
Inhibition of Protein Synthesis Attenuates Formation of Traumatic Memory and Normalizes Fear-Induced c-Fos Expression in a Mouse Model of Posttraumatic Stress Disorder

 ,
, {kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
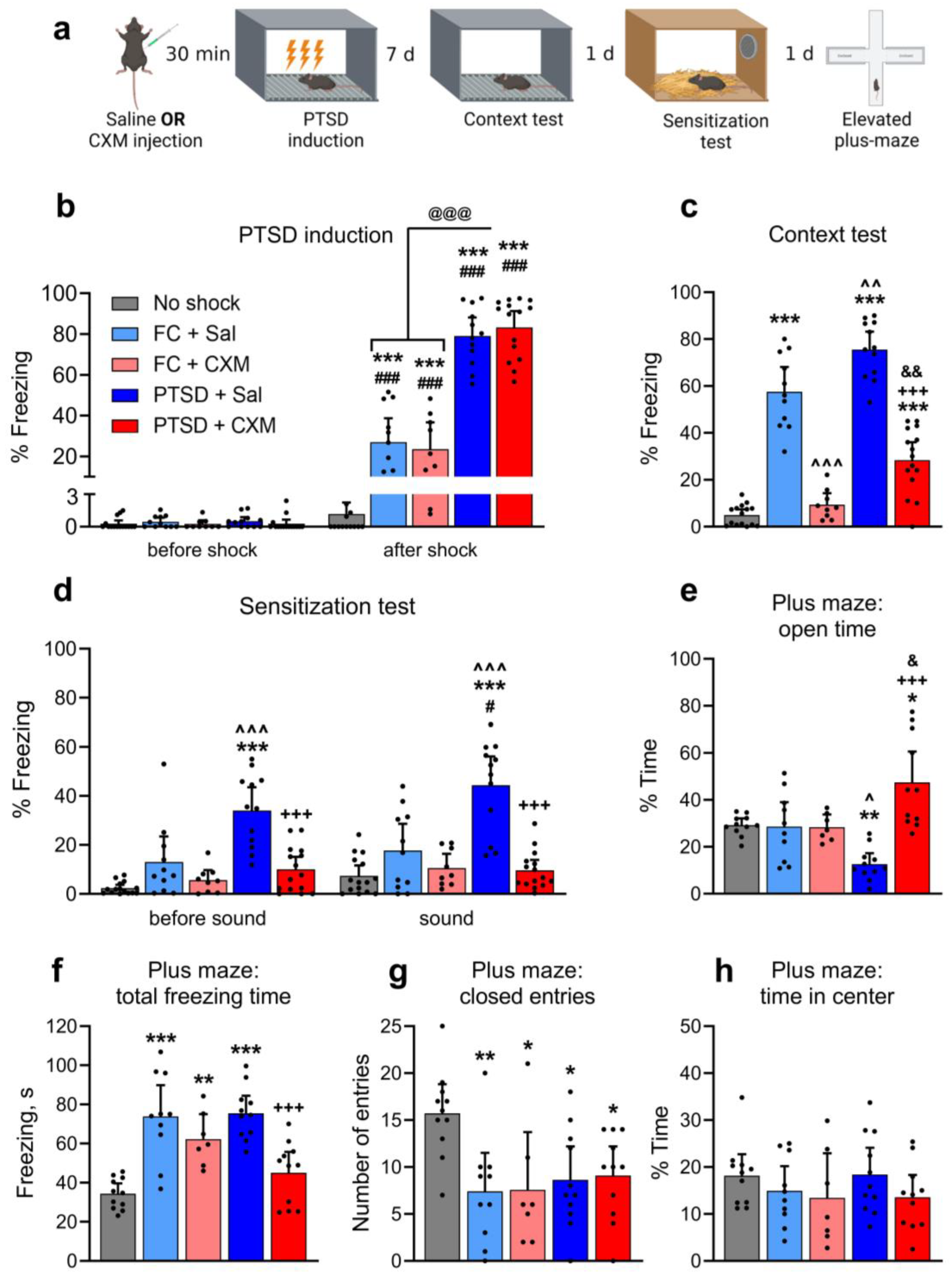
2.1. Effects of Protein Synthesis Inhibition on PTSD-Induced Fear and Anxiety
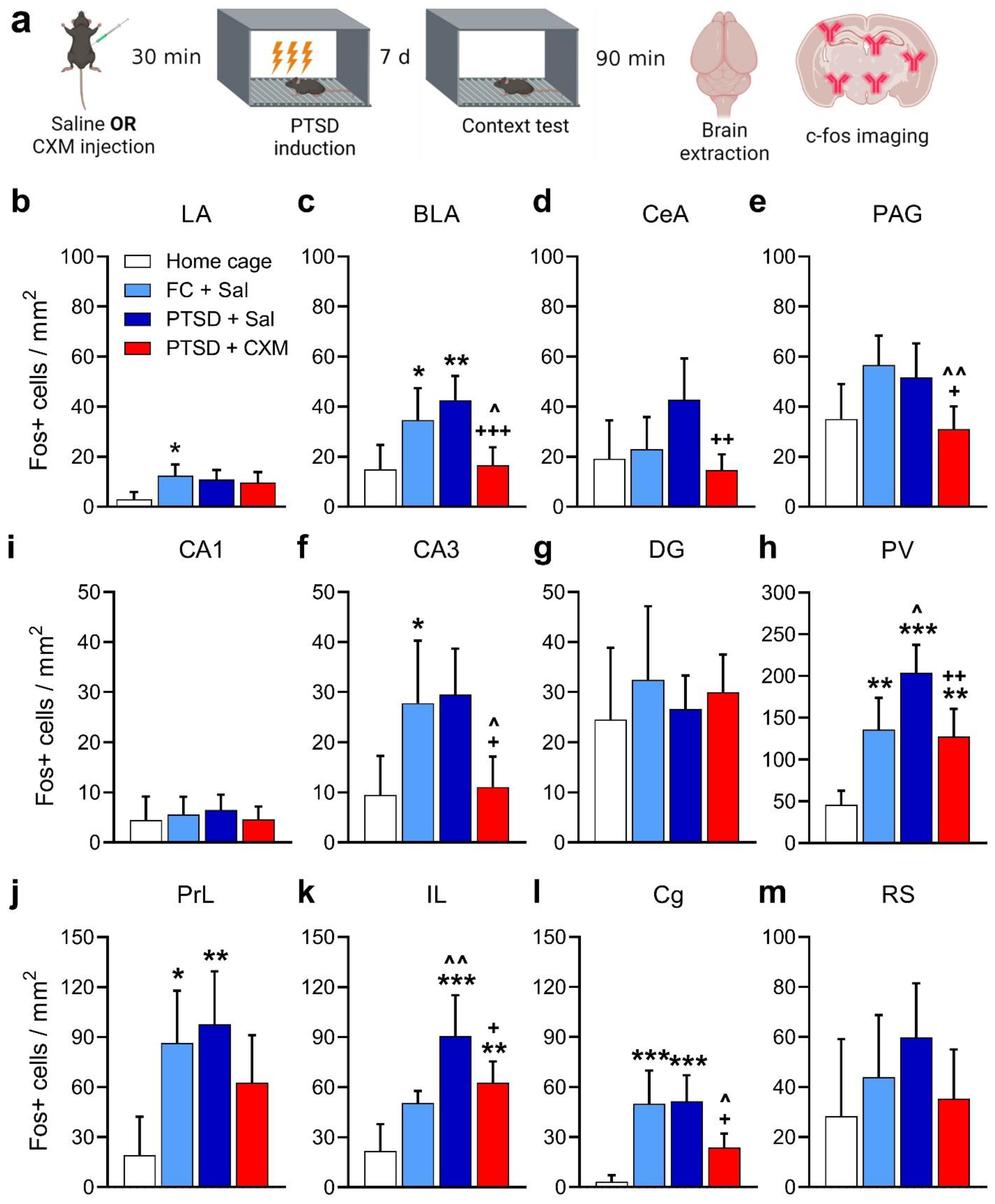
2.2. Patterns of c-Fos Brain Activity following Fear and Traumatic Memory Retrieval and the Effects of Protein Synthesis Inhibition
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Behavior
4.3. Cycloheximide Injection
4.4. Immunohistochemistry
4.5. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Elzinga, B.M.; Bremner, J.D. Are the Neural Substrates of Memory the Final Common Pathway in Posttraumatic Stress Disorder (PTSD)? J. Affect. Disord. 2002, 70, 1–17. [Google Scholar] [CrossRef]
- Cohen, H.; Kozlovsky, N.; Matar, M.A.; Kaplan, Z.; Zohar, J. Mapping the Brain Pathways of Traumatic Memory: Inactivation of Protein Kinase M Zeta in Different Brain Regions Disrupts Traumatic Memory Processes and Attenuates Traumatic Stress Responses in Rats. Eur. Neuropsychopharmacol. 2010, 20, 253–271. [Google Scholar] [CrossRef]
- Johnson, L.R.; McGuire, J.; Lazarus, R.; Palmer, A.A. Pavlovian Fear Memory Circuits and Phenotype Models of PTSD. Neuropharmacology 2012, 62, 638–646. [Google Scholar] [CrossRef]
- Van der Kolk, B.A. The Body Keeps the Score: Memory and the Evolving Psychobiology of Posttraumatic Stress. Harv. Rev. Psychiatry 1994, 1, 253–265. [Google Scholar] [CrossRef]
- Lis, S.; Thome, J.; Kleindienst, N.; Mueller-Engelmann, M.; Steil, R.; Priebe, K.; Schmahl, C.; Hermans, D.; Bohus, M. Generalization of Fear in Post-Traumatic Stress Disorder. Psychophysiology 2020, 57, e13422. [Google Scholar] [CrossRef]
- Thakur, A.; Choudhary, D.; Kumar, B.; Chaudhary, A. A Review on Post-Traumatic Stress Disorder (PTSD): Symptoms, Therapies and Recent Case Studies. Curr. Mol. Pharmacol. 2022, 15, 502–516. [Google Scholar] [CrossRef]
- Ehlers, A.; Hackmann, A.; Michael, T. Intrusive Re-experiencing in Post-traumatic Stress Disorder: Phenomenology, Theory, and Therapy. Memory 2004, 12, 403–415. [Google Scholar] [CrossRef]
- Rothbaum, B.O.; Davis, M. Applying Learning Principles to the Treatment of Post-trauma Reactions. Ann. N. Y. Acad. Sci. 2003, 1008, 112–121. [Google Scholar] [CrossRef]
- Germain, A.; Buysse, D.J.; Nofzinger, E. Sleep-Specific Mechanisms Underlying Posttraumatic Stress Disorder: Integrative Review and Neurobiological Hypotheses. Sleep Med. Rev. 2008, 12, 185–195. [Google Scholar] [CrossRef]
- Milad, M.R.; Orr, S.P.; Lasko, N.B.; Chang, Y.; Rauch, S.L.; Pitman, R.K. Presence and Acquired Origin of Reduced Recall for Fear Extinction in PTSD: Results of a Twin Study. J. Psychiatr. Res. 2008, 42, 515–520. [Google Scholar] [CrossRef]
