
Reactive Oxygen Species Induction by Hepatitis B Virus: Implications for Viral Replication in p53-Positive Human Hepatoma Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
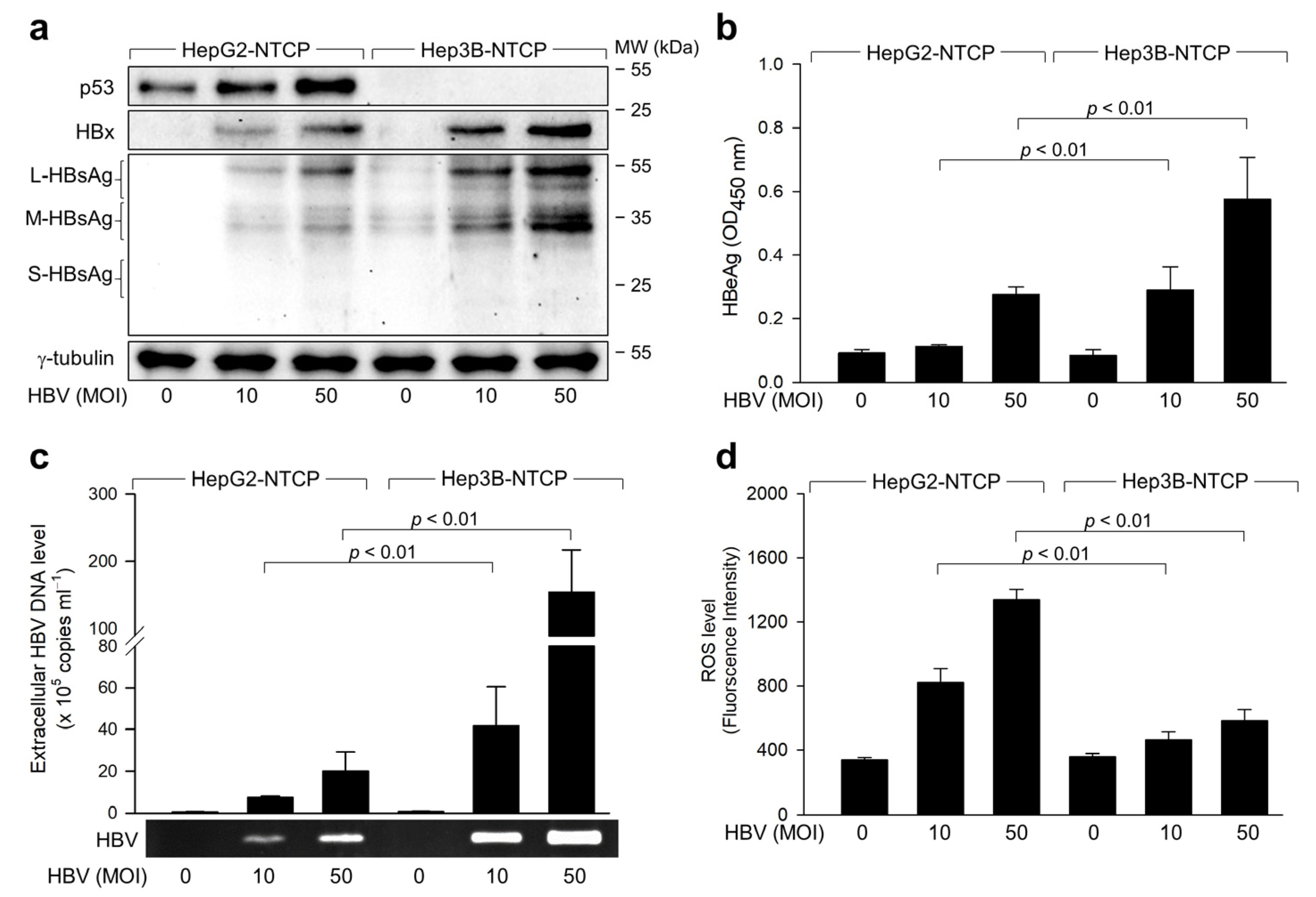
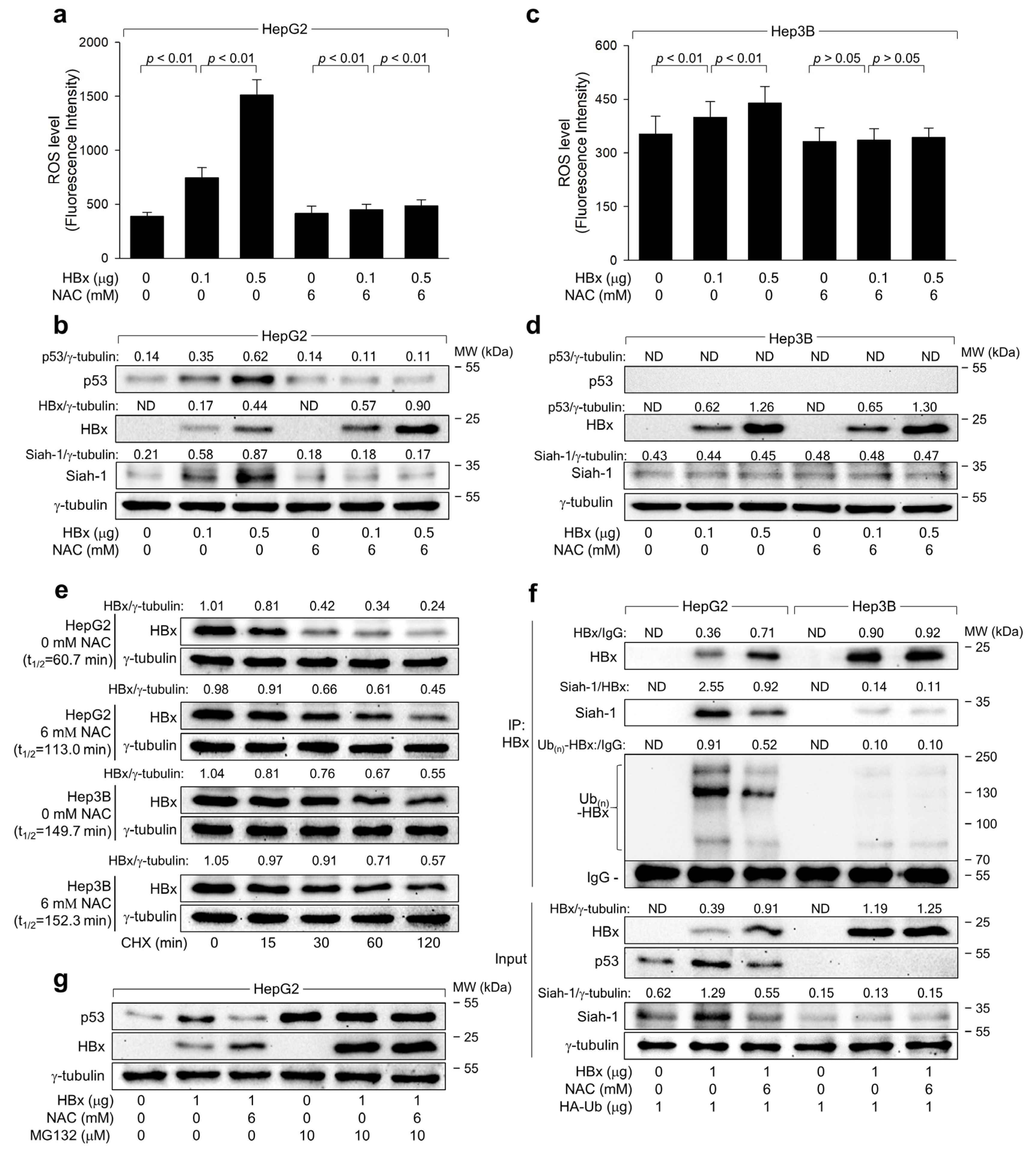
2.1. Exploring the Relationship between ROS Induction and HBV Replication in p53-Positive Human Hepatoma Cells
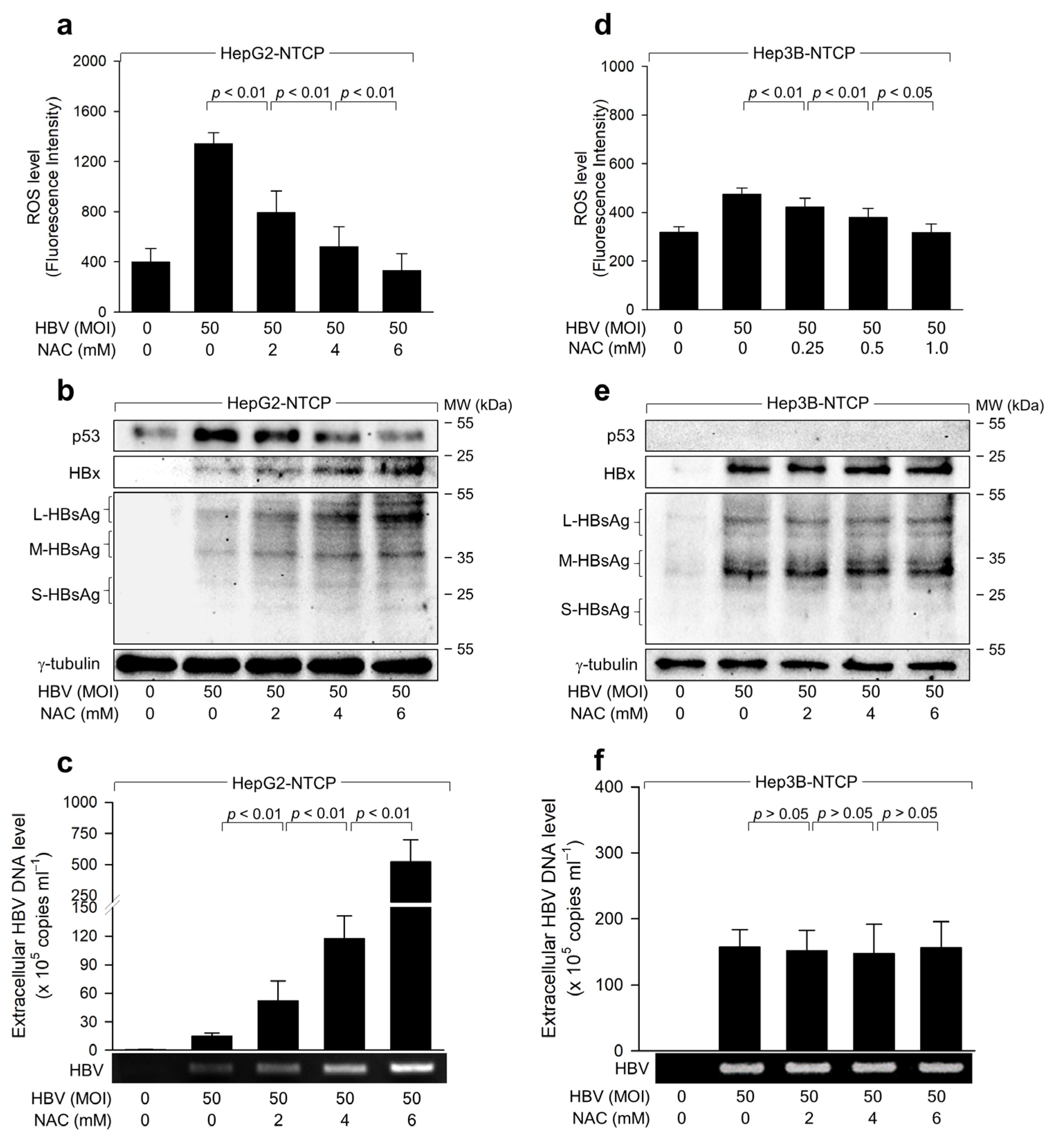
2.2. HBV Induces ROS Generation to Inhibit Virus Replication via a Negative Feedback Loop
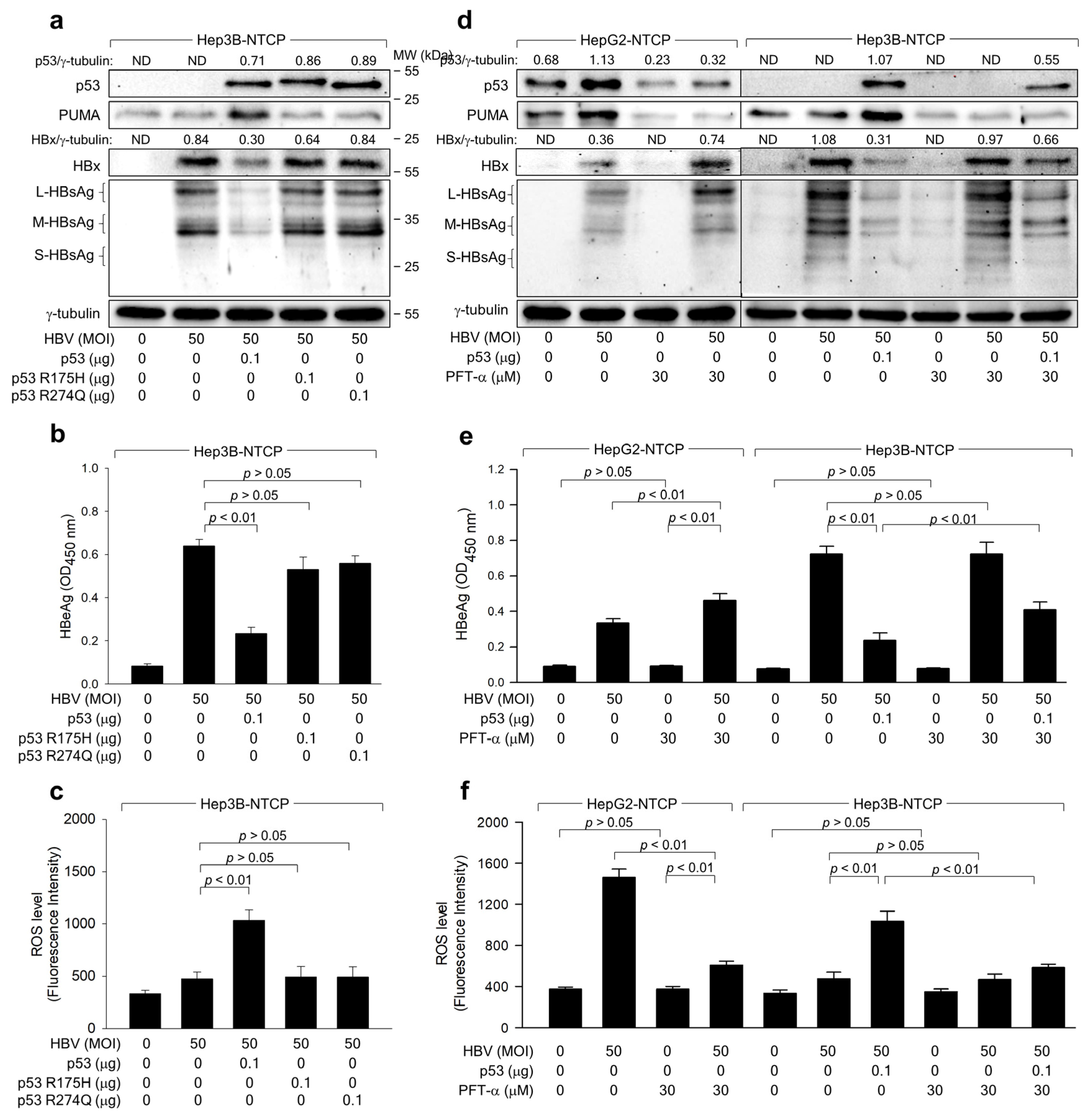
2.3. The Transcriptional Activity of p53 Is Crucial for the ROS-Induced Suppression of HBV Replication
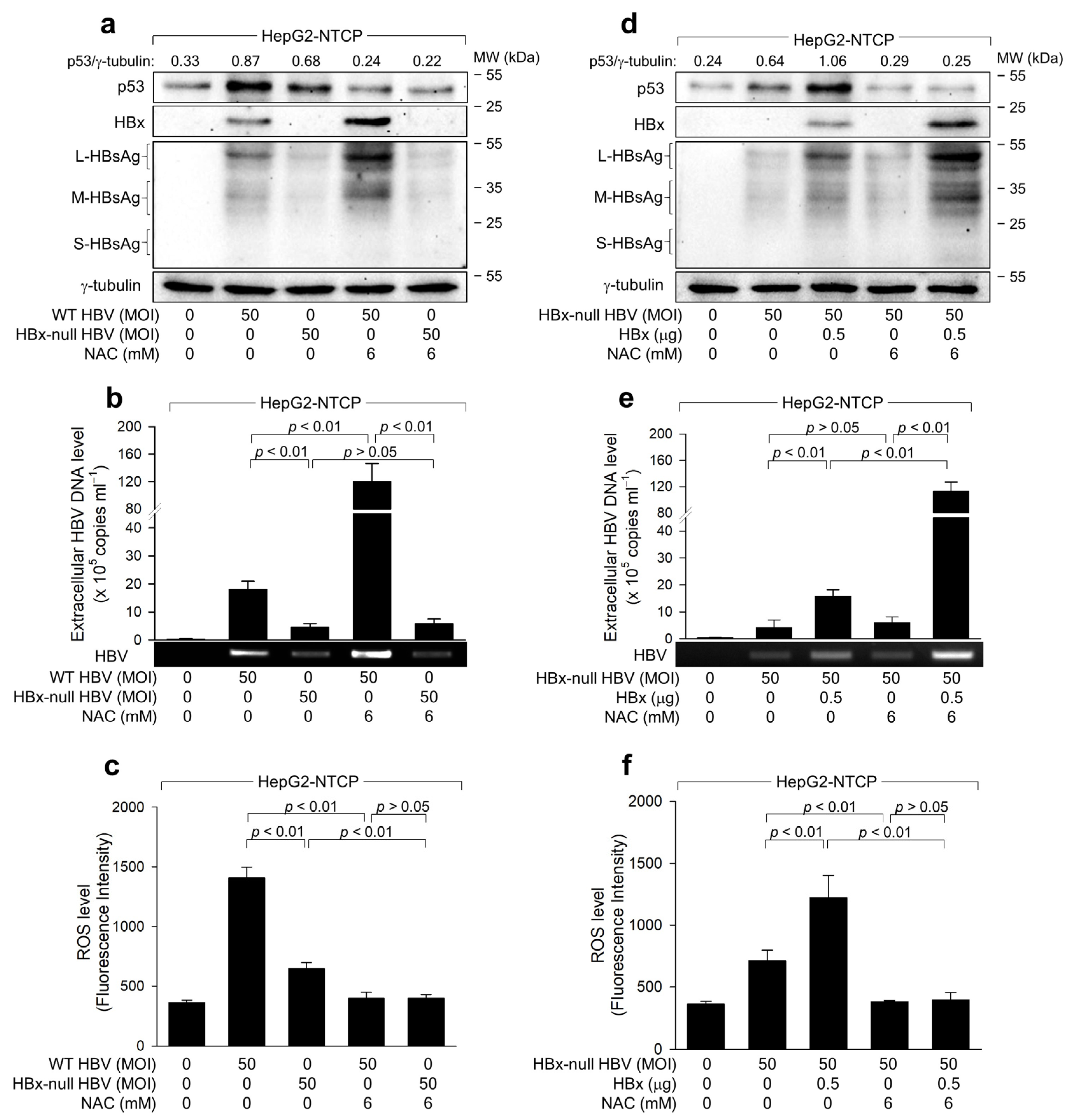
2.4. ROS Reduces HBx Levels to Suppress HBV Replication in Human Hepatoma Cells
2.5. ROS Drives Siah-1-Mediated Proteasomal Degradation of HBx in a p53-Reliant Manner
3. Discussion
4. Materials and Methods
4.1. Plasmid
4.2. Cell Culture and Transfection
4.3. HBV Cell Culture System
4.4. Quantitative Real-Time PCR of HBV DNA
4.5. Measurement of Intracellular ROS Levels
4.6. Western Blot Analysis
4.7. Immunoprecipitation
4.8. Quantification of Western Blot Images
4.9. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ott, J.J.; Stevens, G.A.; Groeger, J.; Wiersma, S.T. Global epidemiology of hepatitis B virus infection: New estimates of age-specific HBsAg seroprevalence and endemicity. Vaccine 2012, 30, 2212–2219. [Google Scholar] [CrossRef] [PubMed]
- Locarnini, S.; Zoulim, F. Molecular genetics of HBV infection. Antivir. Ther. 2010, 15 (Suppl. S3), 3–14. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Koh, S.S.; Lee, C.G. Hepatitis B Virus X Protein and Hepatocarcinogenesis. Int. J. Mol. Sci. 2016, 17, 940. [Google Scholar] [CrossRef] [PubMed]
