldehyde-fixed single cells. Nat. Commun. 2021, 12, 5636. [Google Scholar] [CrossRef] [PubMed]
- Thomsen, E.R.; Mich, J.K.; Yao, Z.; Hodge, R.D.; Doyle, A.M.; Jang, S.; Shehata, S.I.; Nelson, A.M.; Shapovalova, N.V.; Levi, B.P.; et al. Fixed single-cell transcriptomic characterization of human radial glial diversity. Nat. Methods 2016, 13, 87–93. [Google Scholar] [CrossRef]
- Gracia Villacampa, E.; Larsson, L.; Mirzazadeh, R.; Kvastad, L.; Andersson, A.; Mollbrink, A.; Kokaraki, G.; Monteil, V.; Schultz, N.; Appelberg, K.S.; et al. Genome-wide spatial expression profiling in formalin-fixed tissues. Cell Genom. 2021, 1, 100065. [Google Scholar] [CrossRef]
- Helander, K.G. Kinetic Studies of Formaldehyde Binding in Tissue. Biotech. Histochem. 1994, 69, 177–179. [Google Scholar] [CrossRef]
- Civita, P.; Franceschi, S.; Aretini, P.; Ortenzi, V.; Menicagli, M.; Lessi, F.; Pasqualetti, F.; Naccarato, A.G.; Mazzanti, C.M. Laser Capture Microdissection and RNA-Seq Analysis: High Sensitivity Approaches to Explain Histopathological Heterogeneity in Human Glioblastoma FFPE Archived Tissues. Front. Oncol. 2019, 9, 482. [Google Scholar] [CrossRef] [PubMed]
- Foley, J.W.; Zhu, C.; Jolivet, P.; Zhu, S.X.; Lu, P.; Meaney, M.J.; West, R.B. Gene expression profiling of single cells from archival tissue with laser-capture microdissection and Smart-3SEQ. Genome Res. 2019, 29, 1816–1825. [Google Scholar] [CrossRef]
- Evers, D.L.; He, J.; Kim, Y.H.; Mason, J.T.; O’Leary, T.J. Paraffin embedding contributes to RNA aggregation, reduced RNA yield, and low RNA quality. J. Mol. Diagn. 2011, 13, 687–694. [Google Scholar] [CrossRef]
- von Ahlfen, S.; Missel, A.; Bendrat, K.; Schlumpberger, M. Determinants of RNA quality from FFPE samples. PLoS ONE 2007, 2, e1261. [Google Scholar] [CrossRef] [PubMed]
- Sherwood, C.C.; Hof, P.R.; Holloway, R.L.; Semendeferi, K.; Gannon, P.J.; Frahm, H.D.; Zilles, K. Evolution of the brainstem orofacial motor system in primates: A comparative study of trigeminal, facial, and hypoglossal nuclei. J. Hum. Evol. 2005, 48, 45–84. [Google Scholar] [CrossRef]
- McClung, J.R.; Goldberg, S.J. Organization of motoneurons in the dorsal hypoglossal nucleus that innervate the retrusor muscles of the tongue in the rat. Anat. Rec. 1999, 254, 222–230. [Google Scholar] [CrossRef]
- Fischer, W.; Chen, K.S.; Gage, F.H.; Björklund, A. Progressive decline in spatial learning and integrity of forebrain cholinergic neurons in rats during aging. Neurobiol. Aging 1992, 13, 9–23. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, K.; Miranda, A.; Craig, D.W.; Shekhtman, T.; Kmoch, S.; Bleyer, A.; Szelinger, S.; Kato, T.; Kelsoe, J.R. Ntrk1 mutation co-segregating with bipolar disorder and inherited kidney disease in a multiplex family causes defects in neuronal growth and depression-like behavior in mice. Transl. Psychiatry 2020, 10, 407. [Google Scholar] [CrossRef] [PubMed]
- Barwick, K.E.; Wright, J.; Al-Turki, S.; McEntagart, M.M.; Nair, A.; Chioza, B.; Al-Memar, A.; Modarres, H.; Reilly, M.M.; Dick, K.J. Defective presynaptic choline transport underlies hereditary motor neuropathy. Am. J. Hum. Genet. 2012, 91, 1103–1107. [Google Scholar] [CrossRef]
- O’Grady, G.L.; Verschuuren, C.; Yuen, M.; Webster, R.; Menezes, M.; Fock, J.M.; Pride, N.; Best, H.A.; Damm, T.B.; Turner, C. Variants in SLC18A3, vesicular acetylcholine transporter, cause congenital myasthenic syndrome. Neurology 2016, 87, 1442–1448. [Google Scholar] [CrossRef]
- Magno, L.; Barry, C.; Schmidt-Hieber, C.; Theodotou, P.; Häusser, M.; Kessaris, N. NKX2-1 is required in the embryonic septum for cholinergic system development, learning, and memory. Cell Rep. 2017, 20, 1572–1584. [Google Scholar] [CrossRef]
