
Ion Channel and Transporter Involvement in Chemotherapy-Induced Peripheral Neurotoxicity

 ,
,  , ,
, ,  and
and 
Abstract
1. Introduction
2. Biophysical Properties of Transporters/Ion Channels of Interest
2.1. Voltage-Gated Na+ Channels (VGSCs)
2.1.1. Topology and Gating
2.1.2. Classification and Isoforms Involved in Neuropathic Pain
- Nav 1.3
- Nav 1.7
- Nav 1.8
- Nav 1.9
2.2. Voltage-Gated K+ Channels (Kv)
2.2.1. Topology and Gating
2.2.2. Channel Kinetics
- Inactivating K+ Channels
- Delayed Rectifier Currents
2.3. Sodium–Calcium Exchanger (NCX) Family
- NCX2
- NCX3
2.4. Voltage-Gated Ca2+ Channels (VGCCs or Cav)
2.4.1. Topology and Subunits
2.4.2. Classification, Physiological and Pharmacological Properties and Isoforms Involved in Neuropathic Pain
- Cav2.2 (N-Type Channel)
- Cav3.2 (T-Type Channel)
2.5. Transient Receptor Potential Family (TRPA1, TRPM8 and TRPV1)
3. Ion Channels/Transporters in Chemotherapy-Induced Peripheral Neurotoxicity Models
3.1. Voltage-Gated Sodium Channels
3.2. Voltage-Gated Potassium Channels
3.3. Sodium–Calcium Exchanger
3.4. Voltage-Gated Ca2+ Channels
3.5. Transient Receptor Potential Family (TRPA1, TRPM8 and TRPV1)
4. Possible Clinical Translation
5. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Park, S.B.; Cetinkaya-Fisgin, A.; Argyriou, A.A.; Höke, A.; Cavaletti, G.; Alberti, P. Axonal degeneration in chemotherapy-induced peripheral neurotoxicity: Clinical and experimental evidence. J. Neurol. Neurosurg. Psychiatry 2023, 94, 962–972. [Google Scholar] [CrossRef] [PubMed]
- Cavaletti, G.; Cornblath, D.R.; Merkies, I.S.J.; Postma, T.J.; Rossi, E.; Alberti, P.; Bruna, J.; Argyriou, A.A.; Briani, C.; Velasco, R.; et al. Patients’ and physicians’ interpretation of chemotherapy-induced peripheral neurotoxicity. J. Peripher. Nerv. Syst. 2019, 24, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Loprinzi, C.L.; Lacchetti, C.; Bleeker, J.; Cavaletti, G.; Chauhan, C.; Hertz, D.L.; Kelley, M.R.; Lavino, A.; Lustberg, M.B.; Paice, J.A.; et al. Prevention and Management of Chemotherapy-Induced Peripheral Neuropathy in Survivors of Adult Cancers: ASCO Guideline Update. J. Clin. Oncol. 2020, 38, 3325–3348. [Google Scholar] [CrossRef]
- Romanova, D.; Balaban, P.; Nikitin, E. Sodium Channels Involved in the Initiation of Action Potentials in Invertebrate and Mammalian Neurons. Biophysica 2022, 2, 184–193. [Google Scholar] [CrossRef]
- Kariev, A.M.; Green, M.E. Water, Protons, and the Gating of Voltage-Gated Potassium Channels. Membranes 2024, 14, 37. [Google Scholar] [CrossRef]
- Giladi, M.; Shor, R.; Lisnyansky, M.; Khananshvili, D. Structure-Functional Basis of Ion Transport in Sodium-Calcium Exchanger (NCX) Proteins. Int. J. Mol. Sci. 2016, 17, 1949. [Google Scholar] [CrossRef] [PubMed]
- Mochida, S. Presynaptic calcium channels. Neurosci. Res. 2018, 127, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Kalinovskii, A.P.; Utkina, L.L.; Korolkova, Y.V.; Andreev, Y.A. TRPV3 Ion Channel: From Gene to Pharmacology. Int. J. Mol. Sci. 2023, 24, 8601. [Google Scholar] [CrossRef]
- Catterall, W.A.; Goldin, A.L.; Waxman, S.G. International Union of Pharmacology. XLVII. Nomenclature and structure-function relationships of voltage-gated sodium channels. Pharmacol. Rev. 2005, 57, 397–409. [Google Scholar] [CrossRef]
- Bennett, D.L.; Woods, C.G. Painful and painless channelopathies. Lancet Neurol. 2014, 13, 587–599. [Google Scholar] [CrossRef] [PubMed]
- Amir, R.; Argoff, C.E.; Bennett, G.J.; Cummins, T.R.; Durieux, M.E.; Gerner, P.; Gold, M.S.; Porreca, F.; Strichartz, G.R. The role of sodium channels in chronic inflammatory and neuropathic pain. J. Pain 2006, 7, S1–S29. [Google Scholar] [CrossRef]
- Devor, M. Sodium channels and mechanisms of neuropathic pain. J. Pain 2006, 7, S3–S12. [Google Scholar] [CrossRef] [PubMed]
- Hargus, N.J.; Patel, M.K. Voltage-gated Na+ channels in neuropathic pain. Expert Opin. Investig. Drugs 2007, 16, 635–646. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Ou, S.W.; Wang, Y.J. Distribution and function of voltage-gated sodium channels in the nervous system. Channels 2017, 11, 534–554. [Google Scholar] [CrossRef] [PubMed]
- Catterall, W.A. Ion channel voltage sensors: Structure, function, and pathophysiology. Neuron 2010, 67, 915–928. [Google Scholar] [CrossRef]
- Ulbricht, W. Sodium channel inactivation: Molecular determinants and modulation. Physiol. Rev. 2005, 85, 1271–1301. [Google Scholar] [CrossRef] [PubMed]
- Ye, W.; Zhao, H.; Dai, Y.; Wang, Y.; Lo, Y.H.; Jan, L.Y.; Lee, C.H. Activation and closed-state inactivation mechanisms of the human voltage-gated K. Mol. Cell 2022, 82, 2427–2442.e4. [Google Scholar] [CrossRef] [PubMed]
- Lewis, A.H.; Raman, I.M. Resurgent current of voltage-gated Na+ channels. J. Physiol. 2014, 592, 4825–4838. [Google Scholar] [CrossRef] [PubMed]
- Catterall, W.A. From ionic currents to molecular mechanisms: The structure and function of voltage-gated sodium channels. Neuron 2000, 26, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Lampert, A.; Eberhardt, M.; Waxman, S.G. Altered sodium channel gating as molecular basis for pain: Contribution of activation, inactivation, and resurgent currents. Handb. Exp. Pharmacol. 2014, 221, 91–110. [Google Scholar] [CrossRef]
- Cantrell, A.R.; Tibbs, V.C.; Yu, F.H.; Murphy, B.J.; Sharp, E.M.; Qu, Y.; Catterall, W.A.; Scheuer, T. Molecular mechanism of convergent regulation of brain Na+ channels by protein kinase C and protein kinase A anchored to AKAP-15. Mol. Cell. Neurosci. 2002, 21, 63–80. [Google Scholar] [CrossRef] [PubMed]
- Scheuer, T. Regulation of sodium channel activity by phosphorylation. Semin. Cell Dev. Biol. 2011, 22, 160–165. [Google Scholar] [CrossRef] [PubMed]
- Naylor, C.E.; Bagnéris, C.; DeCaen, P.G.; Sula, A.; Scaglione, A.; Clapham, D.E.; Wallace, B.A. Molecular basis of ion permeability in a voltage-gated sodium channel. EMBO J. 2016, 35, 820–830. [Google Scholar] [CrossRef] [PubMed]
- Isom, L.L. Sodium channel beta subunits: Anything but auxiliary. Neuroscientist 2001, 7, 42–54. [Google Scholar] [CrossRef] [PubMed]
- Dib-Hajj, S.D.; Cummins, T.R.; Black, J.A.; Waxman, S.G. Sodium channels in normal and pathological pain. Annu. Rev. Neurosci. 2010, 33, 325–347. [Google Scholar] [CrossRef] [PubMed]
- Cummins, T.R.; Aglieco, F.; Renganathan, M.; Herzog, R.I.; Dib-Hajj, S.D.; Waxman, S.G. Nav1.3 sodium channels: Rapid repriming and slow closed-state inactivation display quantitative differences after expression in a mammalian cell line and in spinal sensory neurons. J. Neurosci. 2001, 21, 5952–5961. [Google Scholar] [CrossRef] [PubMed]
- Hains, B.C.; Klein, J.P.; Saab, C.Y.; Craner, M.J.; Black, J.A.; Waxman, S.G. Upregulation of sodium channel Nav1.3 and functional involvement in neuronal hyperexcitability associated with central neuropathic pain after spinal cord injury. J. Neurosci. 2003, 23, 8881–8892. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Yan, G.J.; Tan, Y.X.; Xue, L.L.; Wang, T.H.; Zhao, H.R.; Lu, M.N.; Zhang, H.X.; Mei, R.; Dong, X.H.; et al. Reduced Expression of Voltage-Gated Sodium Channel Beta 2 Restores Neuronal Injury and Improves Cognitive Dysfunction Induced by A. Neural Plast. 2022, 2022, 3995227. [Google Scholar] [CrossRef] [PubMed]
- Hameed, S. Nav1.7 and Nav1.8: Role in the pathophysiology of pain. Mol. Pain 2019, 15, 1744806919858801. [Google Scholar] [CrossRef] [PubMed]
- Cummins, T.R.; Howe, J.R.; Waxman, S.G. Slow closed-state inactivation: A novel mechanism underlying ramp currents in cells expressing the hNE/PN1 sodium channel. J. Neurosci. 1998, 18, 9607–9619. [Google Scholar] [CrossRef] [PubMed]