- Levrero, M.; Zucman-Rossi, J. Mechanisms of HBV-induced hepatocellular carcinoma. J. Hepatol. 2016, 64 (Suppl. S1), S84–S101. [Google Scholar] [CrossRef] [PubMed]
- Tsuge, M.; Hiraga, N.; Akiyama, R.; Tanaka, S.; Matsushita, M.; Mitsui, F.; Abe, H.; Kitamura, S.; Hatakeyama, T.; Kimura, T.; et al. HBx protein is indispensable for development of viraemia in human hepatocyte chimeric mice. J. Gen. Virol. 2010, 91 Pt 7, 1854–1864. [Google Scholar] [CrossRef] [PubMed]
- Keasler, V.V.; Hodgson, A.J.; Madden, C.R.; Slagle, B.L. Enhancement of hepatitis B virus replication by the regulatory X protein in vitro and in vivo. J. Virol. 2007, 81, 2656–2662. [Google Scholar] [CrossRef] [PubMed]
- Keasler, V.V.; Hodgson, A.J.; Madden, C.R.; Slagle, B.L. Hepatitis B virus HBx protein localized to the nucleus restores HBx-deficient virus replication in HepG2 cells and in vivo in hydrodynamically-injected mice. Virology 2009, 390, 122–129. [Google Scholar] [CrossRef] [PubMed]
- Bouchard, M.J.; Wang, L.H.; Schneider, R.J. Calcium signaling by HBx protein in hepatitis B virus DNA replication. Science 2001, 294, 2376–2378. [Google Scholar] [CrossRef]
- Rawat, S.; Bouchard, M.J. The hepatitis B virus (HBV) HBx protein activates AKT to simultaneously regulate HBV replication and hepatocyte survival. J. Virol. 2015, 89, 999–1012. [Google Scholar] [CrossRef]
- Finkel, T. Oxidant signals and oxidative stress. Curr. Opin. Cell Biol. 2003, 15, 247–254. [Google Scholar] [CrossRef]
- Fujita, N.; Sugimoto, R.; Ma, N.; Tanaka, H.; Iwasa, M.; Kobayashi, Y.; Kawanishi, S.; Watanabe, S.; Kaito, M.; Takei, Y. Comparison of hepatic oxidative DNA damage in patients with chronic hepatitis B and C. J. Viral Hepat. 2008, 15, 498–507. [Google Scholar] [CrossRef]
