
Cytoprotective Role of Autophagy in CDIP1 Expression-Induced Apoptosis in MCF-7 Breast Cancer Cells

Abstract
:1. Introduction
2. Results
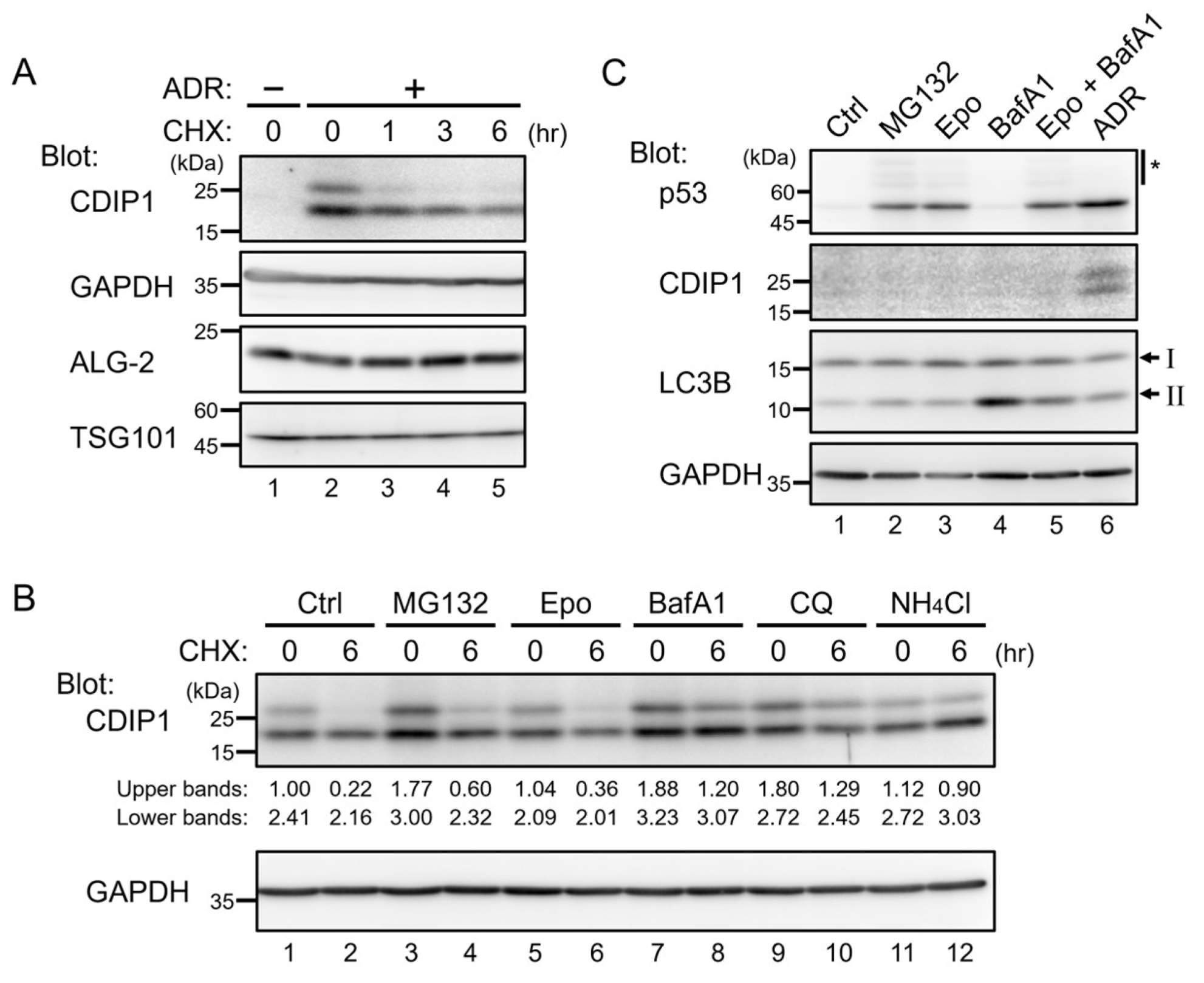
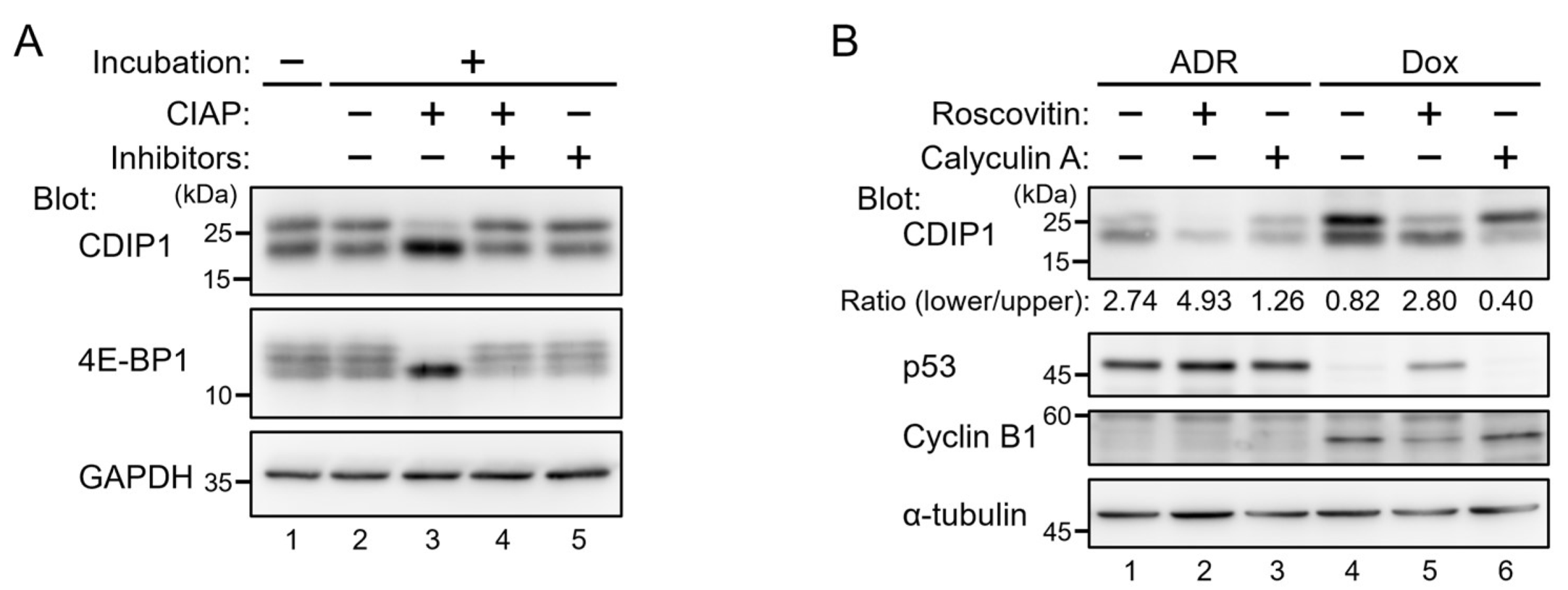
2.1. Lysosomal Degradation of CDIP1
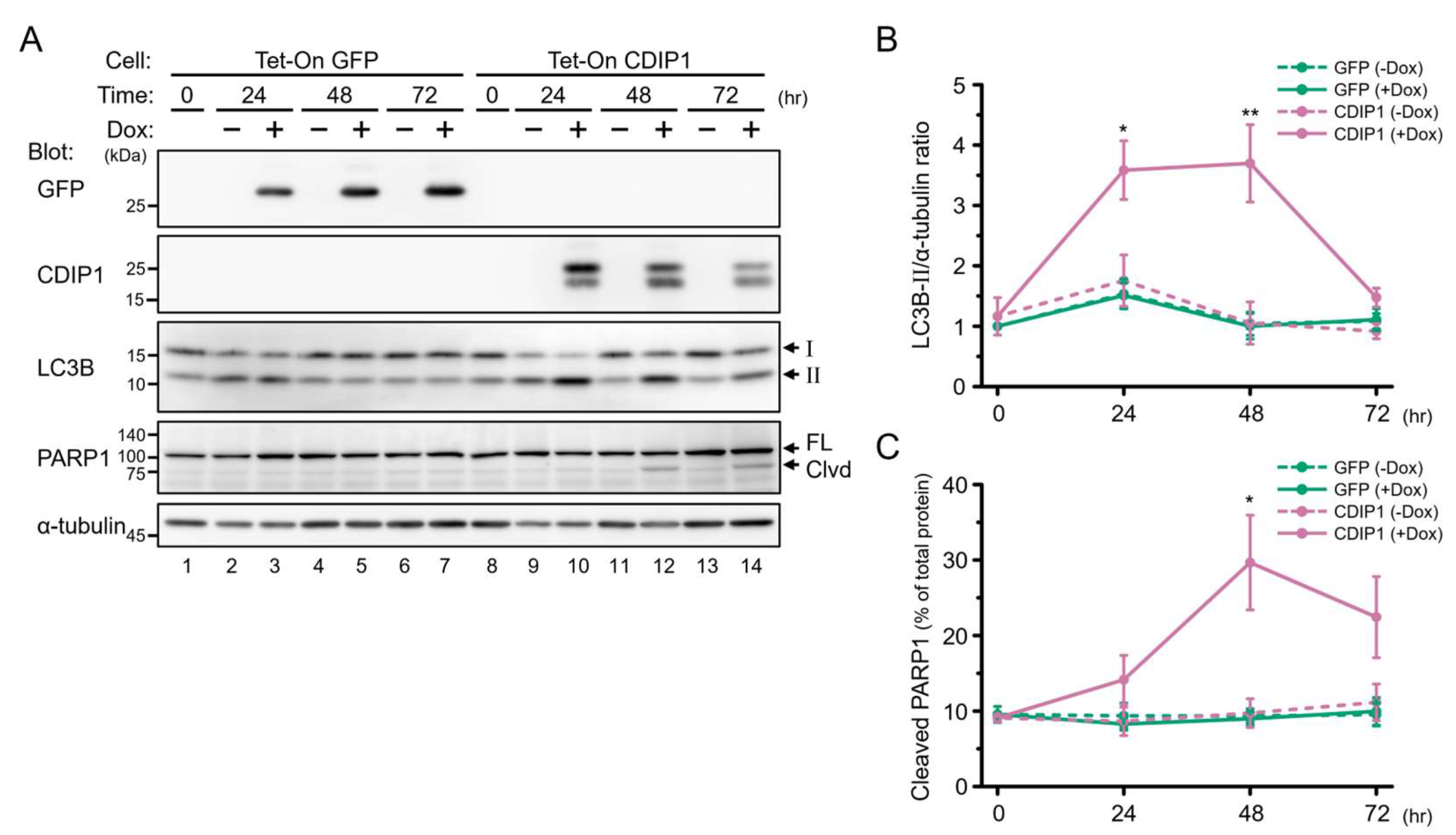
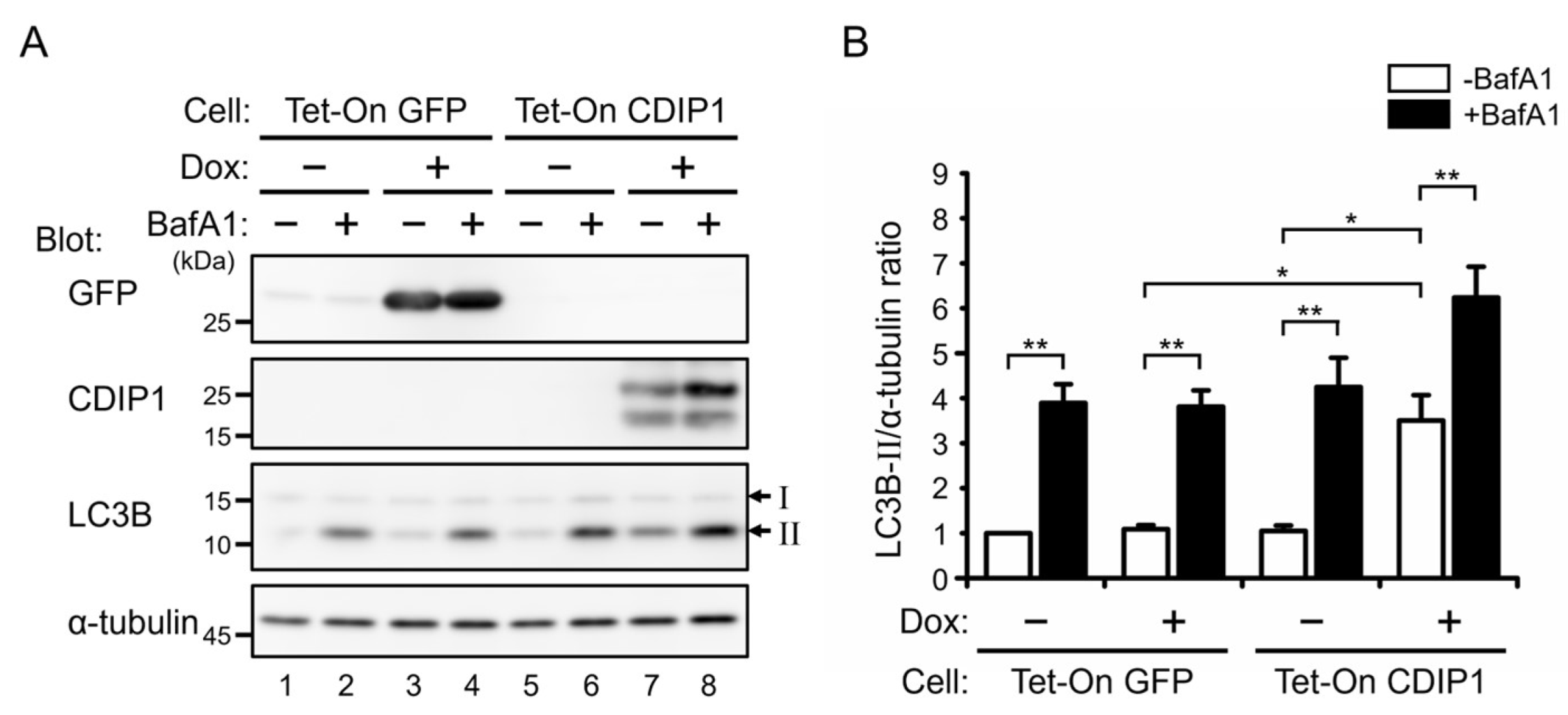
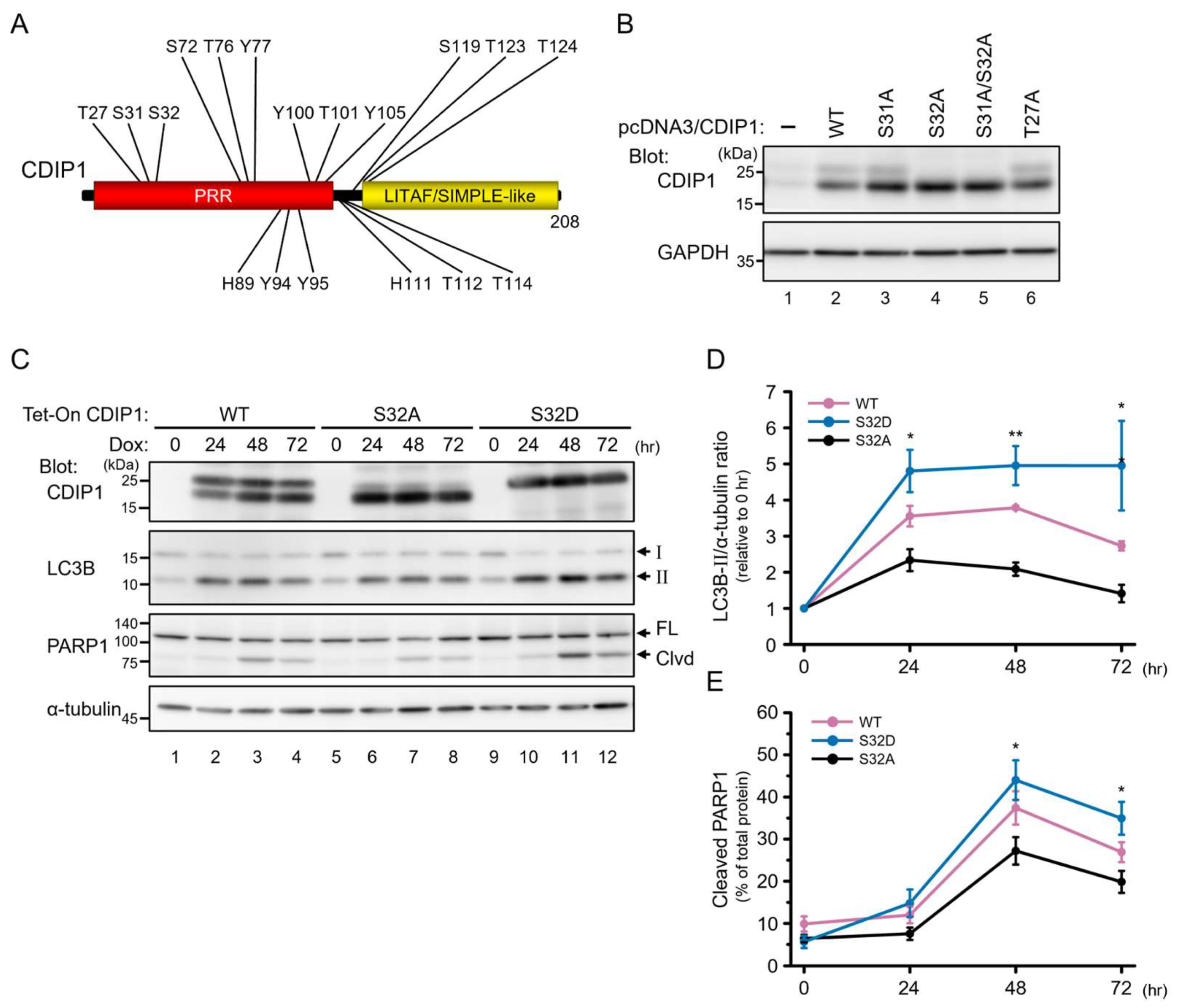
2.2. Induction of Autophagy by CDIP1
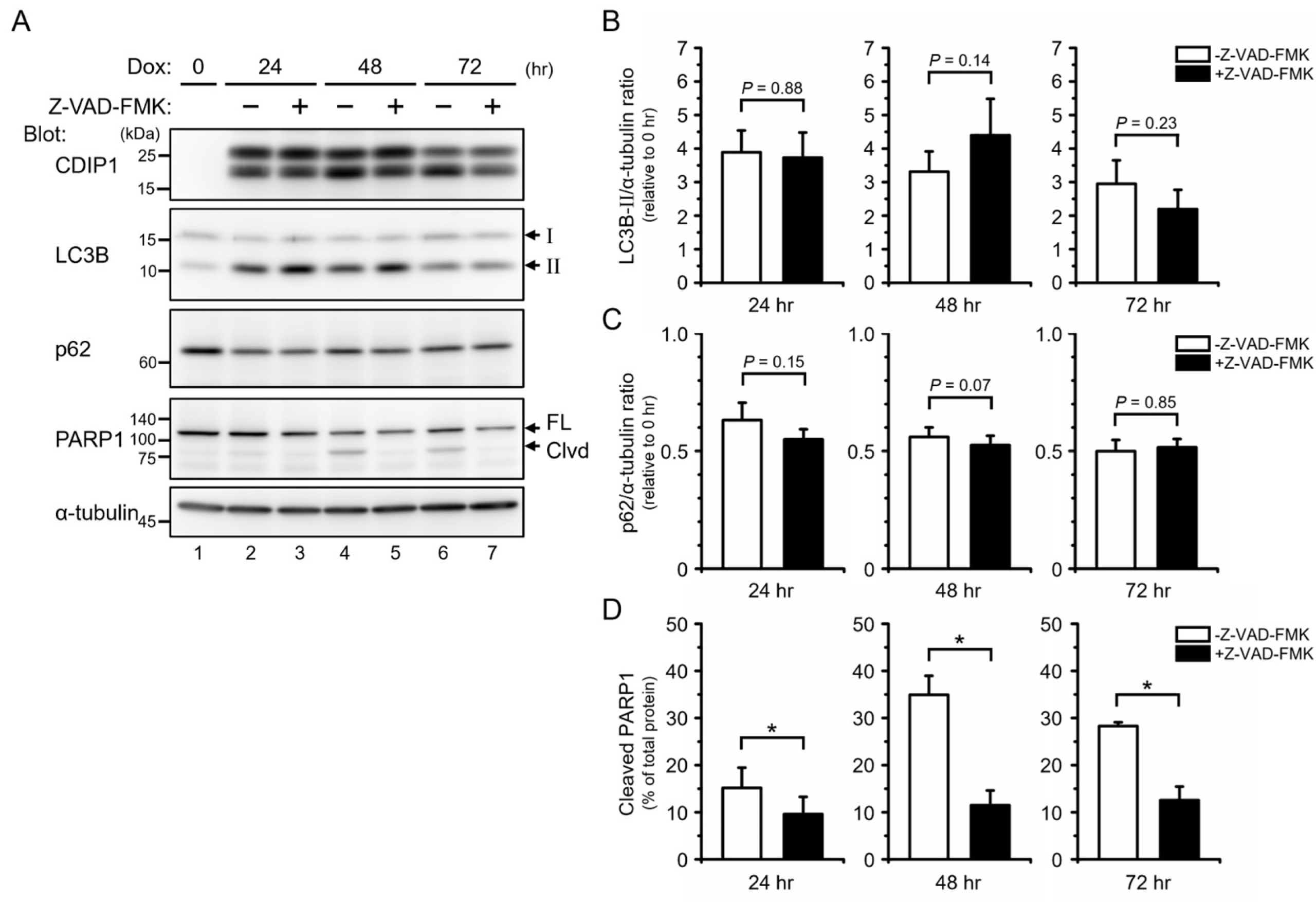
2.3. Protective Role of Autophagy in CDIP1 Expression-Induced Apoptosis
3. Discussion
4. Materials and Methods
4.1. Antibodies and Reagents
4.2. Plasmid Constructions
4.3. Cell Culture, DNA Transfection and Lentiviral Infection
4.4. Cell Lysate Preparation and Western Blotting
4.5. Statistical Analysis
4.6. Research Ethics
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ADR | adriamycin |
| ALG-2 | apoptosis-linked gene 2 |
| BafA1 | bafilomycin A1 |
| BAP31 | B-cell receptor-associated protein 31 |
| CDIP1 | cell death-inducing p53-target protein 1 |
| CHX | cycloheximide |
| CIAP | calf intestine alkaline phosphatase |
| CQ | chloroquine |
| Dox | doxycycline |
| Epo | epoxomicin |
| ER | endoplasmic reticulum |
| GAPDH | glyceraldehyde-3-phosphate dehydrogenase |
| LC3B | microtubule-associated proteins 1A/1B light chain 3B |
| LITAF | lipopolysaccharide-induced tumor necrosis factor-α factor homolog |
| PARP1 | poly[ADP-ribose] polymerase 1 |
| SIMPLE | small integral membrane protein of the late endosome |
| TNF-α | tumor necrosis factor-α |
| TSG101 | tumor susceptibility gene 101 |
References
- Mizushima, N.; Levine, B. Autophagy in Human Diseases. N. Engl. J. Med. 2020, 383, 1564–1576. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Petroni, G.; Amaravadi, R.K.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cadwell, K.; Cecconi, F.; Choi, A.M.K.; et al. Autophagy in major human diseases. EMBO J. 2021, 40, e108863. [Google Scholar] [CrossRef] [PubMed]
- Poillet-Perez, L.; White, E. Role of tumor and host autophagy in cancer metabolism. Genes Dev. 2019, 33, 610–619. [Google Scholar] [CrossRef] [PubMed]