- Rush, A.M.; Cummins, T.R.; Waxman, S.G. Multiple sodium channels and their roles in electrogenesis within dorsal root ganglion neurons. J. Physiol. 2007, 579, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Theile, J.W.; Jarecki, B.W.; Piekarz, A.D.; Cummins, T.R. Nav1.7 mutations associated with paroxysmal extreme pain disorder, but not erythromelalgia, enhance Navbeta4 peptide-mediated resurgent sodium currents. J. Physiol. 2011, 589, 597–608. [Google Scholar] [CrossRef] [PubMed]
- Minett, M.S.; Nassar, M.A.; Clark, A.K.; Passmore, G.; Dickenson, A.H.; Wang, F.; Malcangio, M.; Wood, J.N. Distinct Nav1.7-dependent pain sensations require different sets of sensory and sympathetic neurons. Nat. Commun. 2012, 3, 791. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Dib-Hajj, S.D.; Tyrrell, L.; Wright, D.A.; Fischer, T.Z.; Waxman, S.G. Mutations at opposite ends of the DIII/S4-S5 linker of sodium channel Na V 1.7 produce distinct pain disorders. Mol. Pain 2010, 6, 24. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Dib-Hajj, S.D.; Tyrrell, L.; Waxman, S.G. Mutation I136V alters electrophysiological properties of the Nav1.7 channel in a family with onset of erythromelalgia in the second decade. Mol. Pain 2008, 4, 1. [Google Scholar] [CrossRef] [PubMed]
- Han, C.; Hoeijmakers, J.G.; Ahn, H.S.; Zhao, P.; Shah, P.; Lauria, G.; Gerrits, M.M.; te Morsche, R.H.; Dib-Hajj, S.D.; Drenth, J.P.; et al. Nav1.7-related small fiber neuropathy: Impaired slow-inactivation and DRG neuron hyperexcitability. Neurology 2012, 78, 1635–1643. [Google Scholar] [CrossRef]
- Faber, C.G.; Hoeijmakers, J.G.; Ahn, H.S.; Cheng, X.; Han, C.; Choi, J.S.; Estacion, M.; Lauria, G.; Vanhoutte, E.K.; Gerrits, M.M.; et al. Gain of function Naν1.7 mutations in idiopathic small fiber neuropathy. Ann. Neurol. 2012, 71, 26–39. [Google Scholar] [CrossRef] [PubMed]
- Dib-Hajj, S.D.; Yang, Y.; Black, J.A.; Waxman, S.G. The NaV1.7 sodium channel: From molecule to man. Nat. Rev. Neurosci. 2013, 14, 49–62. [Google Scholar] [CrossRef] [PubMed]
- Blair, N.T.; Bean, B.P. Roles of tetrodotoxin (TTX)-sensitive Na+ current, TTX-resistant Na+ current, and Ca2+ current in the action potentials of nociceptive sensory neurons. J. Neurosci. 2002, 22, 10277–10290. [Google Scholar] [CrossRef] [PubMed]
- Sleeper, A.A.; Cummins, T.R.; Dib-Hajj, S.D.; Hormuzdiar, W.; Tyrrell, L.; Waxman, S.G.; Black, J.A. Changes in expression of two tetrodotoxin-resistant sodium channels and their currents in dorsal root ganglion neurons after sciatic nerve injury but not rhizotomy. J. Neurosci. 2000, 20, 7279–7289. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Santos, S.; Padilla, K.; Printzenhoff, D.; Castle, N.A. Biophysical and Pharmacological Characterization of Nav1.9 Voltage Dependent Sodium Channels Stably Expressed in HEK-293 Cells. PLoS ONE 2016, 11, e0161450. [Google Scholar] [CrossRef]
- Moldovan, M.; Alvarez, S.; Romer Rosberg, M.; Krarup, C. Axonal voltage-gated ion channels as pharmacological targets for pain. Eur. J. Pharmacol. 2013, 708, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Lai, H.C.; Jan, L.Y. The distribution and targeting of neuronal voltage-gated ion channels. Nat. Rev. Neurosci. 2006, 7, 548–562. [Google Scholar] [CrossRef] [PubMed]
- Gutman, G.A.; Chandy, K.G.; Grissmer, S.; Lazdunski, M.; McKinnon, D.; Pardo, L.A.; Robertson, G.A.; Rudy, B.; Sanguinetti, M.C.; Stühmer, W.; et al. International Union of Pharmacology. LIII. Nomenclature and molecular relationships of voltage-gated potassium channels. Pharmacol. Rev. 2005, 57, 473–508. [Google Scholar] [CrossRef] [PubMed]
- Gutman, G.A.; Chandy, K.G.; Adelman, J.P.; Aiyar, J.; Bayliss, D.A.; Clapham, D.E.; Covarriubias, M.; Desir, G.V.; Furuichi, K.; Ganetzky, B.; et al. International Union of Pharmacology. XLI. Compendium of voltage-gated ion channels: Potassium channels. Pharmacol. Rev. 2003, 55, 583–586. [Google Scholar] [CrossRef]
- Rasband, M.N.; Park, E.W.; Vanderah, T.W.; Lai, J.; Porreca, F.; Trimmer, J.S. Distinct potassium channels on pain-sensing neurons. Proc. Natl. Acad. Sci. USA 2001, 98, 13373–13378. [Google Scholar] [CrossRef] [PubMed]
- Fan, L.; Guan, X.; Wang, W.; Zhao, J.Y.; Zhang, H.; Tiwari, V.; Hoffman, P.N.; Li, M.; Tao, Y.X. Impaired neuropathic pain and preserved acute pain in rats overexpressing voltage-gated potassium channel subunit Kv1.2 in primary afferent neurons. Mol. Pain 2014, 10, 8. [Google Scholar] [CrossRef] [PubMed]
- Sokolova, O.; Accardi, A.; Gutierrez, D.; Lau, A.; Rigney, M.; Grigorieff, N. Conformational changes in the C terminus of Shaker K+ channel bound to the rat Kvbeta2-subunit. Proc. Natl. Acad. Sci. USA 2003, 100, 12607–12612. [Google Scholar] [CrossRef] [PubMed]
- MacKinnon, R. Potassium channels. FEBS Lett. 2003, 555, 62–65. [Google Scholar] [CrossRef] [PubMed]
- Dolly, J.O.; Parcej, D.N. Molecular properties of voltage-gated K+ channels. J. Bioenerg. Biomembr. 1996, 28, 231–253. [Google Scholar] [CrossRef] [PubMed]
- Rasband, M.N.; Trimmer, J.S.; Peles, E.; Levinson, S.R.; Shrager, P. K+ channel distribution and clustering in developing and hypomyelinated axons of the optic nerve. J. Neurocytol. 1999, 28, 319–331. [Google Scholar] [CrossRef] [PubMed]
- Busserolles, J.; Tsantoulas, C.; Eschalier, A.; López García, J.A. Potassium channels in neuropathic pain: Advances, challenges, and emerging ideas. Pain 2016, 157 (Suppl. S1), S7–S14. [Google Scholar] [CrossRef] [PubMed]
- Tsantoulas, C.; Zhu, L.; Shaifta, Y.; Grist, J.; Ward, J.P.; Raouf, R.; Michael, G.J.; McMahon, S.B. Sensory neuron downregulation of the Kv9.1 potassium channel subunit mediates neuropathic pain following nerve injury. J. Neurosci. 2012, 32, 17502–17513. [Google Scholar] [CrossRef] [PubMed]
- Johnston, J.; Forsythe, I.D.; Kopp-Scheinpflug, C. Going native: Voltage-gated potassium channels controlling neuronal excitability. J. Physiol. 2010, 588, 3187–3200. [Google Scholar] [CrossRef] [PubMed]
- Chien, L.Y.; Cheng, J.K.; Chu, D.; Cheng, C.F.; Tsaur, M.L. Reduced expression of A-type potassium channels in primary sensory neurons induces mechanical hypersensitivity. J. Neurosci. 2007, 27, 9855–9865. [Google Scholar] [CrossRef] [PubMed]
- Everill, B.; Rizzo, M.A.; Kocsis, J.D. Morphologically identified cutaneous afferent DRG neurons express three different potassium currents in varying proportions. J. Neurophysiol. 1998, 79, 1814–1824. [Google Scholar] [CrossRef] [PubMed]
- Takeda, M.; Tsuboi, Y.; Kitagawa, J.; Nakagawa, K.; Iwata, K.; Matsumoto, S. Potassium channels as a potential therapeutic target for trigeminal neuropathic and inflammatory pain. Mol. Pain 2011, 7, 5. [Google Scholar] [CrossRef] [PubMed]
- Brew, H.M.; Forsythe, I.D. Two voltage-dependent K+ conductances with complementary functions in postsynaptic integration at a central auditory synapse. J. Neurosci. 1995, 15, 8011–8022. [Google Scholar] [CrossRef] [PubMed]
- Carrasquillo, Y.; Nerbonne, J.M. IA channels: Diverse regulatory mechanisms. Neuroscientist 2014, 20, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Jensen, C.S.; Rasmussen, H.B.; Misonou, H. Neuronal trafficking of voltage-gated potassium channels. Mol. Cell. Neurosci. 2011, 48, 288–297. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Liang, C.W.; Muralidharan, S.; Kao, J.P.; Tang, C.M.; Thompson, S.M. Unique roles of SK and Kv4.2 potassium channels in dendritic integration. Neuron 2004, 44, 351–364. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.H.; Liu, Y.; Hoffman, D.A. Identification of Kv4.2 protein complex and modifications by tandem affinity purification-mass spectrometry in primary neurons. Front. Cell. Neurosci. 2022, 16, 1070305. [Google Scholar] [CrossRef] [PubMed]
- Norris, A.J.; Foeger, N.C.; Nerbonne, J.M. Interdependent roles for accessory KChIP2, KChIP3, and KChIP4 subunits in the generation of Kv4-encoded IA channels in cortical pyramidal neurons. J. Neurosci. 2010, 30, 13644–13655. [Google Scholar] [CrossRef] [PubMed]