- Bolukbas, C.; Bolukbas, F.F.; Horoz, M.; Aslan, M.; Celik, H.; Erel, O. Increased oxidative stress associated with the severity of the liver disease in various forms of hepatitis B virus infection. BMC Infect. Dis. 2005, 5, 95. [Google Scholar] [CrossRef]
- Ren, J.-H.; Chen, X.; Zhou, L.; Tao, N.-N.; Zhou, H.-Z.; Liu, B.; Li, W.-Y.; Huang, A.-L.; Chen, J. Protective Role of Sirtuin3 (SIRT3) in Oxidative Stress Mediated by Hepatitis B Virus X Protein Expression. PLoS ONE 2016, 11, e0150961. [Google Scholar] [CrossRef]
- Ivanov, A.V.; Valuev-Elliston, V.T.; Tyurina, D.A.; Ivanova, O.N.; Kochetkov, S.N.; Bartosch, B.; Isaguliants, M.G. Oxidative stress, a trigger of hepatitis C and B virus-induced liver carcinogenesis. Oncotarget 2017, 8, 3895–3932. [Google Scholar] [CrossRef] [PubMed]
- Rahmani, Z.; Huh, K.W.; Lasher, R.; Siddiqui, A. Hepatitis B virus X protein colocalizes to mitochondria with a human voltage-dependent anion channel, HVDAC3, and alters its transmembrane potential. J. Virol. 2000, 74, 2840–2846. [Google Scholar] [CrossRef] [PubMed]
- Clippinger, A.J.; Bouchard, M.J. Hepatitis B virus HBx protein localizes to mitochondria in primary rat hepatocytes and modulates mitochondrial membrane potential. J. Virol. 2008, 82, 6798–6811. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.I.; Hwang, J.M.; Im, J.H.; Lee, Y.I.; Kim, N.S.; Kim, D.G.; Yu, D.Y.; Moon, H.B.; Park, S.K. Human hepatitis B virus-X protein alters mitochondrial function and physiology in human liver cells. J. Biol. Chem. 2004, 279, 15460–15471. [Google Scholar] [CrossRef]
- Xie, H.; Xie, D.; Zhang, J.; Jin, W.; Li, Y.; Yao, J.; Pan, Z.; Xie, D. ROS/NF-kappaB Signaling Pathway-Mediated Transcriptional Activation of TRIM37 Promotes HBV-Associated Hepatic Fibrosis. Mol. Ther. Nucleic Acids 2020, 22, 114–123. [Google Scholar] [CrossRef]
