Abstract
Humans are continuously exposed to various heavy metals including copper, iron, cadmium, and arsenic, which were specifically selected for the current analysis because they are among the most frequently encountered environmental mankind and industrial pollutants potentially causing human health hazards and liver injury. So far, these issues were poorly assessed and remained a matter of debate, also due to inconsistent results. The aim of the actual report is to thoroughly analyze the positive as well as negative effects of these four heavy metals on human health. Copper and iron are correctly viewed as pollutant elements essential for maintaining human health because they are part of important enzymes and metabolic pathways. Healthy individuals are prepared through various genetically based mechanisms to maintain cellular copper and iron homeostasis, thereby circumventing or reducing hazardous liver and organ injury due to excessive amounts of these metals continuously entering the human body. In a few humans with gene aberration, however, liver and organ injury may develop because excessively accumulated copper can lead to Wilson disease and substantial iron deposition to hemochromatosis. At the molecular level, toxicities of some heavy metals are traced back to the Haber Weiss and Fenton reactions involving reactive oxygen species formed in the course of oxidative stress. On the other hand, cellular homeostasis for cadmium and arsenic cannot be provided, causing their life-long excessive deposition in the liver and other organs. Consequently, cadmium and arsenic represent health hazards leading to higher disability-adjusted life years and increased mortality rates due to cancer and non-cancer diseases. For unknown reasons, however, liver injury in humans exposed to cadmium and arsenic is rarely observed. In sum, copper and iron are good for the human health of most individuals except for those with Wilson disease or hemochromatosis at risk of liver injury through radical formation, while cadmium and arsenic lack any beneficial effects but rather are potentially hazardous to human health with a focus on increased disability potential and risk for cancer. Primary efforts should focus on reducing the industrial emission of hazardous heavy metals.
1. Introduction
Heavy metals like copper, iron, cadmium, and arsenic have been formed at various molecular levels from helium and hydrogen in the universe via nuclear fusion in stars during supernova explosions long before they arrived at our globe between ten and twenty billion years ago, likely and more specifically around 13.7 billion years [1,2,3,4,5]. In line with other heavy metals, the chemicals copper, iron, cadmium, and arsenic are defined as metallic elements according to their density of >5 g/cm3 or by considering their atomic weight of >20, but there still is ongoing uncertainty and discussion as to how best to define heavy metals and how toxicity may be incorporated in the definition [6]. In chemistry, arsenic may also be seen as a metalloid, an unprecise term for an element with characteristics between a typical metal and a typical non-metal [6,7]. Metals cannot be broken down and are non-biodegradable unless the organism detoxifies the metal ions by binding the active element with a protein or by depositing them in intracellular granules for long-term storage [6].
Arriving at the earth long predating human existence, heavy metals subsequently became part of the environment preceding and after human evolution. Additional exposure to heavy metals found increasingly in the ecosystem due to mankind and industry activities caused health concerns and led to considerations on how best to meet these ecology issues [6,7]. In addition to providing occupational safety through protecting workers of industry facilities releasing heavy metals, special attention was paid to re-naturize polluted soils by growing plants that take up these heavy metals [6]. However, this approach helped the soil to remove the heavy metals and reduced the local health risk, but at the same time it created the next problem of properly disposing of the contaminated plants. As a result, heavy metals continue to circulate in our ecosystem without the chance of final disposal.
As expected, some heavy metals may impair human health [6,7] and injure the liver as the central metabolic organ, which takes care of many swallowed potentially toxic substances and chemicals like heavy metals. They leave the intestinal tract via the portal system and flow through this organ [8,9]. The human liver has a critical role in maintaining somatic and cellular equilibrium and ensuring hepatic homeostasis necessary for healthy life. The focus of hepatic homeostasis is generally on metabolism [8,9,10,11,12], clearance [11], and immunity [12]. Through these processes, the liver cares for the intermediate metabolism of lipids, carbohydrates, and proteins [13], as well as for drug metabolism [14,15,16] and for the degradation of other exogenous chemicals [17,18,19,20,21,22,23]. Some of these metabolic pathways may be disturbed if the homeostasis of heavy metals is impaired.