- Ariosa, A.R.; Lahiri, V.; Lei, Y.; Yang, Y.; Yin, Z.; Zhang, Z.; Klionsky, D.J. A perspective on the role of autophagy in cancer. Biochim. Biophys. Acta Mol. Basis Dis. 2021, 1867, 166262. [Google Scholar] [CrossRef]
- Amaravadi, R.K.; Kimmelman, A.C.; Debnath, J. Targeting Autophagy in Cancer: Recent Advances and Future Directions. Cancer Discov. 2019, 9, 1167–1181. [Google Scholar] [CrossRef] [PubMed]
- Levy, J.M.M.; Thorburn, A. Autophagy in cancer: Moving from understanding mechanism to improving therapy responses in patients. Cell Death Differ. 2020, 27, 843–857. [Google Scholar] [CrossRef] [PubMed]
- Yun, C.W.; Jeon, J.; Go, G.; Lee, J.H.; Lee, S.H. The Dual Role of Autophagy in Cancer Development and a Therapeutic Strategy for Cancer by Targeting Autophagy. Int. J. Mol. Sci. 2021, 22, 179. [Google Scholar] [CrossRef] [PubMed]
- Brown, L.; Ongusaha, P.P.; Kim, H.G.; Nuti, S.; Mandinova, A.; Lee, J.W.; Khosravi-Far, R.; Aaronson, S.A.; Lee, S.W. CDIP, a novel pro-apoptotic gene, regulates TNFalpha-mediated apoptosis in a p53-dependent manner. EMBO J. 2007, 26, 3410–3422. [Google Scholar] [CrossRef] [PubMed]
- Brown-Endres, L.; Schoenfeld, D.; Tian, F.; Kim, H.G.; Namba, T.; Muñoz-Fontela, C.; Mandinova, A.; Aaronson, S.A.; Lee, S.W. Expression of the p53 target CDIP correlates with sensitivity to TNFα-induced apoptosis in cancer cells. Cancer Res. 2012, 72, 2373–2382. [Google Scholar] [CrossRef]
- Namba, T.; Tian, F.; Chu, K.; Hwang, S.Y.; Yoon, K.W.; Byun, S.; Hiraki, M.; Mandinova, A.; Lee, S.W. CDIP1-BAP31 complex transduces apoptotic signals from endoplasmic reticulum to mitochondria under endoplasmic reticulum stress. Cell Rep. 2013, 5, 331–339. [Google Scholar] [CrossRef]
- Ladin, D.A.; Soliman, E.; Escobedo, R.; Fitzgerald, T.L.; Yang, L.V.; Burns, C.; Van Dross, R. Synthesis and Evaluation of the Novel Prostamide, 15-Deoxy, Δ12,14-Prostamide J2, as a Selective Antitumor Therapeutic. Mol. Cancer Ther. 2017, 16, 838–849. [Google Scholar] [CrossRef] [PubMed]
- Inukai, R.; Mori, K.; Kuwata, K.; Suzuki, C.; Maki, M.; Takahara, T.; Shibata, H. The Novel ALG-2 Target Protein CDIP1 Promotes Cell Death by Interacting with ESCRT-I and VAPA/B. Int. J. Mol. Sci. 2021, 22, 1175. [Google Scholar] [CrossRef] [PubMed]