- An, W.F.; Bowlby, M.R.; Betty, M.; Cao, J.; Ling, H.P.; Mendoza, G.; Hinson, J.W.; Mattsson, K.I.; Strassle, B.W.; Trimmer, J.S.; et al. Modulation of A-type potassium channels by a family of calcium sensors. Nature 2000, 403, 553–556. [Google Scholar] [CrossRef] [PubMed]
- Yang, E.K.; Takimoto, K.; Hayashi, Y.; de Groat, W.C.; Yoshimura, N. Altered expression of potassium channel subunit mRNA and alpha-dendrotoxin sensitivity of potassium currents in rat dorsal root ganglion neurons after axotomy. Neuroscience 2004, 123, 867–874. [Google Scholar] [CrossRef] [PubMed]
- D’Adamo, M.C.; Liantonio, A.; Rolland, J.F.; Pessia, M.; Imbrici, P. Kv1.1 Channelopathies: Pathophysiological Mechanisms and Therapeutic Approaches. Int. J. Mol. Sci. 2020, 21, 2935. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, J.R.; Glassmeier, G.; Cooper, E.C.; Kao, T.C.; Nodera, H.; Tabuena, D.; Kaji, R.; Bostock, H. KCNQ channels mediate IKs, a slow K+ current regulating excitability in the rat node of Ranvier. J. Physiol. 2006, 573, 17–34. [Google Scholar] [CrossRef] [PubMed]
- Devaux, J.J.; Kleopa, K.A.; Cooper, E.C.; Scherer, S.S. KCNQ2 is a nodal K+ channel. J. Neurosci. 2004, 24, 1236–1244. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Kunkel, D.D.; Schwartzkroin, P.A.; Tempel, B.L. Localization of Kv1.1 and Kv1.2, two K channel proteins, to synaptic terminals, somata, and dendrites in the mouse brain. J. Neurosci. 1994, 14, 4588–4599. [Google Scholar] [CrossRef]
- Rasband, M.N. Clustered K+ channel complexes in axons. Neurosci. Lett. 2010, 486, 101–106. [Google Scholar] [CrossRef] [PubMed]
- Hivert, B.; Marien, L.; Agbam, K.N.; Faivre-Sarrailh, C. ADAM22 and ADAM23 modulate the targeting of the Kv1 channel-associated protein LGI1 to the axon initial segment. J. Cell Sci. 2019, 132, jcs219774. [Google Scholar] [CrossRef]
- Zheng, Y.; Liu, H.; Chen, Y.; Dong, S.; Wang, F.; Wang, S.; Li, G.L.; Shu, Y.; Xu, F. Structural insights into the lipid and ligand regulation of a human neuronal KCNQ channel. Neuron 2022, 110, 237–247.e234. [Google Scholar] [CrossRef] [PubMed]
- Abd-Elsayed, A.; Jackson, M.; Gu, S.L.; Fiala, K.; Gu, J. Neuropathic pain and Kv7 voltage-gated potassium channels: The potential role of Kv7 activators in the treatment of neuropathic pain. Mol. Pain 2019, 15, 1744806919864256. [Google Scholar] [CrossRef] [PubMed]
- Battefeld, A.; Tran, B.T.; Gavrilis, J.; Cooper, E.C.; Kole, M.H. Heteromeric Kv7.2/7.3 channels differentially regulate action potential initiation and conduction in neocortical myelinated axons. J. Neurosci. 2014, 34, 3719–3732. [Google Scholar] [CrossRef] [PubMed]
- Ying, Y.; Gong, L.; Tao, X.; Ding, J.; Chen, N.; Yao, Y.; Liu, J.; Chen, C.; Zhu, T.; Jiang, P. Genetic Knockout of TRPM2 Increases Neuronal Excitability of Hippocampal Neurons by Inhibiting Kv7 Channel in Epilepsy. Mol. Neurobiol. 2022, 59, 6918–6933. [Google Scholar] [CrossRef] [PubMed]
- Cooper, E.C.; Aldape, K.D.; Abosch, A.; Barbaro, N.M.; Berger, M.S.; Peacock, W.S.; Jan, Y.N.; Jan, L.Y. Colocalization and coassembly of two human brain M-type potassium channel subunits that are mutated in epilepsy. Proc. Natl. Acad. Sci. USA 2000, 97, 4914–4919. [Google Scholar] [CrossRef] [PubMed]
- Lytton, J. Na+/Ca2+ exchangers: Three mammalian gene families control Ca2+ transport. Biochem. J. 2007, 406, 365–382. [Google Scholar] [CrossRef] [PubMed]
- Golovina, V.A.; Bambrick, L.L.; Yarowsky, P.J.; Krueger, B.K.; Blaustein, M.P. Modulation of two functionally distinct Ca2+ stores in astrocytes: Role of the plasmalemmal Na/Ca exchanger. Glia 1996, 16, 296–305. [Google Scholar] [CrossRef]
- Khananshvili, D. The SLC8 gene family of sodium-calcium exchangers (NCX)—Structure, function, and regulation in health and disease. Mol. Asp. Med. 2013, 34, 220–235. [Google Scholar] [CrossRef] [PubMed]
- On, C.; Marshall, C.R.; Chen, N.; Moyes, C.D.; Tibbits, G.F. Gene structure evolution of the Na+-Ca2+ exchanger (NCX) family. BMC Evol. Biol. 2008, 8, 127. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Lytton, J. The cation/Ca2+ exchanger superfamily: Phylogenetic analysis and structural implications. Mol. Biol. Evol. 2004, 21, 1692–1703. [Google Scholar] [CrossRef] [PubMed]
- Khananshvili, D. Basic and editing mechanisms underlying ion transport and regulation in NCX variants. Cell Calcium 2020, 85, 102131. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.; O’Halloran, D.M. Recent structural and functional insights into the family of sodium calcium exchangers. Genesis 2014, 52, 93–109. [Google Scholar] [CrossRef] [PubMed]
- Hilge, M.; Aelen, J.; Vuister, G.W. Ca2+ regulation in the Na+/Ca2+ exchanger involves two markedly different Ca2+ sensors. Mol. Cell 2006, 22, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Bode, K.; O’Halloran, D.M. NCX-DB: A unified resource for integrative analysis of the sodium calcium exchanger super-family. BMC Neurosci. 2018, 19, 19. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Wen, L.L.; Xie, J.D.; Ouyang, H.D.; Chen, D.T.; Zeng, W.A. Antinociceptive effectiveness of the inhibition of NCX reverse-mode action in rodent neuropathic pain model. Mol. Pain 2019, 15, 1744806919864511. [Google Scholar] [CrossRef] [PubMed]
- Ballarini, E.; Malacrida, A.; Rodriguez-Menendez, V.; Pozzi, E.; Canta, A.; Chiorazzi, A.; Monza, L.; Semperboni, S.; Meregalli, C.; Carozzi, V.A.; et al. Sodium-Calcium Exchanger 2: A Pivotal Role in Oxaliplatin Induced Peripheral Neurotoxicity and Axonal Damage? Int. J. Mol. Sci. 2022, 23, 10063. [Google Scholar] [CrossRef]
- Stys, P.K.; Waxman, S.G.; Ransom, B.R. Ionic mechanisms of anoxic injury in mammalian CNS white matter: Role of Na+ channels and Na+-Ca2+ exchanger. J. Neurosci. 1992, 12, 430–439. [Google Scholar] [CrossRef] [PubMed]
- Reyes, R.C.; Verkhratsky, A.; Parpura, V. Plasmalemmal Na+/Ca2+ exchanger modulates Ca2+-dependent exocytotic release of glutamate from rat cortical astrocytes. ASN Neuro 2012, 4, AN20110059. [Google Scholar] [CrossRef]
- Boscia, F.; Begum, G.; Pignataro, G.; Sirabella, R.; Cuomo, O.; Casamassa, A.; Sun, D.; Annunziato, L. Glial Na+-dependent ion transporters in pathophysiological conditions. Glia 2016, 64, 1677–1697. [Google Scholar] [CrossRef] [PubMed]
- Persson, A.K.; Hoeijmakers, J.G.J.; Estacion, M.; Black, J.A.; Waxman, S.G. Sodium Channels, Mitochondria, and Axonal Degeneration in Peripheral Neuropathy. Trends Mol. Med. 2016, 22, 377–390. [Google Scholar] [CrossRef]
- Quednau, B.D.; Nicoll, D.A.; Philipson, K.D. Tissue specificity and alternative splicing of the Na+/Ca2+ exchanger isoforms NCX1, NCX2, and NCX3 in rat. Am. J. Physiol. 1997, 272, C1250–C1261. [Google Scholar] [CrossRef] [PubMed]
- Thurneysen, T.; Nicoll, D.A.; Philipson, K.D.; Porzig, H. Sodium/calcium exchanger subtypes NCX1, NCX2 and NCX3 show cell-specific expression in rat hippocampus cultures. Brain Res. Mol. Brain Res. 2002, 107, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Canitano, A.; Papa, M.; Boscia, F.; Castaldo, P.; Sellitti, S.; Taglialatela, M.; Annunziato, L. Brain distribution of the Na+/Ca2+ exchanger-encoding genes NCX1, NCX2, and NCX3 and their related proteins in the central nervous system. Ann. N. Y. Acad. Sci. 2002, 976, 394–404. [Google Scholar] [CrossRef]
- Yu, L.; Colvin, R.A. Regional differences in expression of transcripts for Na+/Ca2+ exchanger isoforms in rat brain. Brain Res. Mol. Brain Res. 1997, 50, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Jeon, D.; Yang, Y.M.; Jeong, M.J.; Philipson, K.D.; Rhim, H.; Shin, H.S. Enhanced learning and memory in mice lacking Na+/Ca2+ exchanger 2. Neuron 2003, 38, 965–976. [Google Scholar] [CrossRef] [PubMed]
- Persson, A.K.; Black, J.A.; Gasser, A.; Cheng, X.; Fischer, T.Z.; Waxman, S.G. Sodium-calcium exchanger and multiple sodium channel isoforms in intra-epidermal nerve terminals. Mol. Pain 2010, 6, 84. [Google Scholar] [CrossRef] [PubMed]