Heavy metals are known for their specific environmental issues [24,25,26,27] and clinical challenges related to diseases of the liver [2,28]. More specifically, and in line with the U.S. Food and Drug Administration (US FDA), concern has been expressed that dietary exposure to heavy metals was recognized as a public health issue, focusing among others on arsenic and cadmium [29]. As a consequence, these two chemicals were among the four heavy metals that were intensively discussed in the current analysis. Collectively, various exogenous compounds like heavy metals are classified as risky injurious chemicals if entering the human body, where they may cause health hazards and liver injury that require special attention [2,26,27,28]. In trace amounts, a few heavy metals like chromium, copper, iron, manganese, molybdenum, selenium, and zinc are essential elements [7], initially required for human evolution on earth and subsequently needed to sustain human health. Even at trace levels, other heavy metals are diametrical to human health including to the liver of individuals, who for instance, cannot provide physiological homeostasis due to genetic reasons as evidenced by high amounts of these metals detected in their body.
This article provides a critical analysis and overview on the potential environmental and clinical risks posed by the three heavy metals copper, iron, and cadmium, as well as the metalloid arsenic. These four elements were chosen for this article because they are commonly found as contaminants in the environment and can easily enter the human body via the food chain. Clinical challenges in diagnosis and tentative mechanistic and molecular steps of the cascade of events will be discussed in detail.
2. Basic Considerations
In toxicology studies, the terms of toxicant and toxin are often used interchangeably despite differences in their origin [30,31]. More specifically, toxicants are any injurious substances, which are man-made, occurring in the nature, and found as contaminants in the air, soil, water, or food, whereby heavy metals commonly belong to this category of toxicants [30]. As opposed to this, and without reference to heavy metals, a toxin in a narrower sense is a natural poison classified as a secondary metabolite produced by living organisms themselves, but not just acquired from outside sources, and that typically is not harmful to the organisms themselves but can have an impact on human or animal health when consumed [1,2]. Common sources of such toxins include poisonous plants, fungi, algae, bacteria, and marine biotoxins, while the diversity of these biological systems presents challenges to analytical chemists and food safety implications [30]. In a broader sense, both forms of poisonous chemicals are often termed uniformly as toxins, used also in this article. Evaluating the possible role of heavy metals on human health risks including liver injury requires the consideration of several challenges. Some general aspects regarding heavy metals are additionally worth mentioning.
First, copper, iron, cadmium, and arsenic are commonly found in the environment together with other heavy metals or industrial toxins [7,24,25]. The combined occurrence makes causality assessment for one or the other culprits difficult and requires careful clinical analysis and extension to environments polluted by a single chemical like in the vicinity of a specific industry complex manufacturing products with a predominance of a single chemical.
Second, definitions of health and its associated parameter of disability-adjusted life years (DALYs) are often vague and differ among countries and regions. Respective results may not be applicable throughout the world.
Third, exact quantitative data on exposure to an assumed chemical culprit may hardly be obtainable. This is why in most publications respective results were not reported.
Fourth, a major hurdle is the robust determination of the non-toxic levels of the heavy metal under consideration including arsenic and cadmium [29]. In this report, terms such as daily maximum safe exposure, oral reference dose, provisional total daily intake, or minimal risk level were differently used among various agencies from different countries, and reported reference values substantially differ from one regulatory agency to another and from one country to another [29]. Published values were often adapted by reduction within a few years due to newly incoming data. Overall, there is not enough research to know at what heavy metal levels one should expect non-cancer or cancer health problems of any exposed organ or tissue system.
Fifth, human liver injury due to heavy metals exposure was often insufficiently defined. These challenges apply to all four metals at variable extents and different levels. As a result, to circumvent redundancy in the following chapters, key points ahead are discussed below.