- Qin, W.; Wunderley, L.; Barrett, A.L.; High, S.; Woodman, P.G. The Charcot Marie Tooth disease protein LITAF is a zinc-binding monotopic membrane protein. Biochem. J. 2016, 473, 3965–3978. [Google Scholar] [CrossRef] [PubMed]
- Ho, A.K.; Wagstaff, J.L.; Manna, P.T.; Wartosch, L.; Qamar, S.; Garman, E.F.; Freund, S.M.; Roberts, R.C. The topology, structure and PE interaction of LITAF underpin a Charcot-Marie-Tooth disease type 1C. BMC Biol. 2016, 14, 109. [Google Scholar] [CrossRef] [PubMed]
- Wunderley, L.; Zhang, L.; Yarwood, R.; Qin, W.; Lowe, M.; Woodman, P. Endosomal recycling tubule scission and integrin recycling involve the membrane curvature-supporting protein LITAF. J. Cell Sci. 2021, 134, jcs258549. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.M.; Olzmann, J.A.; Chin, L.S.; Li, L. Mutations associated with Charcot-Marie-Tooth disease cause SIMPLE protein mislocalization and degradation by the proteasome and aggresome-autophagy pathways. J. Cell Sci. 2011, 124, 3319–3331. [Google Scholar] [CrossRef] [PubMed]
- Longva, K.E.; Blystad, F.D.; Stang, E.; Larsen, A.M.; Johannessen, L.E.; Madshus, I.H. Ubiquitination and proteasomal activity is required for transport of the EGF receptor to inner membranes of multivesicular bodies. J. Cell Biol. 2002, 156, 843–854. [Google Scholar] [CrossRef]
- Meng, L.; Mohan, R.; Kwok, B.H.; Elofsson, M.; Sin, N.; Crews, C.M. Epoxomicin, a potent and selective proteasome inhibitor, exhibits in vivo antiinflammatory activity. Proc. Natl. Acad. Sci. USA 1999, 96, 10403–10408. [Google Scholar] [CrossRef] [PubMed]
- Yoshimori, T.; Yamamoto, A.; Moriyama, Y.; Futai, M.; Tashiro, Y. Bafilomycin A1, a specific inhibitor of vacuolar-type H+-ATPase, inhibits acidification and protein degradation in lysosomes of cultured cells. J. Biol. Chem. 1991, 266, 17707–17712. [Google Scholar] [CrossRef]
- Poole, B.; Ohkuma, S. Effect of weak bases on the intralysosomal pH in mouse peritoneal macrophages. J. Cell Biol. 1981, 90, 665–669. [Google Scholar] [CrossRef]
- Solomon, V.R.; Lee, H. Chloroquine and its analogs: A new promise of an old drug for effective and safe cancer therapies. Eur. J. Pharmacol. 2009, 625, 220–233. [Google Scholar] [CrossRef] [PubMed]