- Engelhardt, M.; Vorwald, S.; Sobotzik, J.M.; Bennett, V.; Schultz, C. Ankyrin-B structurally defines terminal microdomains of peripheral somatosensory axons. Brain Struct. Funct. 2013, 218, 1005–1016. [Google Scholar] [CrossRef] [PubMed]
- Persson, A.K.; Kim, I.; Zhao, P.; Estacion, M.; Black, J.A.; Waxman, S.G. Sodium channels contribute to degeneration of dorsal root ganglion neurites induced by mitochondrial dysfunction in an in vitro model of axonal injury. J. Neurosci. 2013, 33, 19250–19261. [Google Scholar] [CrossRef]
- Molinaro, P.; Viggiano, D.; Nisticò, R.; Sirabella, R.; Secondo, A.; Boscia, F.; Pannaccione, A.; Scorziello, A.; Mehdawy, B.; Sokolow, S.; et al. Na+-Ca2+ exchanger (NCX3) knock-out mice display an impairment in hippocampal long-term potentiation and spatial learning and memory. J. Neurosci. 2011, 31, 7312–7321. [Google Scholar] [CrossRef] [PubMed]
- Gribkoff, V.K. The role of voltage-gated calcium channels in pain and nociception. Semin. Cell Dev. Biol. 2006, 17, 555–564. [Google Scholar] [CrossRef] [PubMed]
- Lee, S. Pharmacological Inhibition of Voltage-gated Ca2+ Channels for Chronic Pain Relief. Curr. Neuropharmacol. 2013, 11, 606–620. [Google Scholar] [CrossRef] [PubMed]
- Neher, E.; Sakaba, T. Multiple roles of calcium ions in the regulation of neurotransmitter release. Neuron 2008, 59, 861–872. [Google Scholar] [CrossRef] [PubMed]
- Bagur, R.; Hajnóczky, G. Intracellular Ca2+ Sensing: Its Role in Calcium Homeostasis and Signaling. Mol. Cell 2017, 66, 780–788. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M.; Seagar, M.J.; Jones, J.F.; Reber, B.F.; Catterall, W.A. Subunit structure of dihydropyridine-sensitive calcium channels from skeletal muscle. Proc. Natl. Acad. Sci. USA 1987, 84, 5478–5482. [Google Scholar] [CrossRef] [PubMed]
- Dolphin, A.C. Calcium channel auxiliary α2δ and β subunits: Trafficking and one step beyond. Nat. Rev. Neurosci. 2012, 13, 542–555. [Google Scholar] [CrossRef] [PubMed]
- Li, C.Y.; Zhang, X.L.; Matthews, E.A.; Li, K.W.; Kurwa, A.; Boroujerdi, A.; Gross, J.; Gold, M.S.; Dickenson, A.H.; Feng, G.; et al. Calcium channel alpha2delta1 subunit mediates spinal hyperexcitability in pain modulation. Pain 2006, 125, 20–34. [Google Scholar] [CrossRef] [PubMed]
- Luan, C.; Ye, Y.; Singh, T.; Barghouth, M.; Eliasson, L.; Artner, I.; Zhang, E.; Renström, E. The calcium channel subunit gamma-4 is regulated by MafA and necessary for pancreatic beta-cell specification. Commun. Biol. 2019, 2, 106. [Google Scholar] [CrossRef] [PubMed]
- Cain, S.M.; Snutch, T.P. Voltage-gated calcium channels and disease. Biofactors 2011, 37, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Ertel, E.A.; Campbell, K.P.; Harpold, M.M.; Hofmann, F.; Mori, Y.; Perez-Reyes, E.; Schwartz, A.; Snutch, T.P.; Tanabe, T.; Birnbaumer, L.; et al. Nomenclature of voltage-gated calcium channels. Neuron 2000, 25, 533–535. [Google Scholar] [CrossRef] [PubMed]
- McGivern, J.G.; McDonough, S.I. Voltage-gated calcium channels as targets for the treatment of chronic pain. Curr. Drug Targets CNS Neurol. Disord. 2004, 3, 457–478. [Google Scholar] [CrossRef] [PubMed]
- Cai, S.; Gomez, K.; Moutal, A.; Khanna, R. Targeting T-type/Cav3.2 channels for chronic pain. Transl. Res. 2021, 234, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Altier, C.; Zamponi, G.W. Targeting Ca2+ channels to treat pain: T-type versus N-type. Trends Pharmacol. Sci. 2004, 25, 465–470. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.T.; Cabot, P.J.; Ross, F.B.; Robertson, A.D.; Lewis, R.J. The novel N-type calcium channel blocker, AM336, produces potent dose-dependent antinociception after intrathecal dosing in rats and inhibits substance P release in rat spinal cord slices. Pain 2002, 96, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Saegusa, H.; Matsuda, Y.; Tanabe, T. Effects of ablation of N- and R-type Ca2+ channels on pain transmission. Neurosci. Res. 2002, 43, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Hoppanova, L.; Lacinova, L. Voltage-dependent Cav3.2 and Cav2.2 channels in nociceptive pathways. Pflug. Arch. 2022, 474, 421–434. [Google Scholar] [CrossRef]
- Todorovic, S.M.; Jevtovic-Todorovic, V. Neuropathic pain: Role for presynaptic T-type channels in nociceptive signaling. Pflug. Arch. 2013, 465, 921–927. [Google Scholar] [CrossRef] [PubMed]
- Jacus, M.O.; Uebele, V.N.; Renger, J.J.; Todorovic, S.M. Presynaptic Cav3.2 channels regulate excitatory neurotransmission in nociceptive dorsal horn neurons. J. Neurosci. 2012, 32, 9374–9382. [Google Scholar] [CrossRef] [PubMed]
- Bourinet, E.; Alloui, A.; Monteil, A.; Barrère, C.; Couette, B.; Poirot, O.; Pages, A.; McRory, J.; Snutch, T.P.; Eschalier, A.; et al. Silencing of the Cav3.2 T-type calcium channel gene in sensory neurons demonstrates its major role in nociception. EMBO J. 2005, 24, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Chukyo, A.; Chiba, T.; Kambe, T.; Yamamoto, K.; Kawakami, K.; Taguchi, K.; Abe, K. Oxaliplatin-induced changes in expression of transient receptor potential channels in the dorsal root ganglion as a neuropathic mechanism for cold hypersensitivity. Neuropeptides 2018, 67, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.J.; Sweet, T.B.; Clapham, D.E. International Union of Basic and Clinical Pharmacology. LXXVI. Current progress in the mammalian TRP ion channel family. Pharmacol. Rev. 2010, 62, 381–404. [Google Scholar] [CrossRef] [PubMed]
- Feng, S. TRPC Channel Structure and Properties. Adv. Exp. Med. Biol. 2017, 976, 9–23. [Google Scholar] [CrossRef] [PubMed]
- Seebohm, G.; Schreiber, J.A. Beyond Hot and Spicy: TRPV Channels and their Pharmacological Modulation. Cell. Physiol. Biochem. 2021, 55, 108–130. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Li, X. Role of TRPV4 channel in vasodilation and neovascularization. Microcirculation 2021, 28, e12703. [Google Scholar] [CrossRef] [PubMed]
- Naert, R.; López-Requena, A.; Talavera, K. TRPA1 Expression and Pathophysiology in Immune Cells. Int. J. Mol. Sci. 2021, 22, 11460. [Google Scholar] [CrossRef] [PubMed]
- Zholos, A.; Johnson, C.; Burdyga, T.; Melanaphy, D. TRPM channels in the vasculature. Adv. Exp. Med. Biol. 2011, 704, 707–729. [Google Scholar] [CrossRef] [PubMed]
- Abuammar, H.; Bhattacharjee, A.; Simon-Vecsei, Z.; Blastyák, A.; Csordás, G.; Páli, T.; Juhász, G. Ion Channels and Pumps in Autophagy: A Reciprocal Relationship. Cells 2021, 10, 3537. [Google Scholar] [CrossRef] [PubMed]
- Cavaletti, G.; Alberti, P.; Canta, A.; Carozzi, V.; Cherchi, L.; Chiorazzi, A.; Crippa, L.; Marmiroli, P.; Meregalli, C.; Pozzi, E.; et al. Translation of paclitaxel-induced peripheral neurotoxicity from mice to patients: The importance of model selection. Pain 2024. [Google Scholar] [CrossRef] [PubMed]
- Adelsberger, H.; Quasthoff, S.; Grosskreutz, J.; Lepier, A.; Eckel, F.; Lersch, C. The chemotherapeutic oxaliplatin alters voltage-gated Na+ channel kinetics on rat sensory neurons. Eur. J. Pharmacol. 2000, 406, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Chang, W.; Berta, T.; Kim, Y.H.; Lee, S.; Lee, S.Y.; Ji, R.R. Expression and Role of Voltage-Gated Sodium Channels in Human Dorsal Root Ganglion Neurons with Special Focus on Nav1.7, Species Differences, and Regulation by Paclitaxel. Neurosci. Bull. 2018, 34, 4–12. [Google Scholar] [CrossRef]
- Lee, J.H.; Gang, J.; Yang, E.; Kim, W.; Jin, Y.H. Bee Venom Acupuncture Attenuates Oxaliplatin-Induced Neuropathic Pain by Modulating Action Potential Threshold in A-Fiber Dorsal Root Ganglia Neurons. Toxins 2020, 12, 737. [Google Scholar] [CrossRef] [PubMed]
- Verma, P.; Eaton, M.; Kienle, A.; Flockerzi, D.; Yang, Y.; Ramkrishna, D. Examining Sodium and Potassium Channel Conductances Involved in Hyperexcitability of Chemotherapy-Induced Peripheral Neuropathy: A Mathematical and Cell Culture-Based Study. Front. Comput. Neurosci. 2020, 14, 564980. [Google Scholar] [CrossRef]