2.1. Laboratory Criteria of Liver Injury
In clinical and ambulatory settings, abnormal liver tests (LTs), like slightly increased serum alanine aminotransferase ALT), aspartate aminotransferase (AST), and alkaline phosphatase (ALP) activities, are commonly found and mostly lacking any clinical relevance, viewed as liver adaptation or tolerance [32,33]. In analogy to drug-induced liver injury (DILI) and herb-induced liver injury (HILI), mainstream international consensus among experts exists that the diagnosis of the liver injury requires threshold values of at least two liver values with ALT or AST ≥ 5 times the upper limit of normal (ULN) and/or ALP ≥ 2 times the ULN, both enzymes are viewed as typical diagnostic parameters of liver injury [32,33,34,35,36,37]. On the other hand, total bilirubin as a parameter of liver function when conjugated bilirubin is predominant, is explicitly not part of the diagnostic liver injury algorithm [33] because unconjugated hyperbilirubinemia is not specific for liver injury because it can be seen in patients with the genetic Gilbert syndrome and has a frequency of up to 8% in the general population [38]. Cases with serum ALT or ALP activities below the thresholds are not considered as liver injuries that could carry a risk for patients but are categorized as liver adaptation or liver tolerance [39].
2.2. Types of Liver Injury
The determination of the individual liver injury pattern, also known as phenotype, caused by copper, iron, cadmium, and arsenic is actually derived from criteria previously established for hepatocellular injury and cholestatic or mixed liver injury due to potentially hepatotoxic drugs and herbs [32,33]. This can easily be achieved through the calculation of the ratio (R) by using the multiples of the upper limit of normal (ULN) for serum ALT divided by ALP. Thus, R ≥ 5 classifies the hepatocellular injury, R ≤ 2 the cholestatic injury, and 2 < R < 5 the mixed liver injury. As the determination of the liver injury pattern by the R value requires only the routinely available results of serum ALT and ALP activities, this approach saves financial resources, does not require an invasive and risky liver biopsy, and is helpful for the description of clinical DILI features.
In general, experimental liver injury by intoxicating exogenous heavy metals is dose dependent, and predictable. Respective mechanistic studies in diseased individuals are scarce because the liver is a secret-keeping organ not really accessible for direct analysis. In addition, parameters in the blood that could reflect pathogenetic processes in the liver are largely missing, especially since there is a lack of circulating humoral immune markers like proinflammatory cytokines, including IL-2, IL-4, IL-6, IL-10, TNF-α, and IFN-ɣ in cases of clinical liver injury due to heavy metals [25], which are valuable data like in cases of idiosyncratic DILI [40].
Depending on the type of the heavy metal, the liver injury may be variable and more specific. If applicable, this is outlined in detail for individual heavy metals as described under the respective chapter. Injury specifics mostly prevail in patients with heavy metals stored in their liver like in Wilson disease or hemochromatosis.
2.3. Diagnostic Approaches and Causality Assessment
In each patient with suspected liver injury due to copper, iron, cadmium, and arsenic, a careful clinical evaluation including past medical history, data on exposure to the heavy metal, and the prospective collection of case details including complete laboratory results is essential (Figure 1).
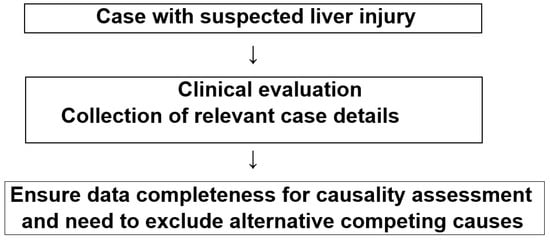
Figure 1.
Flow chart with checklist of differential diagnoses in cases of suspected liver injury due to copper, iron, cadmium, and arsenic.
2.4. Exclusion of Alternative Causes
In any patient with suspected liver injury by toxins like copper, iron, cadmium, and arsenic, although with regrets rarely done, a firm diagnosis of the heavy metal implicated in the liver injury is needed for an adequate evaluation of the case. Increased LTs signify the presence of a liver injury but do not give any information of its cause, because they are found in many other hepatic and extrahepatic as well as systemic diseases. Thus, the exclusion of competing causes is essential (Table 1).

Table 1.
Differential diagnoses of toxic liver injury.