- Kato, Y.; Fusetani, N.; Matsunaga, S.; Hashimoto, K.; Fujita, S.; Furuya, T. Bioactive marine metabolites. Part 16. Calyculin A. A novel antitumor metabolite from the marine sponge Discodermia calyx. J. Am. Chem. Soc. 1986, 108, 2780–2781. [Google Scholar] [CrossRef]
- Ishihara, H.; Martin, B.L.; Brautigan, D.L.; Karaki, H.; Ozaki, H.; Kato, Y.; Fusetani, N.; Watabe, S.; Hashimoto, K.; Uemura, D.; et al. Calyculin A and okadaic acid: Inhibitors of protein phosphatase activity. Biochem. Biophys. Res. Commun. 1989, 159, 871–877. [Google Scholar] [CrossRef] [PubMed]
- Meijer, L.; Borgne, A.; Mulner, O.; Chong, J.P.; Blow, J.J.; Inagaki, N.; Inagaki, M.; Delcros, J.G.; Moulinoux, J.P. Biochemical and cellular effects of roscovitine, a potent and selective inhibitor of the cyclin-dependent kinases cdc2, cdk2 and cdk5. Eur. J. Biochem. 1997, 243, 527–536. [Google Scholar] [CrossRef] [PubMed]
- Kabeya, Y.; Mizushima, N.; Ueno, T.; Yamamoto, A.; Kirisako, T.; Noda, T.; Kominami, E.; Ohsumi, Y.; Yoshimori, T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000, 19, 5720–5728. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Yoshimori, T. How to interpret LC3 immunoblotting. Autophagy 2007, 3, 542–545. [Google Scholar] [CrossRef] [PubMed]
- Hornbeck, P.V.; Kornhauser, J.M.; Latham, V.; Murray, B.; Nandhikonda, V.; Nord, A.; Skrzypek, E.; Wheeler, T.; Zhang, B.; Gnad, F. 15 years of PhosphoSitePlus®: Integrating post-translationally modified sites, disease variants and isoforms. Nucleic Acids Res. 2019, 47, D433–D441. [Google Scholar] [CrossRef] [PubMed]
- Shiromizu, T.; Adachi, J.; Watanabe, S.; Murakami, T.; Kuga, T.; Muraoka, S.; Tomonaga, T. Identification of missing proteins in the neXtProt database and unregistered phosphopeptides in the PhosphoSitePlus database as part of the Chromosome-centric Human Proteome Project. J. Proteome Res. 2013, 12, 2414–2421. [Google Scholar] [CrossRef] [PubMed]
- Petiot, A.; Ogier-Denis, E.; Blommaart, E.F.; Meijer, A.J.; Codogno, P. Distinct classes of phosphatidylinositol 3′-kinases are involved in signaling pathways that control macroautophagy in HT-29 cells. J. Biol. Chem. 2000, 275, 992–998. [Google Scholar] [CrossRef] [PubMed]