- Quirk, G.J.; Garcia, R.; González-Lima, F. Prefrontal Mechanisms in Extinction of Conditioned Fear. Biol. Psychiatry 2006, 60, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Rauch, S.L.; Whalen, P.J.; Shin, L.M.; McInerney, S.C.; Macklin, M.L.; Lasko, N.B.; Orr, S.P.; Pitman, R.K. Exaggerated Amygdala Response to Masked Facial Stimuli in Posttraumatic Stress Disorder: A Functional MRI Study. Biol. Psychiatry 2000, 47, 769–776. [Google Scholar] [CrossRef] [PubMed]
- Bryant, R.A.; Kemp, A.H.; Felmingham, K.L.; Liddell, B.; Olivieri, G.; Peduto, A.; Gordon, E.; Williams, L.M. Enhanced Amygdala and Medial Prefrontal Activation during Nonconscious Processing of Fear in Posttraumatic Stress Disorder: An FMRI Study. Hum. Brain Mapp. 2008, 29, 517–523. [Google Scholar] [CrossRef] [PubMed]
- Carrion, V.G.; Haas, B.W.; Garrett, A.; Song, S.; Reiss, A.L. Reduced Hippocampal Activity in Youth with Posttraumatic Stress Symptoms: An FMRI Study. J. Pediatr. Psychol. 2010, 35, 559–569. [Google Scholar] [CrossRef] [PubMed]
- Werner, N.S.; Meindl, T.; Engel, R.R.; Rosner, R.; Riedel, M.; Reiser, M.; Fast, K. Hippocampal Function during Associative Learning in Patients with Posttraumatic Stress Disorder. J. Psychiatr. Res. 2009, 43, 309–318. [Google Scholar] [CrossRef] [PubMed]
- Shin, L.M.; Whalen, P.J.; Pitman, R.K.; Bush, G.; Macklin, M.L.; Lasko, N.B.; Orr, S.P.; McInerney, S.C.; Rauch, S.L. An FMRI Study of Anterior Cingulate Function in Posttraumatic Stress Disorder. Biol. Psychiatry 2001, 50, 932–942. [Google Scholar] [CrossRef]
- Rougemont-Bücking, A.; Linnman, C.; Zeffiro, T.A.; Zeidan, M.A.; Lebron-Milad, K.; Rodriguez-Romaguera, J.; Rauch, S.L.; Pitman, R.K.; Milad, M.R. Altered Processing of Contextual Information during Fear Extinction in PTSD: An FMRI Study. CNS Neurosci. Ther. 2011, 17, 227–236. [Google Scholar] [CrossRef] [PubMed]
- Dickie, E.W.; Brunet, A.; Akerib, V.; Armony, J.L. An FMRI Investigation of Memory Encoding in PTSD: Influence of Symptom Severity. Neuropsychologia 2008, 46, 1522–1531. [Google Scholar] [CrossRef] [PubMed]
- Siegmund, A.; Wotjak, C.T. Toward an Animal Model of Posttraumatic Stress Disorder. Ann. N. Y. Acad. Sci. 2006, 1071, 324–334. [Google Scholar] [CrossRef]
- Siegmund, A.; Wotjak, C.T. A Mouse Model of Posttraumatic Stress Disorder That Distinguishes between Conditioned and Sensitised Fear. J. Psychiatr. Res. 2007, 41, 848–860. [Google Scholar] [CrossRef]
- Siegmund, A.; Wotjak, C.T. Hyperarousal Does not Depend on Trauma-Related Contextual Memory in an Animal Model of Posttraumatic Stress Disorder. Physiol. Behav. 2007, 90, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Anokhin, K.V.; Sudakov, K. V Genome of Brain Neurons in Organization of Systemic Mechanisms of Behavior. Bull. Exp. Biol. Med. 2003, 135, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Miyashita, T.; Kikuchi, E.; Horiuchi, J.; Saitoe, M. Long-Term Memory Engram Cells Are Established by c-Fos/CREB Transcriptional Cycling. Cell Rep. 2018, 25, 2716–2728. [Google Scholar] [CrossRef] [PubMed]