Under ideal clinical conditions, the updated Roussel Uclaf Causality Assessment Method (RUCAM) should be used to determine causality gradings based on a final RUCAM score: ≤0, excluded causality; 1–2, unlikely; 3–5, possible; 6–8, probable; and ≥9, highly probable [33]. RUCAM was inaugurated to help establish the difficult diagnosis of DILI by a sophisticated diagnostic algorithm considering key elements of DILI that had to be scored [32], an approach in line with principles of artificial intelligence (AI) [41]. So far, RUCAM was applied for drugs causing human liver injury in 81,856 cases of DILI published 1993 until mid-2020 [42] and in a single report on intoxication with mercury and drugs [25,43] but not for any other heavy metal [25].
2.5. Heavy Metals in Plants and Horizontal Transfer
Heavy metals enter the human body via inhalation, skin, and the gastrointestinal tract through both the solid food chain and drinking water [6,44]. With respect to the food chain, contaminated plants are only one category among many sources. Such plants may be used as vegetables or as feeding material for grazing cattle. Plants grown on soils contaminated with heavy metals are a neglected topic but of potential clinical relevance that needs further attention in the future [23], including their use in herbal medicine [24,45,46,47,48,49]. Plants receive heavy metals from contaminated soil via their roots or rhizomes in a similar way to that described for pyrrolizidine alkaloids (PAs), through a process termed horizontal transfer system (Figure 2) [45].
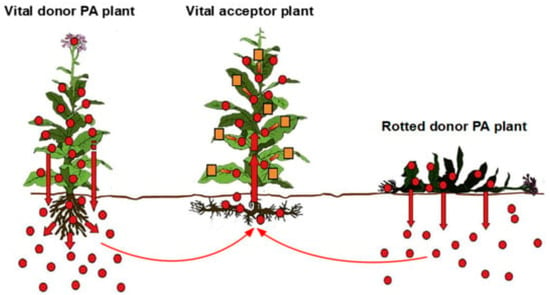
Figure 2.
Horizontal transfer system of chemicals in plants, shown here for pyrrolizidine alkaloids as an example for heavy metals. In this analogy, any vital acceptor plant can uptake heavy metals from polluted soil and potentially serve as a vegetable for food or grain for bread. Derived from a previous open access report [45]. Abbreviation: PA, pyrrolizidine alkaloid. This figure was modified and adapted from previous illustrations and suggestions of the group of Selmar [46,47].
Special care should be addressed to patients under a therapy with Ayurvedic medicine, commonly consisting of several herbs that may be contaminated with arsenic [50]. Contamination is viewed as the intentional adulteration of the medicinal herbs, having been grown on soil previously fortified with arsenic in the erroneous belief that it enhances therapeutic efficacy. Apart from lacking proven efficacy, problems emerge if liver injury is detected, attributable to arsenic or the herbal mixture.
2.6. Issue of Experimental Studies
Mechanistic steps leading to liver injury have largely been investigated in animal models using excessive amounts of the heavy metals, but this approach neglects genetic basics, such as in patients with a hereditary hepatic accumulation of heavy metals, and is therefore less suitable for this special clinical study cohort and in line with considerations on other human liver diseases [51].
3. Copper
Copper (Cu, from the Latin: cuprum) is a typical heavy metal environmentally found on earth [2,52,53]. It is required as a trace element for cellular homeostasis in the human liver and various other organs. This metal is also essential for other living organisms such as animals and plants. In humans, copper levels in the physiologic range are beneficial, but lower or higher amounts commonly create health problems, with the exemption of higher amounts used as a rare therapy for patients with cancer.
3.1. Physiology
Copper enters the body via the intestinal tract, facilitated by a transporter protein in the mucosa cells of the small intestine, known as copper membrane transporter 1 (Ctr1; SLC31A1) [2], that is also important for copper homeostasis in the liver. This intestinal transporter helps carry copper inside the intestinal cells where part of the copper is bound to metallothionein, and part is carried by the antioxidant protein 1 (ATOX1) to an intestinal organelle system known as the trans-Golgi network (TGN). In the case of rising copper levels within intestinal cells, an intestinal protein called ATP7A releases copper into the portal vein to the liver. Liver cells carry the CMT1 protein and metallothionein, and then hepatic ATOX1 binds the copper inside the hepatocytes. Once here, ATP7B links the copper to the hepatocellular TGN, from which the copper is incorporated in cytoplasmic vesicles near the cell periphery to be released into the bloodstream and the bile, thereby removing the excess copper [2]. Apart from the liver, injurious effects of copper in high concentrations may be helpful in future cancer therapy due to a process called cuproptosis, because a correlation was found between cancer angiogenesis, proliferation, growth, and metastasis and the accumulation of copper ions [54,55,56].