- Foo, J.; Bellot, G.; Pervaiz, S.; Alonso, S. Mitochondria-mediated oxidative stress during viral infection. Trends Microbiol. 2022, 30, 679–692. [Google Scholar] [CrossRef]
- Yeom, S.; Kim, S.S.; Jeong, H.; Jang, K.L. Hepatitis B virus X protein activates E3 ubiquitin ligase Siah-1 to control virus propagation via a negative feedback loop. J. Gen. Virol. 2017, 98, 1774–1784. [Google Scholar] [CrossRef]
- Achanta, G.; Huang, P. Role of p53 in sensing oxidative DNA damage in response to reactive oxygen species-generating agents. Cancer Res. 2004, 64, 6233–6239. [Google Scholar] [CrossRef] [PubMed]
- Yoon, H.; Han, J.; Jang, K.L. Hepatitis B Virus X Protein Stimulates Hepatitis C Virus (HCV) Replication by Protecting HCV Core Protein from E6AP-Mediated Proteasomal Degradation. Microbiol. Spectr. 2022, 10, e0143222. [Google Scholar] [CrossRef] [PubMed]
- Michailidis, E.; Pabon, J.; Xiang, K.; Park, P.; Ramanan, V.; Hoffmann, H.H.; Schneider, W.M.; Bhatia, S.N.; de Jong, Y.P.; Shlomai, A.; et al. A robust cell culture system supporting the complete life cycle of hepatitis B virus. Sci. Rep. 2017, 7, 16616. [Google Scholar] [CrossRef] [PubMed]
- Qiu, G.H.; Xie, X.; Xu, F.; Shi, X.; Wang, Y.; Deng, L. Distinctive pharmacological differences between liver cancer cell lines HepG2 and Hep3B. Cytotechnology 2015, 67, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Li, W. The hepatitis B virus receptor. Annu. Rev. Cell Dev. Biol. 2015, 31, 125–147. [Google Scholar] [CrossRef] [PubMed]
- Lim, H.Y.; Han, J.; Yoon, H.; Jang, K.L. Tumor Suppressor p53 Inhibits Hepatitis B Virus Replication by Downregulating HBx via E6AP-Mediated Proteasomal Degradation in Human Hepatocellular Carcinoma Cell Lines. Viruses 2022, 14, 2313. [Google Scholar] [CrossRef] [PubMed]