- Itakura, E.; Kishi, C.; Inoue, K.; Mizushima, N. Beclin 1 forms two distinct phosphatidylinositol 3-kinase complexes with mammalian Atg14 and UVRAG. Mol. Biol. Cell 2008, 19, 5360–5372. [Google Scholar] [CrossRef]
- Liang, C.; Feng, P.; Ku, B.; Dotan, I.; Canaani, D.; Oh, B.H.; Jung, J.U. Autophagic and tumour suppressor activity of a novel Beclin1-binding protein UVRAG. Nat. Cell Biol. 2006, 8, 688–699. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Long, Y.C.; Shen, H.M. Differential regulatory functions of three classes of phosphatidylinositol and phosphoinositide 3-kinases in autophagy. Autophagy 2015, 11, 1711–1728. [Google Scholar] [CrossRef] [PubMed]
- Ronan, B.; Flamand, O.; Vescovi, L.; Dureuil, C.; Durand, L.; Fassy, F.; Bachelot, M.F.; Lamberton, A.; Mathieu, M.; Bertrand, T.; et al. A highly potent and selective Vps34 inhibitor alters vesicle trafficking and autophagy. Nat. Chem. Biol. 2014, 10, 1013–1019. [Google Scholar] [CrossRef] [PubMed]
- Pasquier, B. SAR405, a PIK3C3/Vps34 inhibitor that prevents autophagy and synergizes with mTOR inhibition in tumor cells. Autophagy 2015, 4, 725–726. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Mizushima, N. LC3- and p62-based biochemical methods for the analysis of autophagy progression in mammalian cells. Methods 2015, 75, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, M.; Waguri, S.; Koike, M.; Sou, Y.S.; Ueno, T.; Hara, T.; Mizushima, N.; Iwata, J.; Ezaki, J.; Murata, S.; et al. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell 2007, 131, 1149–1163. [Google Scholar] [CrossRef] [PubMed]
- Pankiv, S.; Clausen, T.H.; Lamark, T.; Brech, A.; Bruun, J.A.; Outzen, H.; Øvervatn, A.; Bjørkøy, G.; Johansen, T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 2007, 282, 24131–24145. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Chang, J.W.; Wang, J.; Kang, S.A.; Thoreen, C.C.; Markhard, A.; Hur, W.; Zhang, J.; Sim, T.; Sabatini, D.M.; et al. Discovery of 1-(4-(4-propionylpiperazin-1-yl)-3-(trifluoromethyl)phenyl)-9-(quinolin-3-yl)benzo[h][1,6]naphthyridin-2(1H)-one as a highly potent, selective mammalian target of rapamycin (mTOR) inhibitor for the treatment of cancer. J. Med. Chem. 2010, 53, 7146–7155. [Google Scholar] [CrossRef] [PubMed]