- Magno, L.; Kretz, O.; Bert, B.; Ersözlü, S.; Vogt, J.; Fink, H.; Kimura, S.; Vogt, A.; Monyer, H.; Nitsch, R. The integrity of cholinergic basal forebrain neurons depends on expression of Nkx2-1. Eur. J. Neurosci. 2011, 34, 1767–1782. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Basak, J.M.; Holtzman, D.M. The Role of Apolipoprotein E in Alzheimer’s Disease. Neuron 2009, 63, 287–303. [Google Scholar] [CrossRef] [PubMed]
- Foster, E.; Dangla-Valls, A.; Lovestone, S.; Ribe, E.; Buckley, N. Clusterin in Alzheimer’s disease: Mechanisms, genetics, and lessons from other pathologies. Front. Neurosci. 2019, 13, 164. [Google Scholar] [CrossRef] [PubMed]
- Muraleva, N.; Kolosova, N.; Stefanova, N. p38 MAPK–dependent alphaB-crystallin phosphorylation in Alzheimer’s disease–like pathology in OXYS rats. Exp. Gerontol. 2019, 119, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Graham, W.V.; Bonito-Oliva, A.; Sakmar, T.P. Update on Alzheimer’s disease therapy and prevention strategies. Annu. Rev. Med. 2017, 68, 413–430. [Google Scholar] [CrossRef] [PubMed]
- Ransohoff, R.M. How neuroinflammation contributes to neurodegeneration. Science 2016, 353, 777–783. [Google Scholar] [CrossRef] [PubMed]
- Jana, A.; Pahan, K. Fibrillar Amyloid—Activated Human Astroglia Kill Primary Human Neurons via Neutral Sphingomyelinase: Implications for Alzheimer’s Disease. J. Neurosci. 2010, 30, 12676–12689. [Google Scholar] [CrossRef] [PubMed]
- Lian, H.; Yang, L.; Cole, A.; Sun, L.; Chiang, A.C.A.; Fowler, S.; Rodriguez-Rivera, J.; Taglialatela, G.; Jankowsky, J.; Zheng, H.; et al. NFκB-Activated Astroglial Release of Complement C3 Compromises Neuronal Morphology and Function Associated with Alzheimer’s Disease. Neuron 2015, 85, 101–115. [Google Scholar] [CrossRef]
- Kenigsbuch, M.; Bost, P.; Halevi, S.; Chang, Y.; Chen, S.; Ma, Q.; Hajbi, R.; Schwikowski, B.; Bodenmiller, B.; Fu, H.; et al. A shared disease-associated oligodendrocyte signature among multiple CNS pathologies. Nat. Neurosci. 2022, 25, 876–886. [Google Scholar] [CrossRef]
- Simms, D.; Cizdziel, P.E.; Chomczynski, P. TRIzol: A new reagent for optimal single-step isolation of RNA. Focus 1993, 15, 532–535. [Google Scholar]
- Amini, P.; Ettlin, J.; Opitz, L.; Clementi, E.; Malbon, A.; Markkanen, E. An optimised protocol for isolation of RNA from small sections of laser-capture microdissected FFPE tissue amenable for next-generation sequencing. BMC Mol. Biol. 2017, 18, 22. [Google Scholar] [CrossRef]
- Wehmas, L.C.; Wood, C.E.; Gagne, R.; Williams, A.; Yauk, C.; Gosink, M.M.; Dalmas, D.; Hao, R.; O’Lone, R.; Hester, S. Demodifying RNA for Transcriptomic Analyses of Archival Formalin-Fixed Paraffin-Embedded Samples. Toxicol. Sci. 2018, 162, 535–547. [Google Scholar] [CrossRef]
- Hamatani, K.; Eguchi, H.; Takahashi, K.; Koyama, K.; Mukai, M.; Ito, R.; Taga, M.; Yasui, W.; Nakachi, K. Improved RT-PCR Amplification for Molecular Analyses with Long-term Preserved Formalin-fixed, Paraffin-embedded Tissue Specimens. J. Histochem. Cytochem. 2006, 54, 773–780. [Google Scholar] [CrossRef] [PubMed]
- Graw, S.; Meier, R.; Minn, K.; Bloomer, C.; Godwin, A.K.; Fridley, B.; Vlad, A.; Beyerlein, P.; Chien, J. Robust gene expression and mutation analyses of RNA-sequencing of formalin-fixed diagnostic tumor samples. Sci. Rep. 2015, 5, 12335. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Fu, C.; Speed, T.P.; Wang, W.; Symmans, W.F. Accurate RNA Sequencing From Formalin-Fixed Cancer Tissue to Represent High-Quality Transcriptome From Frozen Tissue. JCO Precis. Oncol. 2018, 2, 91. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Conley, A.; Zhang, H.; Kim, H.L. Whole-Transcriptome profiling of formalin-fixed, paraffin-embedded renal cell carcinoma by RNA-seq. BMC Genom. 2014, 15, 1087. [Google Scholar] [CrossRef] [PubMed]