Available in the oxidized state as Cu(II) or reduced state as Cu(I), the heavy metal helps play a significant role in cellular physiology as a catalytic cofactor in cell redox systems of enzymes [53]. Copper is needed by the body, functioning as a cofactor for enzymes such as ceruloplasmin, cytochrome c oxidase, dopamine beta-hydroxylase, superoxide dismutase, and tyrosinase [2]. In addition, copper participates in the hepatic mitochondrial respiratory chain and intestinal absorption of iron as another heavy metal [53].
The average daily intake of copper by human adults assessed via a process of balance studies should be regarded with caution because of uncertainties regarding copper concentration in various foods and water as well as concern as a result of the homeostatic mechanisms controlling copper absorption and excretion [57], providing sufficient amounts of copper for blood [58] and liver [59]. Although the beneficial effects of copper as an essential trace element are well known, there is uncertainty with respect to copper reference values for humans due to different recommendations by various national authorities [57]. However, some proposals may be summarized for copper reference ranges as follows [58,59]: blood free serum copper: 1.6–2.4 μmol/L or 10–15 μg/dL [59]; blood total copper: 10–22 μmol/L or 63.7–140.12 μg/dL [58]; serum ceruloplasmin: 2.83–5.50 μmol/L or 18–35 μg/dL [59]; total 24 h urine copper 0.3–0.8 μmol or 20–50 μg [59]; and liver copper 0.3–0.8 μmol/g of tissue or 20–50 μg/g of tissue [59]. On a quantitative basis it has been assumed that normal urinary copper excretion/reabsorption is <70 µg/d and fecal copper output accounts for 1–2 mg/d, originating from the bile [60].
The determination of copper levels can assist in diagnosing several disease processes. These conditions may be monitored by looking at the total copper, the free serum copper, 24 h urine copper, and liver biopsy copper concentrations. Serum ceruloplasmin is also a valuable test and can be used to determine the free serum copper [57,58,59].
3.2. Copper as Pollutant and the Issue of Health Hazards
3.2.1. Sources of Copper as an Environmental Pollutant
Copper is commonly found in the environment [25], such as in and around solid waste fills [23], soil [23,61,62], drinking water [63], and the atmosphere [61]. As an antifungal chemical, copper is widely used in agriculture, explaining its detection in edible plants [64] and in plants used to prepare herbal medicines [24,49]. A prerequisite for the contamination of plants with copper is that they were cultivated on contaminated grounds and had access to contaminated water [64], facilitating copper acquisition by the plants via horizontal copper transfer similar to the uptake of pyrrolizidine by plants (Figure 3). The long-distance transport of the plants may cause external contamination of the plants [64], based on the observation that urban vehicle traffic causes contamination with copper and other heavy metals in road sweeping waste and bottom sediments of retention tanks [65].
Similarly, and most importantly in the clinical context of Wilson disease, products containing high amounts of copper like dark chocolate and cocoa should not be consumed by patients with Wilson disease [66]. This is in line with early observations that chocolate contains copper in amounts up to 16.50 ± 1.29 µg/g of a chocolate with 85% cocoa, and is associated with a linear correlation between the copper content of the chocolate and its cocoa content and a correlation coefficient of 0.89, showing that the cocoa largely contributed to the copper in the chocolate [67]. Raw cocoa beans from plantations in Nigeria had copper contents ranging from 104 µg/g to 642 µg/g, attributed to the use of copper sulfate as a fungicide for disease prevention on these plantations and detected in soil and vegetation components [68]. It seems plausible that the high copper levels detected in cocoa [68] (Owena cocoa, Theobroma cacao L.) are due to direct contact with the fungicide or via uptake from the soil by horizontal transfer similar to plant pyrrolizidine alkaloids (Figure 2).