- Takahara, T.; Amemiya, Y.; Sugiyama, R.; Maki, M.; Shibata, H. Amino acid-dependent control of mTORC1 signaling: A variety of regulatory modes. J. Biomed. Sci. 2020, 27, 87. [Google Scholar] [CrossRef]
- Keyse, S.M.; Tyrrell, A.M. Heme oxygenase is the major 32 kDa stress protein induced in human skin fibroblasts of UVA radiation, hydrogen peroxide and sodium arsenite. Proc. Natl. Acad. Sci. USA 1989, 86, 99–103. [Google Scholar] [CrossRef]
- Maines, M.D. The heme oxygenase system: A regulator of second messenger gases. Annu. Rev. Pharmacol. Toxicol. 1997, 37, 517–554. [Google Scholar] [CrossRef]
- Slee, E.A.; Zhu, H.; Chow, S.C.; MacFarlane, M.; Nicholson, D.W.; Cohen, G.M. Benzyloxycarbonyl-Val-Ala-Asp (OMe) fluoromethylketone (Z-VAD.FMK) inhibits apoptosis by blocking the processing of CPP32. Biochem. J. 1996, 315, 21–24. [Google Scholar] [CrossRef] [PubMed]
- Xiang, X.; Zheng, L.; Li, X. Silencing of Long Noncoding RNA Zinc Finger Antisense 1 Protects Against Hypoxia/Reoxygenation-induced Injury in HL-1 Cells Through Targeting the miR-761/Cell Death Inducing p53 Target 1 Axis. J. Cardiovasc. Pharmacol. 2020, 76, 564–573. [Google Scholar] [CrossRef]
- Lai, C.H.; Barik, P.; Hsieh, D.J.; Day, C.H.; Ho, T.J.; Chen, R.J.; Kuo, W.W.; Padma, V.V.; Shibu, M.A.; Huang, C.Y. Inhibition of cell death-inducing p53 target 1 through miR-210-3p overexpression attenuates reactive oxygen species and apoptosis in rat adipose-derived stem cells challenged with Angiotensin II. Biochem. Biophys. Res. Commun. 2020, 532, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Liao, Z.; Chen, Y.; Duan, C.; Zhu, K.; Huang, R.; Zhao, H.; Hintze, M.; Pu, Q.; Yuan, Z.; Lv, L.; et al. Cardiac telocytes inhibit cardiac microvascular endothelial cell apoptosis through exosomal miRNA-21-5p-targeted cdip1 silencing to improve angiogenesis following myocardial infarction. Theranostics 2021, 11, 268–291. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Wang, H.; Li, H.; Lu, X.; Gao, Y.; Guo, X. Long noncoding RNA BACE1-antisense transcript plays a critical role in Parkinson’s disease via microRNA-214-3p/Cell death-inducing p53-target protein 1 axis. Bioengineered 2022, 13, 10889–10901. [Google Scholar] [CrossRef]