- Cha, M.Y.; Ryu, D.K.; Jung, H.S.; Chang, H.E.; Ryu, W.S. Stimulation of hepatitis B virus genome replication by HBx is linked to both nuclear and cytoplasmic HBx expression. J. Gen. Virol. 2009, 90 Pt 4, 978–986. [Google Scholar] [CrossRef] [PubMed]
- Knowles, B.B.; Howe, C.C.; Aden, D.P. Human hepatocellular carcinoma cell lines secrete the major plasma proteins and hepatitis B surface antigen. Science 1980, 209, 497–499. [Google Scholar] [CrossRef]
- Joschko, N.M. The Role of the Tumour Suppressor p53 in the Elimination of Hepatitis B Virus-infected Hepatocytes-Implications for Viral Clearance. Ph.D. Thesis, Universität Heidelberg, Heidelberg, Germany, 2012. [Google Scholar]
- Oparka, M.; Walczak, J.; Malinska, D.; van Oppen, L.; Szczepanowska, J.; Koopman, W.J.H.; Wieckowski, M.R. Quantifying ROS levels using CM-H(2)DCFDA and HyPer. Methods 2016, 109, 3–11. [Google Scholar] [CrossRef]
- Higgs, M.R.; Chouteau, P.; Lerat, H. ‘Liver let die’: Oxidative DNA damage and hepatotropic viruses. J. Gen. Virol. 2014, 95 Pt 5, 991–1004. [Google Scholar] [CrossRef]
- Yoon, H.; Lee, H.K.; Jang, K.L. Hydrogen Peroxide Inhibits Hepatitis B Virus Replication by Downregulating HBx Levels via Siah-1-Mediated Proteasomal Degradation in Human Hepatoma Cells. Int. J. Mol. Sci. 2023, 24, 13354. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Han, Q.; Zhao, H.; Zhang, J. The Mechanisms of HBV-Induced Hepatocellular Carcinoma. J. Hepatocell. Carcinoma 2021, 8, 435–450. [Google Scholar] [CrossRef] [PubMed]
- Yeom, S.; Jeong, H.; Kim, S.S.; Jang, K.L. Hepatitis B virus X protein activates proteasomal activator 28 gamma expression via upregulation of p53 levels to stimulate virus replication. J. Gen. Virol. 2018, 99, 655–666. [Google Scholar] [CrossRef] [PubMed]