Copper is used in agriculture [69,70], with preference in viticulture to protect grapes from downy mildew [70] and with copper detected in agricultural land focusing on soil surrounding ponds [62]. Industries like copper smelters, iron and steel production, and municipal incinerators contribute to the pollution by copper [61]. Overall environmental copper pollution is well documented using the excellent attic dust approach, whereby dust collected from the attic of old houses is examined, providing an archive of historical air contamination by copper in the urban environment [71]. The observation of air contamination by copper and other heavy metals in the urban environment raised the question of potential health hazards [71], and as expected, there were similar discussions around this issue worldwide.
3.2.2. Elucidating Health Hazards of Environmental Copper
While the existence of environmental copper pollution is well established, there is uncertainty about its effect on human health conditions because of divergent definitions of human health [72,73]. Classical medical research is disease focused and still defines health as the absence of disease [72], while languages associate a positive concept of wholeness with health as does the WHO health definition [73]. Newer medical health definitions emphasize the capacity to adapt to changing external and internal circumstances, and the results of the 2010 Global Burden of Disease study provides keys for quantifiable health metrics by developing statistical tools calculating healthy life expectancy as the sole defined end point [72]. As it currently stands, human health definitions remain vague because scored elements for a quantitative and qualitative evaluation outside of healthy life expectancy are not available.
Elucidating the environmental impact risk of copper on human health, including clinical liver injury, requires a careful and critical analysis of published proposals. Selected sources of relevant copper environmental pollution can be localized to several areas of interest and are briefly described, including critical comments (Table 2) [6,23,24,29,44,48,57,61,62,65,68,69,70,71,72,73,74,75,76,77,78,79,80,81,82,83,84,85,86,87,88,89,90,91].
Table 2.
Copper sources from the natural environment and industry, with proposed impact on human health.
It is interesting to see that the majority of the listed publications already have in the title wordings such as an assessment of health risks, environmental health hazards, or ecological health risk assessment (Table 2). In addition, the relationship of the potentially toxic copper with human health risks seems to be complex (Table 2). It is obvious that copper can be found as a contaminant in many places in the earth’s environment (Table 2).
Copper may enter the human body via gastrointestinal, pulmonal, or dermal uptake. The variable copper amounts leaving the soil or water contaminated with copper and finally arriving at exposed individuals could, theoretically, impair human health at various grades, but this indeed is not the case. In the clinical context, the amounts of copper derived from the environment and entering the human organs seem to be low, raising the question of their impact even at higher amounts on human health and the liver integrity. As expected, and considering the conclusions provided in all publications as listed (Table 2), firm evidence is lacking that contaminating copper may be injurious. Instead, contaminating copper is the best source supplying humans with this essential metal; it is natural and thereby free of costs and preferred to expensive commercial copper-containing supplements.
The mainstream opinion exists among theoretical toxicologists and clinical physicians that, in healthy individuals, perfect conditions of fine-tuned biological systems and mechanisms are available to ensure overall copper homeostasis not only within the liver but also in other organs [2,59]. Collectively speaking, if the uptake of copper is higher than the internal copper demand, excess copper will be excreted via bile or urine to sustain or obtain copper homeostasis, allowing for a healthy life [2,53,54,55,56,57,59]. These conditions apply on a day-by-day basis when humans are exposed to food, water, and air contaminated with copper, but they ultimately represent no health or liver risks that would need further fruitless time- and resource-consuming analytical efforts.
The discussion of a possible health risk by contaminating copper now seemingly comes to a good end: not guilty. However, a cautionary statement is warranted if protective mechanisms providing copper homeostasis are not available in the few patients who suffer from genetic copper-storing diseases such as Wilson disease and Menke disease, or those experiencing intoxications by excess amounts of copper [92,93,94,95,96].