- Tomaszewski, A.; Büsselberg, D. Cisplatin modulates voltage gated channel currents of dorsal root ganglion neurons of rats. Neurotoxicology 2007, 28, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Brenneman, D.E.; Kinney, W.A.; Ward, S.J. Knockdown siRNA Targeting the Mitochondrial Sodium-Calcium Exchanger-1 Inhibits the Protective Effects of Two Cannabinoids Against Acute Paclitaxel Toxicity. J. Mol. Neurosci. 2019, 68, 603–619. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Tatsui, C.E.; Rhines, L.D.; North, R.Y.; Harrison, D.S.; Cassidy, R.M.; Johansson, C.A.; Kosturakis, A.K.; Edwards, D.D.; Zhang, H.; et al. Dorsal root ganglion neurons become hyperexcitable and increase expression of voltage-gated T-type calcium channels (Cav3.2) in paclitaxel-induced peripheral neuropathy. Pain 2017, 158, 417–429. [Google Scholar] [CrossRef] [PubMed]
- Leo, M.; Schmitt, L.I.; Erkel, M.; Melnikova, M.; Thomale, J.; Hagenacker, T. Cisplatin-induced neuropathic pain is mediated by upregulation of N-type voltage-gated calcium channels in dorsal root ganglion neurons. Exp. Neurol. 2017, 288, 62–74. [Google Scholar] [CrossRef]
- Schmitt, L.I.; Leo, M.; Kleinschnitz, C.; Hagenacker, T. Oxaliplatin Modulates the Characteristics of Voltage-Gated Calcium Channels and Action Potentials in Small Dorsal Root Ganglion Neurons of Rats. Mol. Neurobiol. 2018, 55, 8842–8855. [Google Scholar] [CrossRef] [PubMed]
- Tomita, S.; Sekiguchi, F.; Deguchi, T.; Miyazaki, T.; Ikeda, Y.; Tsubota, M.; Yoshida, S.; Nguyen, H.D.; Okada, T.; Toyooka, N.; et al. Critical role of Cav3.2 T-type calcium channels in the peripheral neuropathy induced by bortezomib, a proteasome-inhibiting chemotherapeutic agent, in mice. Toxicology 2019, 413, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Materazzi, S.; Fusi, C.; Benemei, S.; Pedretti, P.; Patacchini, R.; Nilius, B.; Prenen, J.; Creminon, C.; Geppetti, P.; Nassini, R. TRPA1 and TRPV4 mediate paclitaxel-induced peripheral neuropathy in mice via a glutathione-sensitive mechanism. Pflug. Arch. 2012, 463, 561–569. [Google Scholar] [CrossRef] [PubMed]
- Nassini, R.; Gees, M.; Harrison, S.; De Siena, G.; Materazzi, S.; Moretto, N.; Failli, P.; Preti, D.; Marchetti, N.; Cavazzini, A.; et al. Oxaliplatin elicits mechanical and cold allodynia in rodents via TRPA1 receptor stimulation. Pain 2011, 152, 1621–1631. [Google Scholar] [CrossRef] [PubMed]
- Sánchez, J.C.; Muñoz, L.V.; Ehrlich, B.E. Modulating TRPV4 channels with paclitaxel and lithium. Cell Calcium 2020, 91, 102266. [Google Scholar] [CrossRef] [PubMed]
- Ta, L.E.; Bieber, A.J.; Carlton, S.M.; Loprinzi, C.L.; Low, P.A.; Windebank, A.J. Transient Receptor Potential Vanilloid 1 is essential for cisplatin-induced heat hyperalgesia in mice. Mol. Pain 2010, 6, 15. [Google Scholar] [CrossRef]
- Trevisan, G.; Materazzi, S.; Fusi, C.; Altomare, A.; Aldini, G.; Lodovici, M.; Patacchini, R.; Geppetti, P.; Nassini, R. Novel therapeutic strategy to prevent chemotherapy-induced persistent sensory neuropathy by TRPA1 blockade. Cancer Res. 2013, 73, 3120–3131. [Google Scholar] [CrossRef] [PubMed]
- Ertilav, K.; Nazıroğlu, M.; Ataizi, Z.S.; Yıldızhan, K. Melatonin and Selenium Suppress Docetaxel-Induced TRPV1 Activation, Neuropathic Pain and Oxidative Neurotoxicity in Mice. Biol. Trace Elem. Res. 2021, 199, 1469–1487. [Google Scholar] [CrossRef] [PubMed]
- Anand, U.; Otto, W.R.; Anand, P. Sensitization of capsaicin and icilin responses in oxaliplatin treated adult rat DRG neurons. Mol. Pain 2010, 6, 82. [Google Scholar] [CrossRef] [PubMed]
- Leo, M.; Schmitt, L.I.; Küsterarent, P.; Kutritz, A.; Rassaf, T.; Kleinschnitz, C.; Hendgen-Cotta, U.B.; Hagenacker, T. Platinum-Based Drugs Cause Mitochondrial Dysfunction in Cultured Dorsal Root Ganglion Neurons. Int. J. Mol. Sci. 2020, 21. [Google Scholar] [CrossRef] [PubMed]
- Sánchez, J.C.; Muñoz, L.V.; Galindo-Márquez, M.L.; Valencia-Vásquez, A.; García, A.M. Paclitaxel Regulates TRPA1 Function and Expression Through PKA and PKC. Neurochem. Res. 2023, 48, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Caudle, R.M.; Neubert, J.K. Effects of Oxaliplatin on Facial Sensitivity to Cool Temperatures and TRPM8 Expressing Trigeminal Ganglion Neurons in Mice. Front. Pain Res. 2022, 3, 868547. [Google Scholar] [CrossRef] [PubMed]
- Nieto, F.R.; Entrena, J.M.; Cendán, C.M.; Del Pozo, E.; Vela, J.M.; Baeyens, J.M. Tetrodotoxin inhibits the development and expression of neuropathic pain induced by paclitaxel in mice. Pain 2008, 137, 520–531. [Google Scholar] [CrossRef] [PubMed]
- Makker, P.G.S.; White, D.; Lees, J.G.; Parmar, J.; Goldstein, D.; Park, S.B.; Howells, J.; Moalem-Taylor, G. Acute changes in nerve excitability following oxaliplatin treatment in mice. J. Neurophysiol. 2020, 124, 232–244. [Google Scholar] [CrossRef] [PubMed]
- Alberti, P.; Canta, A.; Chiorazzi, A.; Fumagalli, G.; Meregalli, C.; Monza, L.; Pozzi, E.; Ballarini, E.; Rodriguez-Menendez, V.; Oggioni, N.; et al. Topiramate prevents oxaliplatin-related axonal hyperexcitability and oxaliplatin induced peripheral neurotoxicity. Neuropharmacology 2020, 164, 107905. [Google Scholar] [CrossRef]
- Braden, K.; Stratton, H.J.; Salvemini, D.; Khanna, R. Small molecule targeting NaV1.7 via inhibition of the CRMP2-Ubc9 interaction reduces and prevents pain chronification in a mouse model of oxaliplatin-induced neuropathic pain. Neurobiol. Pain 2022, 11, 100082. [Google Scholar] [CrossRef] [PubMed]
- Di Cesare Mannelli, L.; Lucarini, E.; Micheli, L.; Mosca, I.; Ambrosino, P.; Soldovieri, M.V.; Martelli, A.; Testai, L.; Taglialatela, M.; Calderone, V.; et al. Effects of natural and synthetic isothiocyanate-based H. Neuropharmacology 2017, 121, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Jia, S.; Wei, G.; Bono, J.; Pan, Z.; Zheng, B.; Wang, B.; Adaralegbe, A.; Tenorio, C.; Bekker, A.; Tao, Y.X. TET1 overexpression attenuates paclitaxel-induced neuropathic pain through rescuing K. Life Sci. 2022, 297, 120486. [Google Scholar] [CrossRef]
- Kagiava, A.; Tsingotjidou, A.; Emmanouilides, C.; Theophilidis, G. The effects of oxaliplatin, an anticancer drug, on potassium channels of the peripheral myelinated nerve fibres of the adult rat. Neurotoxicology 2008, 29, 1100–1106. [Google Scholar] [CrossRef] [PubMed]
- Kanbara, T.; Nakamura, A.; Shibasaki, M.; Mori, T.; Suzuki, T.; Sakaguchi, G.; Kanemasa, T. Morphine and oxycodone, but not fentanyl, exhibit antinociceptive effects mediated by G-protein inwardly rectifying potassium (GIRK) channels in an oxaliplatin-induced neuropathy rat model. Neurosci. Lett. 2014, 580, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Lucarini, E.; Micheli, L.; Trallori, E.; Citi, V.; Martelli, A.; Testai, L.; De Nicola, G.R.; Iori, R.; Calderone, V.; Ghelardini, C.; et al. Effect of glucoraphanin and sulforaphane against chemotherapy-induced neuropathic pain: Kv7 potassium channels modulation by H2S release in vivo. Phytother. Res. 2018, 32, 2226–2234. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, E.; Gold, M.S. Paclitaxel-induced increase in NCX activity in subpopulations of nociceptive afferents: A protective mechanism against chemotherapy-induced peripheral neuropathy? Cell Calcium 2016, 60, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Nodera, H.; Spieker, A.; Sung, M.; Rutkove, S. Neuroprotective effects of Kv7 channel agonist, retigabine, for cisplatin-induced peripheral neuropathy. Neurosci. Lett. 2011, 505, 223–227. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, K.; Chiba, T.; Katagiri, N.; Saduka, M.; Abe, K.; Utsunomiya, I.; Hama, T.; Taguchi, K. Paclitaxel increases high voltage-dependent calcium channel current in dorsal root ganglion neurons of the rat. J. Pharmacol. Sci. 2012, 120, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, M.; Inoue, M.; Hald, A.; Xie, W.; Ueda, H. Inhibition of paclitaxel-induced A-fiber hypersensitization by gabapentin. J. Pharmacol. Exp. Ther. 2006, 318, 735–740. [Google Scholar] [CrossRef] [PubMed]