- Offutt, T.L.; Ieong, P.U.; Demir, Ö.; Amaro, R.E. Dynamics and Molecular Mechanisms of p53 Transcriptional Activation. Biochemistry 2018, 57, 6528–6537. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Singh, M.; Selivanova, G.; Peuget, S. Pifithrin-α alters p53 post-translational modifications pattern and differentially inhibits p53 target genes. Sci. Rep. 2020, 10, 1049. [Google Scholar] [CrossRef] [PubMed]
- Harris, S.L.; Levine, A.J. The p53 pathway: Positive and negative feedback loops. Oncogene 2005, 24, 2899–2908. [Google Scholar] [CrossRef]
- Lee, S.G.; Rho, H.M. Transcriptional repression of the human p53 gene by hepatitis B viral X protein. Oncogene 2000, 19, 468–471. [Google Scholar] [CrossRef]
- Zhao, J.; Wang, C.; Wang, J.; Yang, X.; Diao, N.; Li, Q.; Wang, W.; Xian, L.; Fang, Z.; Yu, L. E3 ubiquitin ligase Siah-1 facilitates poly-ubiquitylation and proteasomal degradation of the hepatitis B viral X protein. FEBS Lett. 2011, 585, 2943–2950. [Google Scholar] [CrossRef] [PubMed]
- Belikov, A.V.; Schraven, B.; Simeoni, L. T cells and reactive oxygen species. J. Biomed. Sci. 2015, 22, 85. [Google Scholar] [CrossRef]
- Tan, C.; Guo, H.; Zheng, M.; Chen, Y.; Huang, W. Involvement of mitochondrial permeability transition in hepatitis B virus replication. Virus Res. 2009, 145, 307–311. [Google Scholar] [CrossRef]
- Kim, S.; Lee, H.-S.; Ji, J.-H.; Cho, M.-Y.; Yoo, Y.-S.; Park, Y.-Y.; Cha, H.-J.; Lee, Y.; Kim, Y.; Cho, H. Hepatitis B virus X protein activates the ATM–Chk2 pathway and delays cell cycle progression. J. Gen. Virol. 2015, 96, 2242–2251. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.M.; Lee, S.Y.; Kim, B.J. Naturally Occurring Hepatitis B Virus Mutations Leading to Endoplasmic Reticulum Stress and Their Contribution to the Progression of Hepatocellular Carcinoma. Int. J. Mol. Sci. 2019, 20, 597. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Chen, Y.; St. Clair, D.K. ROS and p53: A versatile partnership. Free Radic. Biol. Med. 2008, 44, 1529–1535. [Google Scholar] [CrossRef] [PubMed]