3.3. Acute Human Liver Injury by Exogenous Copper
Reports are lacking a description of acute liver injury in connection with exposure to environmental copper [25]. Acute copper poisoning is more often observed in South Asian countries where it is more prevalent in rural populations [92], while uncommon in Western countries [93]. Copper sulfate is easily available worldwide, and used in agriculture, the leather industry, and at home to make glue [92]. In addition, the burning of copper sulfate at home or in shops, seen as a good luck charm and also used for religious reasons, is a widespread practice in countries like India where Buddhists and Hindus live [92,94]. Copper intoxications have been reported worldwide following accidental or intentional exposure through various routes including oral [92,93,95,96] and inhalation [61]. Toxic amounts occur after the ingestion of as little as 1 g [93], while the lethal dose of ingested copper is 10 to 20 g [94]. About 60% of the ingested dose is absorbed in the gastrointestinal tract and attached mostly to ceruloplasmin (95%), whereas free copper binds to albumin, forming its toxic form [94,95,96,97].
The clinical manifestation of acute copper intoxication in one patient was substantial liver injury, with a serum AST of 2340 U/L and ALT of 780 U/L and a ratio of AST/ALT of 3; there were no ALP results with which to determine the R value to determine the liver injury pattern [93]. In another patient, serum ALT activity was 10 U/L with ALP activity of 406 U/L [94]. This was in line with a cholestatic liver injury as based on laboratory data [33]. Acute liver failure can develop due to direct copper toxicity [94,95,96,97]. In line with expectations, mechanistic steps leading to human liver injury due to acute intoxications by copper were not studied in acutely intoxicated patients [93,94,95,96,97]. However, it has been noted that copper toxicity is due to the generation of free oxygen radicals in the cells, causing severe intra-nuclear and cytoplasmic injury and cell death [96].
Clinical characteristics may include jaundice due to non-immune hemolysis as evidenced by a low blood hemoglobin level of 6.9 g/dL, low serum haptoglobin of <10 mg/dL, high serum activity of lactate dehydrogenase of 2148 U/L, increased serum unconjugated bilirubin of 4.5 mg/dL, and negative Coombs test [93], which classifies the hemolysis similar to the known hemolysis of genetic Wilson disease with a negative Coombs test [66], as opposed to autoimmune hemolysis with a positive Coombs test [98]. Copper can also lead to methemoglobinemia, in which the diagnosis of acute copper intoxication is confirmed by a total serum copper of 874 µg/dL and urine copper of 356 µg/24 h [93]. Intravascular hemolysis commonly starts as early as within the first 24 h of ingestion and is due to the direct oxidative damage to erythrocyte membranes [94]. The Cu2+ ion oxidizes the Fe2+ ion in hemoglobin to Fe3+ resulting in its conversion to methemoglobin [94,96]. This manifests as cyanosis and a loss of the oxygen carrying capacity of blood [94]. Copper causes direct injury to the proximal renal tubules and acute tubular necrosis with increased serum values of creatinine, with a maximum of 621 µmol/L as a sign of renal impairment, attributable also to dehydration, hemoglobinuria, rhabdomyolysis, and sepsis [94].
Evidence-based recommendations for the treatment of acute oral intoxications by copper are not available. This is due to the small numbers of affected patients that do not allow for valid randomized controlled trials (RCTs) evaluating efficacy and risks of therapeutic approaches. As a result, therapeutic measures are currently based on experiences as described in anecdotal reports. Gastric lavage is viewed as usually unnecessary due to persistent vomiting [93]. Particular care is certainly needed for spontaneous vomiting due to the risk of aspiration, especially in unconscious patients, who may require preventive intubation analogous to acute oral intoxications by aliphatic halogenated hydrocarbons like carbon tetrachloride or chloroform [20,21,22]. There is no place for intentional forced vomiting, and the use of activated charcoal was anecdotally reported despite a lack of efficacy [93]. The drinking of milk and physiological saline was occasionally recommended [94] but a better option may be the intestinal lavage to quickly remove the copper from the intestine, a procedure successfully applied in patients intoxicated by the ingestion of aliphatic halogenated hydrocarbons [20,21,22]. Chelating agents like D-penicillamine are commonly applied and recommended in severe poisoning, although pharmacokinetic data are scarce to guide their use [93]. Zinc occasionally was used [99]. The hemolysis-induced anemia is to be corrected by a transfusion of packed red blood cells [94,100], and hemodialysis may be indicated for renal insufficiency [94,100]. Renal failure will require hemodialysis to temporarily substitute the injured non-functional kidneys without the intention to remove the toxic copper from the blood [94]. Of note, some reports claim that through contaminated hemodialysis fluid infusions, the copper may be freed from the dialysis device and given to the dialyzed patients [92].