- Okubo, K.; Nakanishi, H.; Matsunami, M.; Shibayama, H.; Kawabata, A. Topical application of disodium isostearyl 2-O-L-ascorbyl phosphate, an amphiphilic ascorbic acid derivative, reduces neuropathic hyperalgesia in rats. Br. J. Pharmacol. 2012, 166, 1058–1068. [Google Scholar] [CrossRef] [PubMed]
- Sekiguchi, F.; Kawara, Y.; Tsubota, M.; Kawakami, E.; Ozaki, T.; Kawaishi, Y.; Tomita, S.; Kanaoka, D.; Yoshida, S.; Ohkubo, T.; et al. Therapeutic potential of RQ-00311651, a novel T-type Ca2+ channel blocker, in distinct rodent models for neuropathic and visceral pain. Pain 2016, 157, 1655–1665. [Google Scholar] [CrossRef] [PubMed]
- Meregalli, C.; Maricich, Y.; Cavaletti, G.; Canta, A.; Carozzi, V.A.; Chiorazzi, A.; Newbold, E.; Marmiroli, P.; Ceresa, C.; Diani, A.; et al. Reversal of Bortezomib-Induced Neurotoxicity by Suvecaltamide, a Selective T-Type Ca-Channel Modulator, in Preclinical Models. Cancers 2021, 13, 5013. [Google Scholar] [CrossRef] [PubMed]
- Sharma, J.; Maslov, L.N.; Singh, N.; Jaggi, A.S. Pain attenuating actions of vincristinet-preconditioning in chemotherapeutic agent-induced neuropathic pain: Key involvement of T-type calcium channels. Fundam. Clin. Pharmacol. 2020, 34, 336–344. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Yang, C.; Wang, Z.J. Proteinase-activated receptor 2 sensitizes transient receptor potential vanilloid 1, transient receptor potential vanilloid 4, and transient receptor potential ankyrin 1 in paclitaxel-induced neuropathic pain. Neuroscience 2011, 193, 440–451. [Google Scholar] [CrossRef] [PubMed]
- Hori, K.; Ozaki, N.; Suzuki, S.; Sugiura, Y. Upregulations of P2X3 and ASIC3 involve in hyperalgesia induced by cisplatin administration in rats. Pain 2010, 149, 393–405. [Google Scholar] [CrossRef] [PubMed]
- Quartu, M.; Carozzi, V.A.; Dorsey, S.G.; Serra, M.P.; Poddighe, L.; Picci, C.; Boi, M.; Melis, T.; Del Fiacco, M.; Meregalli, C.; et al. Bortezomib treatment produces nocifensive behavior and changes in the expression of TRPV1, CGRP, and substance P in the rat DRG, spinal cord, and sciatic nerve. Biomed Res. Int. 2014, 2014, 180428. [Google Scholar] [CrossRef] [PubMed]
- Mao, Q.; Wu, S.; Gu, X.; Du, S.; Mo, K.; Sun, L.; Cao, J.; Bekker, A.; Chen, L.; Tao, Y.X. DNMT3a-triggered downregulation of K. Int. J. Cancer 2019, 145, 2122–2134. [Google Scholar] [CrossRef] [PubMed]
- Pereira, V.; Busserolles, J.; Christin, M.; Devilliers, M.; Poupon, L.; Legha, W.; Alloui, A.; Aissouni, Y.; Bourinet, E.; Lesage, F.; et al. Role of the TREK2 potassium channel in cold and warm thermosensation and in pain perception. Pain 2014, 155, 2534–2544. [Google Scholar] [CrossRef]
- Rapacz, A.; Obniska, J.; Koczurkiewicz, P.; Wójcik-Pszczoła, K.; Siwek, A.; Gryboś, A.; Rybka, S.; Karcz, A.; Pękala, E.; Filipek, B. Antiallodynic and antihyperalgesic activity of new 3,3-diphenyl-propionamides with anticonvulsant activity in models of pain in mice. Eur. J. Pharmacol. 2018, 821, 39–48. [Google Scholar] [CrossRef]
- Sałat, K.; Furgała-Wojas, A.; Sałat, R. The Microglial Activation Inhibitor Minocycline, Used Alone and in Combination with Duloxetine, Attenuates Pain Caused by Oxaliplatin in Mice. Molecules 2021, 26, 3577. [Google Scholar] [CrossRef] [PubMed]
- Alberti, P. Platinum-drugs induced peripheral neurotoxicity: Clinical course and preclinical evidence. Expert Opin. Drug Metab. Toxicol. 2019, 15, 487–497. [Google Scholar] [CrossRef] [PubMed]
- Lucchetta, M.; Lonardi, S.; Bergamo, F.; Alberti, P.; Velasco, R.; Argyriou, A.A.; Briani, C.; Bruna, J.; Cazzaniga, M.; Cortinovis, D.; et al. Incidence of atypical acute nerve hyperexcitability symptoms in oxaliplatin-treated patients with colorectal cancer. Cancer Chemother. Pharmacol. 2012, 70, 899–902. [Google Scholar] [CrossRef] [PubMed]
- Alberti, P.; Salvalaggio, A.; Argyriou, A.A.; Bruna, J.; Visentin, A.; Cavaletti, G.; Briani, C. Neurological Complications of Conventional and Novel Anticancer Treatments. Cancers 2022, 14, 6088. [Google Scholar] [CrossRef] [PubMed]
- Chiorazzi, A.; Semperboni, S.; Marmiroli, P. Current View in Platinum Drug Mechanisms of Peripheral Neurotoxicity. Toxics 2015, 3, 304–321. [Google Scholar] [CrossRef] [PubMed]
- Monza, L.; Fumagalli, G.; Chiorazzi, A.; Alberti, P. Addressing the Need of a Translational Approach in Peripheral Neuropathy Research: Morphology Meets Function. Brain Sci. 2021, 11, 139. [Google Scholar] [CrossRef] [PubMed]
- Monza, L.; Fumagalli, G.; Chiorazzi, A.; Alberti, P. Translating morphology from bench side to bed side via neurophysiology: 8-min protocol for peripheral neuropathy research. J. Neurosci. Methods 2021, 363, 109323. [Google Scholar] [CrossRef] [PubMed]
- Bostock, H.; Cikurel, K.; Burke, D. Threshold tracking techniques in the study of human peripheral nerve. Muscle Nerve 1998, 21, 137–158. [Google Scholar] [CrossRef]
- Boërio, D.; Greensmith, L.; Bostock, H. Excitability properties of motor axons in the maturing mouse. J. Peripher. Nerv. Syst. 2009, 14, 45–53. [Google Scholar] [CrossRef]
- Arnold, R.; Moldovan, M.; Rosberg, M.R.; Krishnan, A.V.; Morris, R.; Krarup, C. Nerve excitability in the rat forelimb: A technique to improve translational utility. J. Neurosci. Methods 2017, 275, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Park, S.B.; Goldstein, D.; Lin, C.S.; Krishnan, A.V.; Friedlander, M.L.; Kiernan, M.C. Acute abnormalities of sensory nerve function associated with oxaliplatin-induced neurotoxicity. J. Clin. Oncol. 2009, 27, 1243–1249. [Google Scholar] [CrossRef] [PubMed]
- McHugh, J.C.; Tryfonopoulos, D.; Fennelly, D.; Crown, J.; Connolly, S. Electroclinical biomarkers of early peripheral neurotoxicity from oxaliplatin. Eur. J. Cancer Care 2012, 21, 782–789. [Google Scholar] [CrossRef] [PubMed]
- Eckel, F.; Schmelz, R.; Adelsberger, H.; Erdmann, J.; Quasthoff, S.; Lersch, C. Prevention of oxaliplatin-induced neuropathy by carbamazepine. A pilot study. Dtsch. Med. Wochenschr. 2002, 127, 78–82. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Shao, J.; Wang, J.; Liu, Y.; Zhang, Y.; Zhang, M.; Zhang, J.; Ren, X.; Su, S.; Li, Y.; et al. MiR-30b-5p attenuates oxaliplatin-induced peripheral neuropathic pain through the voltage-gated sodium channel Na. Neuropharmacology 2019, 153, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Azad, I.; Khan, T.; Ahmad, N.; Khan, A.R.; Akhter, Y. Updates on drug designing approach through computational strategies: A review. Future Sci. OA 2023, 9, FSO862. [Google Scholar] [CrossRef] [PubMed]
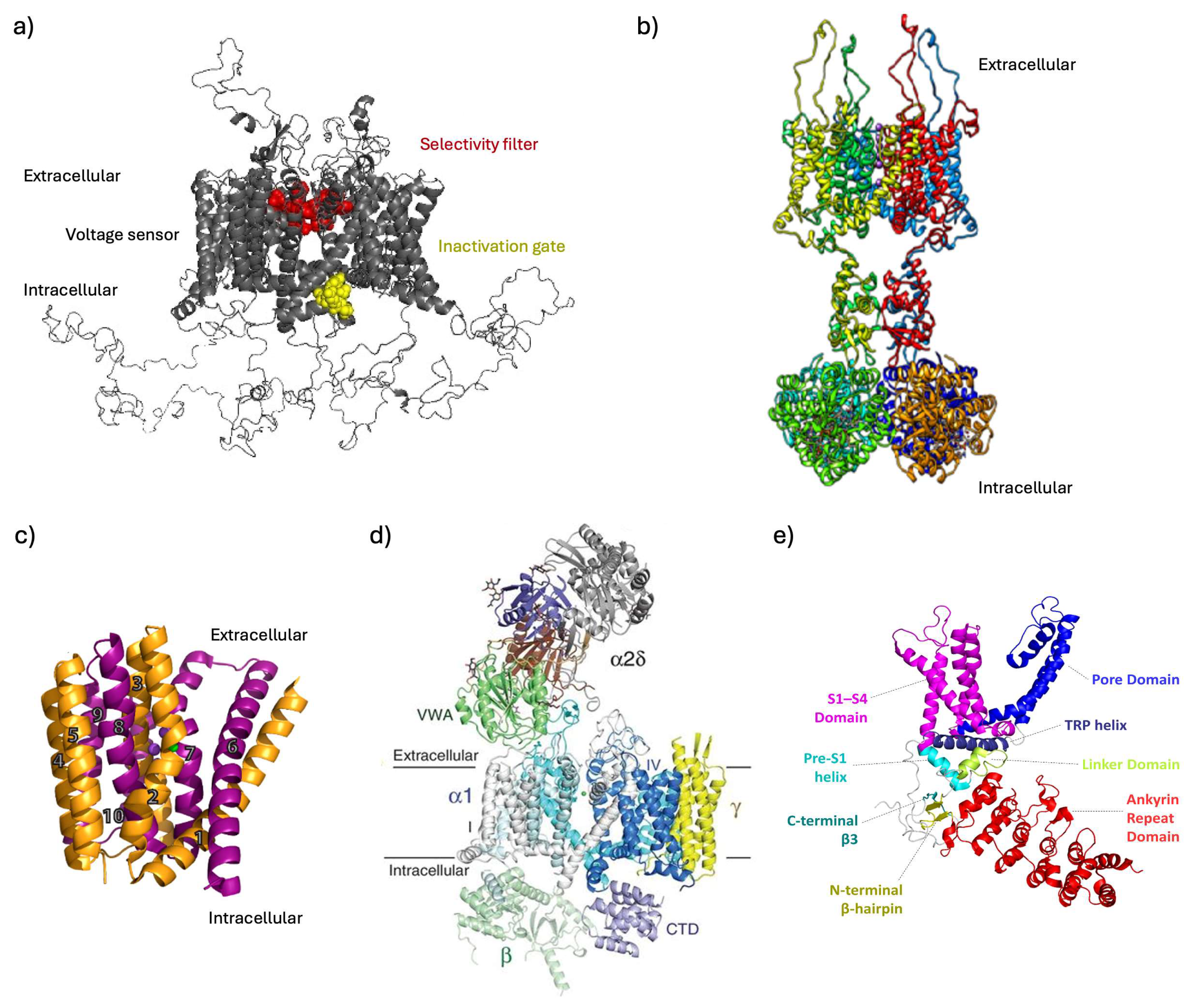

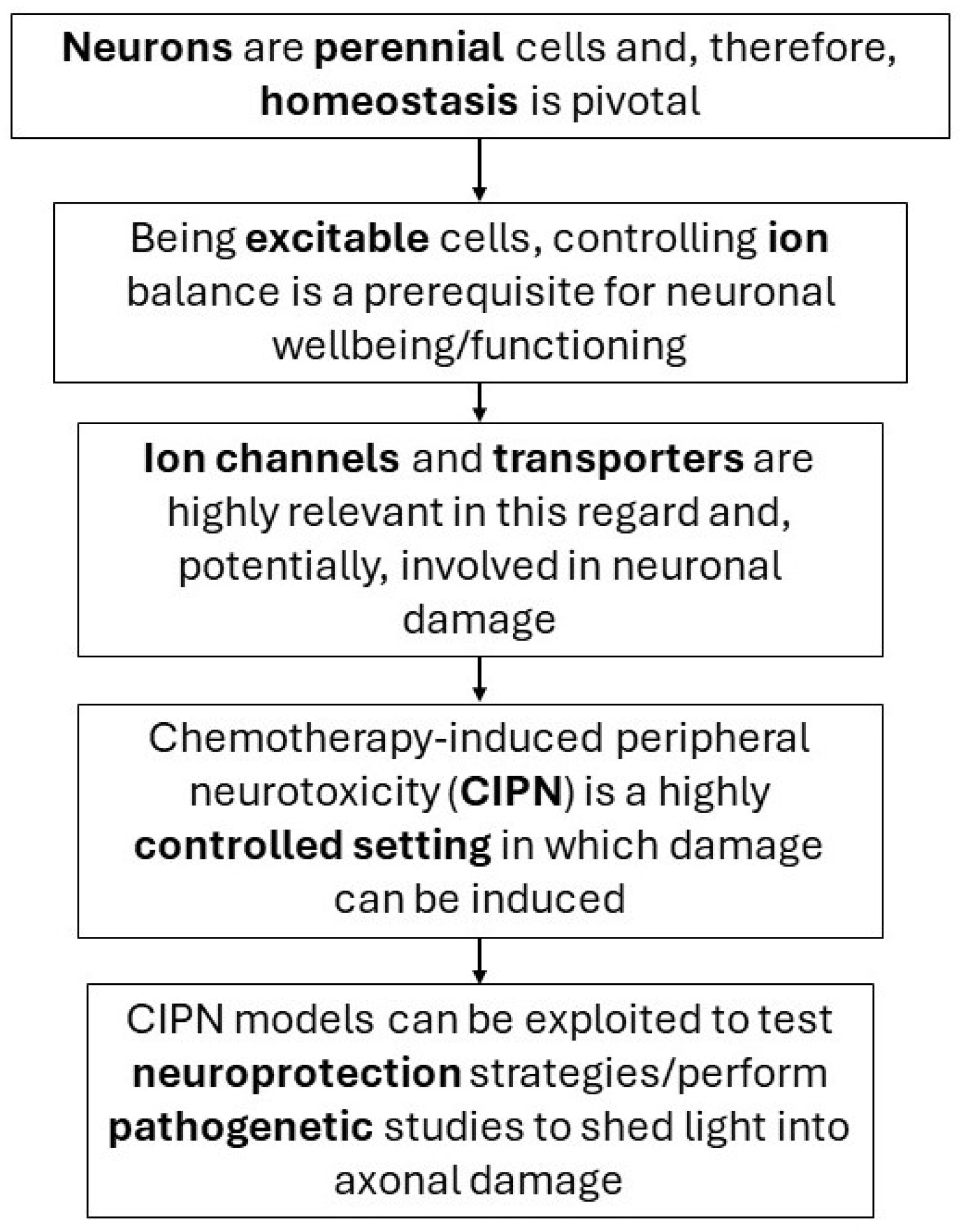

{kind=link}
{kind=link}
{kind=link}
| Authors | Target | Cell Culture and Treatment | Neurotoxicity Assessment | Observations |
|---|---|---|---|---|
| Ballarini et al. [87] | NCX2 | OHP 7.5 µM. Rat DRG neurons | Neurite elongation | Protection of neurite outgrowth with a selective NCX blocker |
| Adelsberger et al. [129] | VGSCs | OHP 250 µM. Rat DRG and hippocampal neurons | Patch clamp recordings | Increase in the Na+ current on DRG neurons but not on hippocampal neurons |
| Chang et al. [130] | VGSCs | PTX 0.1–1 µM. Human DRG neurons | Immunohistochemistry, qRT-PCR, transient Na+ currents and action potential frequency | Increase in Nav 1.7 mRNA expression but not Nav 1.8. Increased transient Na+ currents amplitude and action potential firing frequency |
| Lee et al. [131] | VGSCs | OHP 10 and 100 µM. Rat DRG neurons | Patch clamp recordings | Alteration of VGSC conductance towards negative membrane potentials in A-fibers of DRGs |