- Liochev, S.I.; Fridovich, I. The effects of superoxide dismutase on H2O2 formation. Free Radic. Biol. Med. 2007, 42, 1465–1469. [Google Scholar] [CrossRef]
- Komarov, P.G.; Komarova, E.A.; Kondratov, R.V.; Christov-Tselkov, K.; Coon, J.S.; Chernov, M.V.; Gudkov, A.V. A chemical inhibitor of p53 that protects mice from the side effects of cancer therapy. Science 1999, 285, 1733–1737. [Google Scholar] [CrossRef] [PubMed]
- Culmsee, C.; Zhu, X.; Yu, Q.S.; Chan, S.L.; Camandola, S.; Guo, Z.; Greig, N.H.; Mattson, M.P. A synthetic inhibitor of p53 protects neurons against death induced by ischemic and excitotoxic insults, and amyloid beta-peptide. J. Neurochem. 2001, 77, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Jang, K.L. All-trans Retinoic Acid Inhibits Hepatitis B Virus Replication by Downregulating HBx Levels via Siah-1-Mediated Proteasomal Degradation. Viruses 2023, 15, 1456. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.W.; Yen, T.S. Negative regulation of hepatitis B virus gene expression and replication by oxidative stress. J. Biol. Chem. 1994, 269, 8857–8862. [Google Scholar] [CrossRef] [PubMed]
- Park, S.G.; Min, J.Y.; Chung, C.; Hsieh, A.; Jung, G. Tumor suppressor protein p53 induces degradation of the oncogenic protein HBx. Cancer Lett. 2009, 282, 229–237. [Google Scholar] [CrossRef]
- Matsuzawa, S.-I.; Takayama, S.; Froesch, B.A.; Zapata, J.M.; Reed, J.C. p53-inducible human homologue of Drosophila seven in absentia (Siah) inhibits cell growth: Suppression by BAG-1. EMBO J. 1998, 17, 2736–2747. [Google Scholar] [CrossRef]