- Yan, G.; Lei, W. Role of ELK1 in regulating colorectal cancer progression: miR-31-5p/CDIP1 axis in CRC pathogenesis. PeerJ 2023, 11, e15602. [Google Scholar] [CrossRef]
- Avinery, L.; Gahramanov, V.; Hesin, A.; Sherman, M.Y. Hsp70–Bag3 Module Regulates Macrophage Motility and Tumor Infiltration via Transcription Factor LITAF and CSF1. Cancers 2022, 14, 4168. [Google Scholar] [CrossRef]
- Liu, J.; Zuo, Z.; Sastalla, I.; Liu, C.; Jang, J.Y.; Sekine, Y.; Li, Y.; Pirooznia, M.; Leppla, S.H.; Finkel, T.; et al. Sequential CRISPR-Based Screens Identify LITAF and CDIP1 as the Bacillus cereus Hemolysin BL Toxin Host Receptors. Cell Host Microbe 2020, 28, 402–410.e5. [Google Scholar] [CrossRef]
- Farkas, Á.; Bohnsack, K.E. Capture and delivery of tail-anchored proteins to the endoplasmic reticulum. J. Cell Biol. 2021, 220, e202105004. [Google Scholar] [CrossRef]
- Bórquez, D.A.; Olmos, C.; Álvarez, S.; Di Genova, A.; Maass, A.; González-Billault, C. Bioinformatic survey for new physiological substrates of Cyclin-dependent kinase 5. Genomics 2013, 101, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Bach, S.; Knockaert, M.; Reinhardt, J.; Lozach, O.; Schmitt, S.; Baratte, B.; Koken, M.; Coburn, S.P.; Tang, L.; Jiang, T.; et al. Roscovitine targets, protein kinases and pyridoxal kinase. J. Biol. Chem. 2005, 280, 31208–31219. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeldt, M.T.; O’Prey, J.; Morton, J.P.; Nixon, C.; MacKay, G.; Mrowinska, A.; Au, A.; Rai, T.S.; Zheng, L.; Ridgway, R.; et al. p53 status determines the role of autophagy in pancreatic tumour development. Nature 2013, 504, 296–300. [Google Scholar] [CrossRef] [PubMed]
- Shibata, H.; Suzuki, H.; Yoshida, H.; Maki, M. ALG-2 directly binds Sec31A and localizes at endoplasmic reticulum exit sites in a Ca2+-dependent manner. Biochem. Biophys. Res. Commun. 2007, 353, 756–763. [Google Scholar] [CrossRef]