- Adamec, R.; Strasser, K.; Blundell, J.; Burton, P.; McKay, D.W. Protein Synthesis and the Mechanisms of Lasting Change in Anxiety Induced by Severe Stress. Behav. Brain Res. 2006, 167, 270–286. [Google Scholar] [CrossRef] [PubMed]
- Cohen, H.; Kaplan, Z.; Matar, M.A.; Loewenthal, U.; Kozlovsky, N.; Zohar, J. Anisomycin, a Protein Synthesis Inhibitor, Disrupts Traumatic Memory Consolidation and Attenuates Posttraumatic Stress Response in Rats. Biol. Psychiatry 2006, 60, 767–776. [Google Scholar] [CrossRef] [PubMed]
- Kozlovsky, N.; Kaplan, Z.; Zohar, J.; Matar, M.A.; Shimon, H.; Cohen, H. Protein Synthesis Inhibition before or after Stress Exposure Results in Divergent Endocrine and BDNF Responses Disassociated from Behavioral Responses. Depress. Anxiety 2008, 25, E24–E34. [Google Scholar] [CrossRef]
- Johansen, J.P.; Cain, C.K.; Ostroff, L.E.; LeDoux, J.E. Molecular Mechanisms of Fear Learning and Memory. Cell 2011, 147, 509–524. [Google Scholar] [CrossRef]
- Schafe, G.E.; Nader, K.; Blair, H.T.; LeDoux, J.E. Memory Consolidation of Pavlovian Fear Conditioning: A Cellular and Molecular Perspective. Trends Neurosci. 2001, 24, 540–546. [Google Scholar] [CrossRef]
- Toropova, K.A.; Anokhin, K.V. Modeling of Post-Traumatic Stress Disorder in Mice: Nonlinear Relationship with the Strength of the Traumatic Event. Neurosci. Behav. Physiol. 2019, 49, 875–886. [Google Scholar] [CrossRef]
- Sharma, A.V.; Nargang, F.E.; Dickson, C.T. Neurosilence: Profound Suppression of Neural Activity Following Intracerebral Administration of the Protein Synthesis Inhibitor Anisomycin. J. Neurosci. 2012, 32, 2377–2387. [Google Scholar] [CrossRef]
- Squire, L.R.; Smith, G.A.; Barondes, S.H. Cycloheximide Affects Memory within Minutes after the Onset of Training. Nature 1973, 242, 201–202. [Google Scholar] [CrossRef] [PubMed]
- Gutwein, B.M.; Quartermain, D.; McEwen, B.S. Dissociation of Cycloheximide’s Effects on Activity from Its Effects on Memory. Pharmacol. Biochem. Behav. 1974, 2, 753–756. [Google Scholar] [CrossRef] [PubMed]
- Kozlovsky, N.; Matar, M.A.; Kaplan, Z.; Kotler, M.; Zohar, J.; Cohen, H. The Immediate Early Gene Arc Is Associated with Behavioral Resilience to Stress Exposure in an Animal Model of Posttraumatic Stress Disorder. Eur. Neuropsychopharmacol. 2008, 18, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Gold, P.E. Protein Synthesis Inhibition and Memory: Formation vs Amnesia. Neurobiol. Learn. Mem. 2008, 89, 201–211. [Google Scholar] [CrossRef]
- Frankland, P.W.; Bontempi, B. The Organization of Recent and Remote Memories. Nat. Rev. Neurosci. 2005, 6, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Diekelmann, S.; Born, J. One Memory, Two Ways to Consolidate? Nat. Neurosci. 2007, 10, 1085–1086. [Google Scholar] [CrossRef] [PubMed]
- Born, J.; Wilhelm, I. System Consolidation of Memory during Sleep. Psychol. Res. 2012, 76, 192–203. [Google Scholar] [CrossRef]