- Quasdorff, M.; Protzer, U. Control of hepatitis B virus at the level of transcription. J. Viral Hepat. 2010, 17, 527–536. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Delgermaa, L.; Huang, F.; Oishi, N.; Liu, L.; He, F.; Zhao, L.; Murakami, S. The transcriptional transactivation function of HBx protein is important for its augmentation role in hepatitis B virus replication. J. Virol. 2005, 79, 5548–5556. [Google Scholar] [CrossRef] [PubMed]
- Kwun, H.J.; Jang, K.L. Natural variants of hepatitis B virus X protein have differential effects on the expression of cyclin-dependent kinase inhibitor p21 gene. Nucleic Acids Res. 2004, 32, 2202–2213. [Google Scholar] [CrossRef] [PubMed]
- Davarinejad, H. Quantifications of Western Blots with ImageJ; University of York: York, UK, 2015. [Google Scholar]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jeong, Y.; Han, J.; Jang, K.L. Reactive Oxygen Species Induction by Hepatitis B Virus: Implications for Viral Replication in p53-Positive Human Hepatoma Cells. Int. J. Mol. Sci. 2024, 25, 6606. https://doi.org/10.3390/ijms25126606
Jeong Y, Han J, Jang KL. Reactive Oxygen Species Induction by Hepatitis B Virus: Implications for Viral Replication in p53-Positive Human Hepatoma Cells. International Journal of Molecular Sciences. 2024; 25(12):6606. https://doi.org/10.3390/ijms25126606
Chicago/Turabian StyleJeong, Yuna, Jiwoo Han, and Kyung Lib Jang. 2024. "Reactive Oxygen Species Induction by Hepatitis B Virus: Implications for Viral Replication in p53-Positive Human Hepatoma Cells" International Journal of Molecular Sciences 25, no. 12: 6606. https://doi.org/10.3390/ijms25126606
APA StyleJeong, Y., Han, J., & Jang, K. L. (2024). Reactive Oxygen Species Induction by Hepatitis B Virus: Implications for Viral Replication in p53-Positive Human Hepatoma Cells. International Journal of Molecular Sciences, 25(12), 6606. https://doi.org/10.3390/ijms25126606