- Okegawa, Y.; Motohashi, K. A simple and ultra-low cost homemade seamless ligation cloning extract (SLiCE) as an alternative to a commercially available seamless DNA cloning kit. Biochem. Biophys. Rep. 2015, 4, 148–151. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mutations | Sence Primer Sequences | Antisense Primer Sequences |
|---|---|---|
| T27A | 5′-agccccgcccgccccaggccgtt-3′ | 5′-aacggcctggggcgggcggggct-3′ |
| S31A | 5′-cccaggccgtgcctccccagc-3′ | 5′-gctggggaggcacggcctggg-3′ |
| S32A | 5′-caggccgttccgccccagctgtg-3′ | 5′-cacagctggggcggaacggcctg-3′ |
| S32D | 5′-caggccgttccgacccagctgtg-3′ | 5′-cacagctgggtcggaaccgcctg-3′ |
| S31A/S32A | 5′-cccaggccgtgccgccccagctgtg-3′ | 5′-cacagctggggcggcacggcctggg-3′ |
| S72A/T76A/Y77A | 5′-caccacacatggctgcagatggcgccgccatgcctccg-3′ | 5′-cggaggcatggcggcgccatctcagccatgtgtggtg-3′ |
| H89A | 5′-ctccaggccccgccccacccatg-3′ | 5′-catgggtggggcggggcctggag-3′ |
| Y94A/Y95A | 5′-cacccatgggcgccgcccccccaggg-3′ | 5′-ccctgggggggcggcgcccatgggtg-3′ |
| Y100A/Y105A | 5′-ccccagggcccgccacgccagggcccgcccctggccctg-3′ | 5′-cagggccaggggcgggccctggcgtggcgggccctgggg-3′ |
| H111A | 5′-cctgggggcgccacagccacag-3′ | 5′-ctgtggctgtggcgcccccagg-3′ |
| T112A/T114A | 5′-ctgggggccacgcagccgcagtcctggtc-3′ | 5′-gaccaggactgcggctgcgtggcccccag-3′ |
| S119A | 5′-gtcctggtccctgcaggagctgccac-3′ | 5′-gtggcagctcctgcagggaccaggac-3′ |
| T123A/T124A/T126A | 5′-caggagctgccgccgcagtggcagtgctgcag-3′ | 5′-ctgcagcactgccactgcggcggcaggtcctg-3′ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Inukai, R.; Mori, K.; Maki, M.; Takahara, T.; Shibata, H. Cytoprotective Role of Autophagy in CDIP1 Expression-Induced Apoptosis in MCF-7 Breast Cancer Cells. Int. J. Mol. Sci. 2024, 25, 6520. https://doi.org/10.3390/ijms25126520
Inukai R, Mori K, Maki M, Takahara T, Shibata H. Cytoprotective Role of Autophagy in CDIP1 Expression-Induced Apoptosis in MCF-7 Breast Cancer Cells. International Journal of Molecular Sciences. 2024; 25(12):6520. https://doi.org/10.3390/ijms25126520
Chicago/Turabian StyleInukai, Ryuta, Kanako Mori, Masatoshi Maki, Terunao Takahara, and Hideki Shibata. 2024. "Cytoprotective Role of Autophagy in CDIP1 Expression-Induced Apoptosis in MCF-7 Breast Cancer Cells" International Journal of Molecular Sciences 25, no. 12: 6520. https://doi.org/10.3390/ijms25126520