- Takehara-Nishiuchi, K. Neurobiology of Systems Memory Consolidation. Eur. J. Neurosci. 2021, 54, 6850–6863. [Google Scholar] [CrossRef] [PubMed]
- Pynoos, R.S.; Ritzmann, R.F.; Steinberg, A.M.; Goenjian, A.; Prisecaru, I. A Behavioral Animal Model of Posttraumatic Stress Disorder Featuring Repeated Exposure to Situational Reminders. Biol. Psychiatry 1996, 39, 129–134. [Google Scholar] [CrossRef]
- Hymel, K.A.; Eans, S.O.; Sitchenko, K.L.; Gomes, S.M.; Lukowsky, A.L.; Medina, J.M.; Sypek, E.I.; Carey, A.N.; McLaughlin, J.P. Stress-Induced Increases in Depression-like and Cocaine Place-Conditioned Behaviors Are Reversed by Disruption of Memories during Reconsolidation. Behav. Pharmacol. 2014, 25, 599–608. [Google Scholar] [CrossRef]
- Debiec, J.; LeDoux, J.E. Contribution of Noradrenergic Transmission to Memory Reconsolidation in Animals and Humans: Implications for PTSD. In Biological Psychiatry; Dennis, S., Charney, M.D., Eds.; Elsevier: New York, NY, USA, 2006; Volume 59, pp. 14s–15s. [Google Scholar]
- Dunsmoor, J.E.; Cisler, J.M.; Fonzo, G.A.; Creech, S.K.; Nemeroff, C.B. Laboratory Models of Post-Traumatic Stress Disorder: The Elusive Bridge to Translation. Neuron 2022, 110, 1754–1776. [Google Scholar] [CrossRef]
- Nadel, L.; Hupbach, A.; Gomez, R.; Newman-Smith, K. Memory Formation, Consolidation and Transformation. Neurosci. Biobehav. Rev. 2012, 36, 1640–1645. [Google Scholar] [CrossRef] [PubMed]
- Nordman, J.; Ma, X.; Li, Z. Traumatic Stress Induces Prolonged Aggression Increase through Synaptic Potentiation in the Medial Amygdala Circuits. eNeuro 2020, 7, 1–14. [Google Scholar] [CrossRef]
- Adamec, R.; Kent, P.; Anisman, H.; Shallow, T.; Merali, Z. Neural Plasticity, Neuropeptides and Anxiety in Animals—Implications for Understanding and Treating Affective Disorder Following Traumatic Stress in Humans. Neurosci. Biobehav. Rev. 1998, 23, 301–318. [Google Scholar] [CrossRef] [PubMed]
- Dahlhoff, M.; Siegmund, A.; Golub, Y.; Wolf, E.; Holsboer, F.; Wotjak, C.T. AKT/GSK-3β/β-Catenin Signalling within Hippocampus and Amygdala Reflects Genetically Determined Differences in Posttraumatic Stress Disorder like Symptoms. Neuroscience 2010, 169, 1216–1226. [Google Scholar] [CrossRef]
- Azevedo, M.; Martinho, R.; Oliveira, A.; Correia-de-Sá, P.; Moreira-Rodrigues, M. Molecular Pathways Underlying Sympathetic Autonomic Overshooting Leading to Fear and Traumatic Memories: Looking for Alternative Therapeutic Options for Post-Traumatic Stress Disorder. Front. Mol. Neurosci. 2024, 16, 1332348. [Google Scholar] [CrossRef]
- Adamec, R.E.; Blundell, J.; Burton, P. Phosphorylated Cyclic AMP Response Element Binding Protein Expression Induced in the Periaqueductal Gray by Predator Stress: Its Relationship to the Stress Experience, Behavior and Limbic Neural Plasticity. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2003, 27, 1243–1267. [Google Scholar] [CrossRef]