| Verma et al. [132] | VGSCs Nav 1.7 and Nav 1.8, KDR, KA, leak channel | PTX 250 nM. Rat DRG neurons | Micro/multielectrode array recordings | Decrease in PTX-induced hyperexcitability by a Nav 1.8 blocker and a KDR agonist treatments |
| Tomaszewski et al. [133] | VGCCs, VGKCs, VGSCs | CDDP 1, 5, 10, 50 and 100 µM. Rat DRG neurons | Patch clamp recordings | Decrease in Ca2+ and K+ currents in small DRG neurons but only a trend toward reduction in Na+ currents |
| Brenneman et al. [134] | mNCX-1 | PTX 3 µM. Rat DRG neurons | Cell viability assays, IR cell bodies and neuritic areas | mNCX-1 siRNA decreases CBD protection from PTX toxicity, decrease in IR neuronal cell bodies and neuritic IR areas |
| Li et al. [135] | VGCC T-type | PTX 1 µM. Human DRG neurons | Patch clamp recordings, immunohistochemistry | Increase in Ca2+ current, increase in DRG excitability |
| Leo et al. [136] | VGCCs | CDDP 0.5 or 5 µM. Rat DRG neurons | Patch clamp recordings, immunostaining, calpain activity assay | Decrease in Ca2+ current in L-type, P-/Q-type and T-type channels but increase in N-type VGCC currents. Increased expression of N-type VGCC proteins. DRG neuroprotection by N-type VGCC blocker |
| Schmitt et al. [137] | VGCCs | OHP 1, 10, 100, 250 and 500 µM. Rat DRG neurons | Patch clamp recordings, immunocytochemistry, Western blot, calpain activity assay | Decrease in L-type, P/Q-type and T-type VGCCs currents. Prolonged treatment increased current density. Increase in L-type and T-type VGCCs protein expression. Increase in the action potential amplitude through modulation of T-type and L-type VGCCs |
| Tomita et al. [138] | VGCCs T type | BTZ 0.1 nM. Mouse neuroblastoma x rat DRG neuron hybrid cells | Western blot, qRT-PCR, patch clamp recordings | Increase in T-type VGCC protein expression. Increase in Ca2+ currents |
| Materazzi et al. [139] | TRPA1, TRPV4 | PTX 10, 30 and 50 µM. Mice DRG neurons or esophagus slices | Ca2+ imaging, neuropeptides release assay | Modulation of TRPA1 and TRPV4 by Ca2+-dependent CGRP secretion |
| Nassini et al. [140] | TRPA1 | OHP or CDDP 100 µM, guinea pig pulmonary artery. TRPA1+ CHO cells expressing mouse (10 to 300 µM OHP/CDDP) | Guinea pig pulmonary artery assay of neurogenic relaxation. DRG and CHO Ca2+ response to OHP or CDDP | OHP and CDDP activate TRPA1 channel on nociceptive nerve terminals. The activation of TRPA1 is mediated by oxidative stress |
| Sanchez et al. [141] | TRPV4 | PTX 1 µM. Human SH-SY5Y cells | qRT-PCR, Western blot, patch clamp recordings, cytosolic Ca2+ measurement | Increase in TRPV4 protein and mRNA expression. Increase in outward and inward current density. Increase in cytosolic Ca2+ concentrations |
| Ta et al. [142] | TRPV1, TRPM8, TRPA1 | CDDP or OHP 6.7 µM. Rat DRG neurons | qRT-PCR | TRPV1, TRPM8 and TRPA1 mRNA expressions are differently upregulated by CDDP and OHP |
| Trevisan et al. [143] | TRPA1 | BTZ 10 or 100 µM. Mouse DRG neurons | Ca2+ imaging | BTZ did not evoke Ca2+ responses in TRPA1+ neurons |
| Ertilav et al. [144] | TRPV1 | DT 10 nM. TRPV1 transfected SH-SY5Y cells | Ca2+ fluorescence, Western blot | Activation of TRPV1 |
| Anand et al. [145] | TRPV1, TRPA1, TRPM8 | OHP 12–120 µM. Rat DRG neurons | Neurite elongation and density, cell viability assay, cAMP assay, Ca2+ imaging | TRPV1 and TRPA1 sensitization but not for TRPM8 |
| Leo et al. [146] | TRPA1, TRPV1 | CDDP and OHP 10 µM. Rat DRG neurons | Cell viability assay, immunocytochemical staining, cytosolic and intramitochondrial Ca2+ measurement | Increase in cytosolic Ca2+ concentration and decrease in intramitochondrial Ca2+ concentration in TRPA1+ and TRPV1+ DRG neurons |
| Sanchez et al. [147] | TRPA1 | PTX 1 µM. Human SH-SY5Y cells | qRT-PCR, Western blot, patch clamp recordings, cytosolic Ca2+ measurement | Increase in TRPA1 protein expression, TRPA1 current density and TRPA1-mediated Ca2+ concentrations |
| Authors | Target | Animal Model | Neurotoxicity Assessment | Observations |
|---|---|---|---|---|
| Ballarini et al. [87] | NCX2 | OHP 7 mg/kg in mice, i.v., once a week for 8 weeks | NCS and NET recordings, mechanical allodynia test, immunohistochemistry, Western blot, caudal nerve morphology and morphometry, IENFD | Decrease in NCX2 protein expression in DRGs |
| Chukyo et al. [120] | TRPV1, TRPA1, TRPM8 | OHP 6 mg/kg in rats, single i.p. | Acetone spray test, immunohistochemistry, in situ hybridization | Increase in TRPA1, TRPV1 and TRPM8 protein expression in DRGs. Increase in TRPA1 and TRPV1 mRNA coexpression in DRGs |
| Caudle and Neubert [148] | HCN, VGSCs, menthol, TRPM8 | OHP 10 mg/kg in mice, i.p., two administrations; PTX 26 mg/kg in mice, i.p., four administrations. Dissociated TRG neurons * | Orofacial Pain Assessment Devices, patch clamp recordings | Increase in HCN, VGSCs and menthol evoked TRPM8 currents but not of VGKCs |
| Nieto et al. [149] | VGSCs TTX sensitive | PTX 2 mg/kg in mice, i.p., 5 days | Heat hyperalgesia test, acetone cold allodynia test, mechanical allodynia test, rotarod test | Decrease in heat hyperalgesia, mechanical and cold allodynia by TTX administration |
| Makker et al. [150] | VGSCs and VGKCs | OHP 10 mg/kg i.p. or 7.5 and 15 mg/kg i.m. in mice, single dose; 5 mg/kg i.p. on days 0, 2, 4, 6 | CMAP and SNAP recording, mathematical modeling of axonal excitability | Change of the depolarization phase and creation of afterdischarges, inactivation of VGCCs, reduction in fast K+ conductance in motor axons. Increase in hyperpolarization and decrease in peak amplitude in sensory axons |
| Alberti et al. [151] | VGSC | OHP 5 mg/kg in rats, twice a week for 4 weeks | NCS and NET recordings, mechanical allodynia test, caudal nerve morphology and morphometry, IENFD | Modulating VGSC with topiramate (100 mg/kg per os, daily, starting 5 days before first OHP administration and continuing up to chemotherapy completion) complete neurotoxicity prevention was observed via neurophysiology, neuropathology and behavioral tests |
| Braden et al. [152] | VGSC Nav 1.7 | OHP 3 mg/kg in mice, i.p., on days 0–4 and 10–14 | Von Frey test | Decrease in mechanical allodynia through indirect inhibition of Nav 1.7 |
| Di Cesare Mannelli et al. [153] | VGKCs Kv7 | PTX 2 mg/kg in mice, i.p., on days 1, 3, 5 and 7; OHP 2.4 mg/kg in mice, i.p., on days 1–2, 5–9, 12–14 | Cold plate test, Von Frey test, hot plate test | Kv7 channel blocker XE991 antagonized the pain-relieving activity of H2S donors, demonstrating the role of Kv7 in neuropathic pain |
| Jia et al. [154] | K+ channel 1.1 (K2p 1.1) | PTX 4 mg/kg in rats, i.p., every other day for a total of four injections, on days 0, 2, 4, and 6 | Mechanical allodynia heat, heat hyperalgesia test and cold hyperalgesia test | Reduction in K+ channel 1.1 |
| Kagiava et al. [155] | VGKCs | OHP 25, 100 and 500 µM. Rat isolated sciatic nerve * | Evoked CAP recordings | Induce alterations in CAP waveform, firing frequency and repolarization phase through VGKCs but not VGSCs |
| Kanbara et al. [156] | GIRK1 | OHP 2 mg/kg in rats, i.p., twice a week for 4 weeks | Von Frey test | GIRK1 activation contributes to MOR antinociception |
| Lucarini et al. [157] | VGKC Kv7 | OHP 2.4 mg/kg in mice, i.p., on days 1–2, 5–9 and 12–14 | Cold plate test | Modulating Kv7 channels, a reduction in painful features is observed |
| Yilmaz et al. [158] | NCX | PTX 2 mg/kg in rats, on days 0, 2, 4 and 6. Dissociated DRG neurons * | Ca2+ imaging | PTX-induced inhibition of Ca2+ transients is not modulated by NCX activity |
| Li et al. [135] | VGCC T type | PTX 2 mg/kg in rats, i.p., on days 0, 2, 4 and 6 | Von Frey test, patch clamp recordings, Ca2+ imaging, immunohistochemistry, Western blot | Increase in Ca2+ current, increase of DRG excitability, increase in T-type VGCC expression in DRGs and spinal cord. Decrease in mechanical allodynia by T-type VGCC blocker |