- Hester, S.D.; Bhat, V.; Chorley, B.N.; Carswell, G.; Jones, W.; Wehmas, L.C.; Wood, C.E. Editor’s Highlight: Dose–Response Analysis of RNA-Seq Profiles in Archival Formalin-Fixed Paraffin-Embedded Samples. Toxicol. Sci. 2016, 154, 202–213. [Google Scholar] [CrossRef] [PubMed]
- Pena, J.T.; Sohn-Lee, C.; Rouhanifard, S.H.; Ludwig, J.; Hafner, M.; Mihailovic, A.; Lim, C.; Holoch, D.; Berninger, P.; Zavolan, M.; et al. miRNA in situ hybridization in formaldehyde and EDC-fixed tissues. Nat. Methods 2009, 6, 139–141. [Google Scholar] [CrossRef]
- Sylwestrak, E.L.; Rajasethupathy, P.; Wright, M.A.; Jaffe, A.; Deisseroth, K. Multiplexed Intact-Tissue Transcriptional Analysis at Cellular Resolution. Cell 2016, 164, 792–804. [Google Scholar] [CrossRef]
- Alldred, M.J.; Penikalapati, S.C.; Lee, S.H.; Heguy, A.; Roussos, P.; Ginsberg, S.D. Profiling Basal Forebrain Cholinergic Neurons Reveals a Molecular Basis for Vulnerability Within the Ts65Dn Model of Down Syndrome and Alzheimer’s Disease. Mol. Neurobiol. 2021, 58, 5141–5162. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yu, H.; Zhang, B.; Li, L.; Chen, W.; Yu, Q.; Huang, X.; Ke, X.; Wang, Y.; Jing, W.; et al. Molecularly defined and functionally distinct cholinergic subnetworks. Neuron 2022, 110, 3774–3788.e3777. [Google Scholar] [CrossRef]
- Chen, J.; Suo, S.; Tam, P.P.; Han, J.-D.J.; Peng, G.; Jing, N. Spatial transcriptomic analysis of cryosectioned tissue samples with Geo-seq. Nat. Protoc. 2017, 12, 566–580. [Google Scholar] [CrossRef] [PubMed]
- Gong, H.; Xu, D.; Yuan, J.; Li, X.; Guo, C.; Peng, J.; Li, Y.; Schwarz, L.A.; Li, A.; Hu, B.; et al. High-throughput dual-colour precision imaging for brain-wide connectome with cytoarchitectonic landmarks at the cellular level. Nat. Commun. 2016, 7, 12142. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Wang, J.; Wu, H.; Zhang, L.; Zhou, J.; Ye, Q.; Shao, X.; Guan, C.; Xu, J.; Yang, Y.; et al. Low-temperature glycol methacrylate resin embedding method: A protocol suitable for bone marrow immunohistochemistry, PCR, and fish analysis. Microsc. Res. Tech. 2010, 73, 1067–1071. [Google Scholar] [CrossRef]
- Torgersen, J.S.; Takle, H.; Andersen, Ø. Localization of mRNAs and Proteins in Methyl Methacrylate-embedded Tissues. J. Histochem. Cytochem. 2009, 57, 825–830. [Google Scholar] [CrossRef]
- Jiang, T.; Long, B.; Gong, H.; Xu, T.; Li, X.; Duan, Z.; Li, A.; Deng, L.; Zhong, Q.; Peng, X.; et al. A platform for efficient identification of molecular phenotypes of brain-wide neural circuits. Sci. Rep. 2017, 7, 13891. [Google Scholar] [CrossRef]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. GigaScience 2021, 10, giab008. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Wang, L.; Wang, S.; Li, W. RSeQC: Quality control of RNA-seq experiments. Bioinformatics 2012, 28, 2184–2185. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; Van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS 2012, 16, 284–287. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ji, B.; Chen, J.; Gong, H.; Li, X. Streamlined Full-Length Total RNA Sequencing of Paraformaldehyde-Fixed Brain Tissues. Int. J. Mol. Sci. 2024, 25, 6504. https://doi.org/10.3390/ijms25126504
Ji B, Chen J, Gong H, Li X. Streamlined Full-Length Total RNA Sequencing of Paraformaldehyde-Fixed Brain Tissues. International Journal of Molecular Sciences. 2024; 25(12):6504. https://doi.org/10.3390/ijms25126504
Chicago/Turabian StyleJi, Bingqing, Jiale Chen, Hui Gong, and Xiangning Li. 2024. "Streamlined Full-Length Total RNA Sequencing of Paraformaldehyde-Fixed Brain Tissues" International Journal of Molecular Sciences 25, no. 12: 6504. https://doi.org/10.3390/ijms25126504