- Blundell, J.; Adamec, R. Elevated PCREB in the PAG after Exposure to the Elevated plus Maze in Rats Previously Exposed to a Cat. Behav. Brain Res. 2006, 175, 285–295. [Google Scholar] [CrossRef]
- Chang, S.-H.; Chang, Y.-M.; Chen, H.-Y.; Shaw, F.-Z.; Shyu, B.-C. Time-Course Analysis of Frontal Gene Expression Profiles in the Rat Model of Posttraumatic Stress Disorder and a Comparison with the Conditioned Fear Model. Neurobiol. Stress 2023, 27, 100569. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhou, R.; Xing, G.; Hough, C.J.; Li, X.; Li, H. Identification of Gene Markers Based on Well Validated and Subcategorized Stressed Animals for Potential Clinical Applications in PTSD. Med. Hypotheses 2006, 66, 309–314. [Google Scholar] [CrossRef]
- Girgenti, M.J.; Hare, B.D.; Ghosal, S.; Duman, R.S. Molecular and Cellular Effects of Traumatic Stress: Implications for PTSD. Curr. Psychiatry Rep. 2017, 19, 85. [Google Scholar] [CrossRef]
- Fenster, R.J.; Lebois, L.A.M.; Ressler, K.J.; Suh, J. Brain Circuit Dysfunction in Post-Traumatic Stress Disorder: From Mouse to Man. Nat. Rev. Neurosci. 2018, 19, 535–551. [Google Scholar] [CrossRef] [PubMed]
- Kunimatsu, A.; Yasaka, K.; Akai, H.; Kunimatsu, N.; Abe, O. MRI Findings in Posttraumatic Stress Disorder. J. Magn. Reson. Imaging 2020, 52, 380–396. [Google Scholar] [CrossRef] [PubMed]
- Zilcha-Mano, S.; Zhu, X.; Lazarov, A.; Suarez-Jimenez, B.; Helpman, L.; Kim, Y.; Maitlin, C.; Neria, Y.; Rutherford, B.R. Structural Brain Features Signaling Trauma, PTSD, or Resilience? A Systematic Exploration. Depress. Anxiety 2022, 39, 695–705. [Google Scholar] [CrossRef] [PubMed]
- Leite, L.; Esper, N.B.; Junior, J.R.M.L.; Lara, D.R.; Buchweitz, A. An Exploratory Study of Resting-State Functional Connectivity of Amygdala Subregions in Posttraumatic Stress Disorder Following Trauma in Adulthood. Sci. Rep. 2022, 12, 9558. [Google Scholar] [CrossRef] [PubMed]
- Morey, R.A.; Clarke, E.K.; Haswell, C.C.; Phillips, R.D.; Clausen, A.N.; Mufford, M.S.; Saygin, Z.; Wagner, H.R.; LaBar, K.S. Amygdala Nuclei Volume and Shape in Military Veterans with Posttraumatic Stress Disorder. Biol. Psychiatry. Cogn. Neurosci. Neuroimaging 2020, 5, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Kredlow, A.M.; Fenster, R.J.; Laurent, E.S.; Ressler, K.J.; Phelps, E.A. Prefrontal Cortex, Amygdala, and Threat Processing: Implications for PTSD. Neuropsychopharmacology 2022, 47, 247–259. [Google Scholar] [CrossRef] [PubMed]
- Del Casale, A.; Ferracuti, S.; Barbetti, A.S.; Bargagna, P.; Zega, P.; Iannuccelli, A.; Caggese, F.; Zoppi, T.; De Luca, G.P.; Parmigiani, G. Grey Matter Volume Reductions of the Left Hippocampus and Amygdala in PTSD: A Coordinate-Based Meta-Analysis of Magnetic Resonance Imaging Studies. Neuropsychobiology 2022, 81, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Sharp, B.M. Basolateral Amygdala and Stress-Induced Hyperexcitability Affect Motivated Behaviors and Addiction. Transl. Psychiatry 2017, 7, e1194. [Google Scholar] [CrossRef]