| Leo et al. [136] | VGCCs | CDDP 1.5 mg/kg in rats, i.p., two cycles of four daily administrations with four days rest | Von Frey test, hot plate test, rotarod test, Western blot, qRT-PCR | Increased expression of N-type VGCC proteins, but not mRNA in DRGs. Decrease in thermal hyperalgesia and mechanical allodynia by N-type VGCC blocker |
| Tomita et al. [138] | VGCCs T type | BTZ 0.4 mg/kg in mice, i.p., six administrations in 2 weeks | Western blot, Von Frey test | Increase in T-type VGCCs’ protein expression in DRGs. Decrease in mechanical hyperalgesia through T-type VGGC blockers and gene silencing |
| Nodera et al. [159] | Kv7 VDKCs | CDDP 2.3 mg/kg in mice, i.p., on days 1–5 and 13–17 | SNAP recording, NET recording, NCS recording | Axonal protection, preserved membrane potential through increase in K+ currents with treatment Kv7 agonist retigabine |
| Kawakami et al. [160] | VGCCs | PTX 2 and 4 mg/kg in rats, i.p., on days 0, 2, 4 and 6. Dissociated DRG neurons | Von Frey test, patch clamp recordings | Increase in Ca2+ currents. GBP, a Ca2+ channel blocker, reverses mechanical hyperalgesia |
| Matsumoto et al. [161] | VGCCs α2δ-1 subunit | PTX 4 mg/kg in mice, single i.p. or i.v., or i.p. on days 0, 2, 4 and 6 | Heat hyperalgesia test, electrical hyperalgesia test, qRT-PCR, Western blot, immunohistochemistry | Increase in DRGs’ expression of α2δ-1 subunit. GBP blockade of VGCCs decreases mechanical allodynia and sensitization of myelinated A-fibers |
| Okubo et al. [162] | VGCCs T type | PTX 2 mg/kg in rats, i.p., on days 0, 2, 4 and 6 | Paw pressure test | Decrease in hyperalgesia through administration of T-type VGCCs selective blockers |
| Sekiguchi et al. [163] | T-type VGCCs | PTX 4 mg/kg in mice or 2 mg/kg in rats, i.p., on days 0, 2, 4 and 6 | Von Frey test, paw pressure test, open field test, rotarod test | T-type VGCC blockers reduce neuropathic mechanical allodynia |
| Meregalli et al. [164] | VGCCs T type | BTZ 0.2 mg/kg in rats, i.v., three times a week for 4 weeks | NCV measurement, mechanical allodynia test, β-tubulin polymerization assay, IENFD, proteasome inhibition assay | Suvecaltamide modulation of T-type VGCCs reverses NCV and IENFD neuropathy, reverses β-tubulin polymerization increase but does not affect proteasome inhibition by BTZ |
| Sharma et al. [165] | VGCCs L type | VCR 50 μg/kg in rats, i.p., 10 days administration | acetone drop test, pin-prick test, hot plate test | Decrease in the protective effect of VCR pretreatment on allodynia and hyperalgesia following treatment with T-type VGCC blocker |
| Materazzi et al. [139] | TRPA1, TRPV4 | PTX 6 mg/kg in WT and TRPA1 KO mice, single i.p. | Von Frey test, acetone cold stimulation test | Decrease in mechanical allodynia by TRPA1 and TRPV4 blockers. Decrease in cold hypersensitivity by TRPA1 but not TRPV4 blocker |
| Nassini et al. [140] | TRPA1 | OHP 2 mg/kg i.v., CDDP 2 mg/kg i.p. in C57/BL6, Trpa1+/+ or Trpa1−/− mice | Von Frey test, cold plate test, qRT-PCR | TRPA1 modulation decreases painful features related to OHP and CDDP single administration |
| Ta et al. [142] | TRPV1, TRPM8, TRPA1 | CDDP 2.3 mg/kg or OHP 3 mg/kg in WT or TRPV1 KO mice, i.p., 5 days, 5 days rest and 5 days treatment | Von Frey test, radiant heat test, cold plate test, tail immersion test, qRT-PCR, immunohistochemistry | Upregulation of TRPV1 and TRPA1 mRNA following CDDP treatment, but only TRPA1 upregulation following OHP treatment in TGs. Decrease in mechanical allodynia following CDDP and OHP treatment in TRPV1 KO mice. Decrease in CDDP-induced thermal hypersensitivity in TRPV1 KO mice |
| Trevisan et al. [143] | TRPA1 | BTZ 0.2, 0.5 or 1 mg/kg in WT or TRPA1 KO mice, single i.p. | Von Frey test, hot plate test, cold stimulation, chemical hyperalgesia test, rotarod test, Western blot | BTZ did not modify TRPA1 expression level in DRGs. Decrease in mechanical and cold hyperalgesia through TRPA1 agonist treatment and in TRPA1 KO mice |
| Chen et al. [166] | TRPV1, TRPV4, TRPA1 | PTX 1 mg/kg in mice, i.p., on days 0, 2, 4 and 6 | Von Frey test, hot plate test, cold hyperalgesia test | Reduction in heat hyperalgesia, but not mechanical allodynia and cold hyperalgesia, through TRPV1 blocking. Reduction in mechanical and heat, but not cold, hyperalgesia through TRPV4 blocking. Reduction in mechanical allodynia, heat and cold hyperalgesia through TRPA1 blocking |
| Ertilav et al. [144] | TRPV1 | DT 30 mg/kg in mice, single i.p. Dissociated DRG neurons * | Von Frey test, hot plate test, Western blot, patch clamp recordings, Ca2+ fluorescence, cell viability assay | Increase in cytosolic Ca2+ concentration through TRPV1 channel agonist stimulation. Increase in TRPV1 expression level |
| Hori et al. [167] | TRPV1, TRPV2, P2 × 3 and ASIC3 | CDDP 3 mg/kg in rats, i.p., once per week for five weeks | Von Frey test, pin-prick test, mechanical allodynia test, grid force test, rotarod test and immunohistochemistry | Increase in TRPV2, P2 × 3 and ASIC3 expression, but not in TRPV1 in DRGs |
| Quartu et al. [168] | TRPV1 | BTZ 0.20 mg/kg in rats, single i.v., or three administrations for 8 weeks | Caudal NCV recordings, mechanical allodynia test, thermal hyperalgesia test, immunohistochemistry, morphometry, qRT-PCR and Western blot | Reduction in caudal NCV, increase in mechanical allodynia but not of thermal hyperalgesia. Increase in TRPV1 protein expression, but decrease in TRPV1 and CGRP mRNA level, in DRGs and spinal cord |
| Mao et al. [169] | K2p1.1 channel | PTX 4 mg/kg in mice, i.p., on days 0, 2, 4 and 6 | Von Frey test, heat hyperalgesia, conditioned place preference, patch clamp recordings, qRT-PCR, Western blot, immunohistochemistry | PTX induces a decrease of K2P1.1 expression, contributing to chemotherapy-induced neuropathic pain |
| Pereira et al. [170] | TREK2 | OHP 6 mg/kg in WT and TREK2 KO mice, single i.p. | Von Frey test, flinch test, immersion tests, hot plate test, thermal preference test, dynamic cold plate test; qRT-PCR; single C-fibers recordings | Decrease in TREK2 expression in DRGs. TREK2 mediates neuropathic hyperalgesia, regulates heat sensitivity of C-fibers, but does not play a role in noxious thermal hypersensitivity |
| Rapacz et al. [171] | VGSCs and L-type VGCCs | OHP 10 mg/kg in mice, i.p. | Von Frey test, hot plate test, formalin test | Decrease in mechanical allodynia by VGSCs and VGCCs blocking |
| Salat et al. [172] | VGSCs | OHP 10 mg/kg in mice, single i.p. | Von Frey test, cold plate tests, rotarod test | Reduced mechanical allodynia following a VGSC blocker |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pozzi, E.; Terribile, G.; Cherchi, L.; Di Girolamo, S.; Sancini, G.; Alberti, P. Ion Channel and Transporter Involvement in Chemotherapy-Induced Peripheral Neurotoxicity. Int. J. Mol. Sci. 2024, 25, 6552. https://doi.org/10.3390/ijms25126552
Pozzi E, Terribile G, Cherchi L, Di Girolamo S, Sancini G, Alberti P. Ion Channel and Transporter Involvement in Chemotherapy-Induced Peripheral Neurotoxicity. International Journal of Molecular Sciences. 2024; 25(12):6552. https://doi.org/10.3390/ijms25126552
Chicago/Turabian StylePozzi, Eleonora, Giulia Terribile, Laura Cherchi, Sara Di Girolamo, Giulio Sancini, and Paola Alberti. 2024. "Ion Channel and Transporter Involvement in Chemotherapy-Induced Peripheral Neurotoxicity" International Journal of Molecular Sciences 25, no. 12: 6552. https://doi.org/10.3390/ijms25126552
APA StylePozzi, E., Terribile, G., Cherchi, L., Di Girolamo, S., Sancini, G., & Alberti, P. (2024). Ion Channel and Transporter Involvement in Chemotherapy-Induced Peripheral Neurotoxicity. International Journal of Molecular Sciences, 25(12), 6552. https://doi.org/10.3390/ijms25126552