- Adamec, R.E.; Burton, P.; Shallow, T.; Budgell, J. NMDA Receptors Mediate Lasting Increases in Anxiety-Like Behavior Produced by the Stress of Predator Exposure—Implications for Anxiety Associated with Posttraumatic Stress Disorder. Physiol. Behav. 1998, 65, 723–737. [Google Scholar] [CrossRef]
- Pitman, R.K.; Rasmusson, A.M.; Koenen, K.C.; Shin, L.M.; Orr, S.P.; Gilbertson, M.W.; Milad, M.R.; Liberzon, I. Biological Studies of Post-Traumatic Stress Disorder. Nat. Rev. Neurosci. 2012, 13, 769–787. [Google Scholar] [CrossRef] [PubMed]
- Ehring, T.; Quack, D. Emotion Regulation Difficulties in Trauma Survivors: The Role of Trauma Type and PTSD Symptom Severity. Behav. Ther. 2010, 41, 587–598. [Google Scholar] [CrossRef] [PubMed]
- Felmingham, K.L.; Rennie, C.; Manor, B.; Bryant, R.A. Eye Tracking and Physiological Reactivity to Threatening Stimuli in Posttraumatic Stress Disorder. J. Anxiety Disord. 2011, 25, 668–673. [Google Scholar] [CrossRef] [PubMed]
- Bian, X.-L.; Qin, C.; Cai, C.-Y.; Zhou, Y.; Tao, Y.; Lin, Y.-H.; Wu, H.-Y.; Chang, L.; Luo, C.-X.; Zhu, D.-Y. Anterior Cingulate Cortex to Ventral Hippocampus Circuit Mediates Contextual Fear Generalization. J. Neurosci. 2019, 39, 5728–5739. [Google Scholar] [CrossRef] [PubMed]
- Milad, M.R.; Pitman, R.K.; Ellis, C.B.; Gold, A.L.; Shin, L.M.; Lasko, N.B.; Zeidan, M.A.; Handwerger, K.; Orr, S.P.; Rauch, S.L. Neurobiological Basis of Failure to Recall Extinction Memory in Posttraumatic Stress Disorder. Biol. Psychiatry 2009, 66, 1075–1082. [Google Scholar] [CrossRef] [PubMed]
- Uniyal, A.; Singh, R.; Akhtar, A.; Dhaliwal, J.; Kuhad, A.; Sah, S.P. Pharmacological Rewriting of Fear Memories: A Beacon for Post-Traumatic Stress Disorder. Eur. J. Pharmacol. 2020, 870, 172824. [Google Scholar] [CrossRef] [PubMed]
- Bukalo, O.; Nonaka, M.; Weinholtz, C.A.; Mendez, A.; Taylor, W.W.; Holmes, A. Effects of Optogenetic Photoexcitation of Infralimbic Cortex Inputs to the Basolateral Amygdala on Conditioned Fear and Extinction. Behav. Brain Res. 2021, 396, 112913. [Google Scholar] [CrossRef] [PubMed]
- Do-Monte, F.H.; Manzano-Nieves, G.; Quiñones-Laracuente, K.; Ramos-Medina, L.; Quirk, G.J. Revisiting the Role of Infralimbic Cortex in Fear Extinction with Optogenetics. J. Neurosci. 2015, 35, 3607–3615. [Google Scholar] [CrossRef] [PubMed]
- Bloodgood, D.W.; Sugam, J.A.; Holmes, A.; Kash, T.L. Fear Extinction Requires Infralimbic Cortex Projections to the Basolateral Amygdala. Transl. Psychiatry 2018, 8, 60. [Google Scholar] [CrossRef]
- Qin, C.; Bian, X.-L.; Wu, H.-Y.; Xian, J.-Y.; Cai, C.-Y.; Lin, Y.-H.; Zhou, Y.; Kou, X.-L.; Chang, L.; Luo, C.-X. Dorsal Hippocampus to Infralimbic Cortex Circuit Is Essential for the Recall of Extinction Memory. Cereb. Cortex 2021, 31, 1707–1718. [Google Scholar] [CrossRef]
- Kirouac, G.J. The Paraventricular Nucleus of the Thalamus as an Integrating and Relay Node in the Brain Anxiety Network. Front. Behav. Neurosci. 2021, 15, 627633. [Google Scholar] [CrossRef] [PubMed]
- Penzo, M.A.; Gao, C. The Paraventricular Nucleus of the Thalamus: An Integrative Node Underlying Homeostatic Behavior. Trends Neurosci. 2021, 44, 538–549. [Google Scholar] [CrossRef] [PubMed]
- Dirven, B.C.J.; Botan, A.; van der Geugten, D.; Kraakman, B.; van Melis, L.; Merjenburgh, S.; van Rijn, R.; Waajen, L.; Homberg, J.R.; Kozicz, T. Longitudinal Assessment of Amygdala Activity in Mice Susceptible to Trauma. Psychoneuroendocrinology 2022, 145, 105912. [Google Scholar] [CrossRef] [PubMed]
- Shin, L.M.; Orr, S.P.; Carson, M.A.; Rauch, S.L.; Macklin, M.L.; Lasko, N.B.; Peters, P.M.; Metzger, L.J.; Dougherty, D.D.; Cannistraro, P.A. Regional Cerebral Blood Flow in the Amygdala and Medial Prefrontalcortex during Traumatic Imagery in Male and Female Vietnam Veterans with Ptsd. Arch. Gen. Psychiatry 2004, 61, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Kalin, N.H.; Shelton, S.E.; Davidson, R.J. The Role of the Central Nucleus of the Amygdala in Mediating Fear and Anxiety in the Primate. J. Neurosci. 2004, 24, 5506–5515. [Google Scholar] [CrossRef] [PubMed]
- Whittle, N.; Fadok, J.; MacPherson, K.P.; Nguyen, R.; Botta, P.; Wolff, S.B.E.; Müller, C.; Herry, C.; Tovote, P.; Holmes, A.; et al. Central Amygdala Micro-Circuits Mediate Fear Extinction. Nat. Commun. 2021, 12, 4156. [Google Scholar] [CrossRef] [PubMed]
- Aspesi, D.; Pinna, G. Animal Models of Post-Traumatic Stress Disorder and Novel Treatment Targets. Behav. Pharmacol. 2019, 30, 130–150. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, J.; Huang, G.-D.; Xue, Y.-X.; Jia, X.-J. The neural circuits and molecular mechanisms underlying fear dysregulation in posttraumatic stress disorder. Front. Neurosci. 2023, 17, 1281401. [Google Scholar] [CrossRef] [PubMed]
- Cohen, H.; Zohar, J.; Matar, M.A.; Zeev, K.; Loewenthal, U.; Richter-Levin, G. Setting Apart the Affected: The Use of Behavioral Criteria in Animal Models of Post Traumatic Stress Disorder. Neuropsychopharmacology 2004, 29, 1962–1970. [Google Scholar] [CrossRef]
- Shinba, T.; Shinozaki, T.; Mugishima, G. Clonidine Immediately after Immobilization Stress Prevents Long-Lasting Locomotion Reduction in the Rat. Prog. Neuropsychopharmacol. Biol. Psychiatry 2001, 25, 1629–1640. [Google Scholar] [CrossRef]
- Cohen, H.; Zohar, J. An Animal Model of Posttraumatic Stress Disorder: The Use of Cut-Off Behavioral Criteria. Ann. N. Y. Acad. Sci. 2004, 1032, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Lister, R.G. The Use of a Plus-Maze to Measure Anxiety in the Mouse. Psychopharmacology 1987, 92, 180–185. [Google Scholar] [CrossRef] [PubMed]
- Rodgers, R.J.; Johnson, N.J.T. Factor Analysis of Spatiotemporal and Ethological Measures in the Murine Elevated Plus-Maze Test of Anxiety. Pharmacol. Biochem. Behav. 1995, 52, 297–303. [Google Scholar] [CrossRef] [PubMed]
- Walf, A.A.; Frye, C.A. The Use of the Elevated plus Maze as an Assay of Anxiety-Related Behavior in Rodents. Nat. Protoc. 2007, 2, 322–328. [Google Scholar] [CrossRef] [PubMed]
- Rodgers, R.J.; Lee, C.; Shepherd, J.K. Effects of Diazepam on Behavioural and Antinociceptive Responses to the Elevated Plus-Maze in Male Mice Depend upon Treatment Regimen and Prior Maze Experience. Psychopharmacology 1992, 106, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Trullas, R.; Skolnick, P. Differences in Fear Motivated Behaviors among Inbred Mouse Strains. Psychopharmacology 1993, 111, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Cruz, A.P.d.M.; Frei, F.; Graeff, F.G. Ethopharmacological Analysis of Rat Behavior on the Elevated Plus-Maze. Pharmacol. Biochem. Behav. 1994, 49, 171–176. [Google Scholar] [CrossRef]
- Randt, C.T.; Barnett, B.M.; McEwen, B.S.; Quartermain, D. Amnesic Effects of Cycloheximide on Two Strains of Mice with Different Memory Characteristics. Exp. Neurol. 1971, 30, 467–474. [Google Scholar] [CrossRef] [PubMed]
- Gotthard, G.H.; Bashford, A.R.; Bsales, D.A.; Golbitz, J.-A.; Shear, R. Effects of Cycloheximide on Recent and Remote Appetitive Odor Discrimination Memory in Rats. Learn. Motiv. 2020, 72, 101670. [Google Scholar] [CrossRef]
- Vogt, B.A.; Paxinos, G. Cytoarchitecture of Mouse and Rat Cingulate Cortex with Human Homologies. Brain Struct. Funct. 2014, 219, 185–192. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zamorina, T.A.; Ivashkina, O.I.; Toropova, K.A.; Anokhin, K.V. Inhibition of Protein Synthesis Attenuates Formation of Traumatic Memory and Normalizes Fear-Induced c-Fos Expression in a Mouse Model of Posttraumatic Stress Disorder. Int. J. Mol. Sci. 2024, 25, 6544. https://doi.org/10.3390/ijms25126544
Zamorina TA, Ivashkina OI, Toropova KA, Anokhin KV. Inhibition of Protein Synthesis Attenuates Formation of Traumatic Memory and Normalizes Fear-Induced c-Fos Expression in a Mouse Model of Posttraumatic Stress Disorder. International Journal of Molecular Sciences. 2024; 25(12):6544. https://doi.org/10.3390/ijms25126544
Chicago/Turabian StyleZamorina, Tatyana A., Olga I. Ivashkina, Ksenia A. Toropova, and Konstantin V. Anokhin. 2024. "Inhibition of Protein Synthesis Attenuates Formation of Traumatic Memory and Normalizes Fear-Induced c-Fos Expression in a Mouse Model of Posttraumatic Stress Disorder" International Journal of Molecular Sciences 25, no. 12: 6544. https://doi.org/10.3390/ijms25126544
APA StyleZamorina, T. A., Ivashkina, O. I., Toropova, K. A., & Anokhin, K. V. (2024). Inhibition of Protein Synthesis Attenuates Formation of Traumatic Memory and Normalizes Fear-Induced c-Fos Expression in a Mouse Model of Posttraumatic Stress Disorder. International Journal of Molecular Sciences, 25(12), 6544. https://doi.org/10.3390/ijms25126544