Prognosis is poor unless patients receive quick treatment with chelation and supportive measures in the face of lethality rates from 14% to 36% [92]. There is no supporting evidence that acute copper ingestion increases the risk of hepatocellular carcinoma. Most importantly, stopping the over-the-counter sale of copper sulfate and restricting the sale to authorized agents is strongly recommended to decrease the risk of acute copper intoxication, as is providing copper sulfate as large crystals, rather than as a fine powder easily dissolved [92,94].
3.4. Chronic Human Liver Injury by Exogenous Copper
In a patient with prolonged exogenous copper exposure for 10 years, the serum activity of ALT was 51 U/L, associated with an AST of 190 U/L and ALP of 68 U/L, but the absence of ULN data prevented a calculation of the R value [79]. The patient used up to 8 mg of copper as a daily dose for treating copper deficiency, known as human Swayback disease, whereby this iatrogenic copper overload led to liver transplantation due to compensated cirrhosis. LT values have not been reported in patients with acute renal failure following copper sulfate intoxication [101]. Similarly, no ALT or AST values are available as they were not analyzed in copper workers.
In the patient with prolonged copper overdose, transjugular liver biopsy demonstrated, upon light microscopy, ongoing portal and segmental inflammation with a ballooning degeneration of the hepatocytes as well as diffuse hepatocellular copper accumulation on rhodamine [99]. The diagnosis of cirrhosis was first established at the occasion of a laparotomy for umbilical hernia repair as evidenced by macroscopical morphology and later confirmed at the time of liver transplantation, showing both micronodules and macronodules of the liver surface. An analysis of the hepatic content of copper provided extremely high values, suggesting copper as the causative agent in this patient [99]. Otherwise, convincing data of a direct mechanistic cascade of events leading to this liver injury due to a prolonged intake of exogenous copper are lacking but likely are similar to those described for Wilson disease, which is caused by genetic modifications, conditions that do not apply to the other forms of prolonged copper uptake. As a consequence, Wilson disease mimics chronic intoxication by copper only partially; the similarity is restricted to the liver injury and does not include the non-hepatic organs that are affected in Wilson disease due to local genetically impaired actions, circumstances that do not apply to non-Wilson diseases, such as in those chronically exposed to copper.
Experimental studies in animals showed mostly unchanged, or in rare cases, slightly increased serum ALT and AST activities after the application of high copper amounts [102,103,104]. In animals, however, overdosed copper administration caused no liver injury as assessed by light microscopy [102,103,105] or only minimal, partially dose-dependent changes such as small vacuoles of hepatocytes, hepatocyte swelling, inflammatory cells, or sinusoidal congestion [104]. Electron microscopy data on the liver of the patient exposed to high amounts of copper were not available [99,101] but have been reported in animal studies as irregularly shaped nuclei, abundant mitochondria, and displayed cristae, and hepatocytes with an inclusion of secondary lysosomes [82]. Prolonged intoxication with exogenous copper has to be differentiated clinically from Wilson disease [66,105,106], a genetic disorder of the liver leading to hepatic copper accumulation [107,108].
The earlier termed Indian childhood cirrhosis (ICC) was attributed, in previous reports, preferentially to exogenous copper in drinking water or milk [109], which was obviously an assumption error [63,110,111,112]. In more detail, ICC was initially considered preventable and to be clearly distinguishable in Indian children from other chronic liver disorders including Wilson disease [109]. Grossly increased hepatic, urinary, and serum copper concentrations were described as characteristic findings of ICC. These increased concentrations were easily demonstrated histologically with orcein-rhodamine staining. The environmental ingestion of copper was the most plausible explanation for ICC, as shown by feeding histories, the prevention of ICC in siblings and in the Pune district by a change in feeding vessels, and the dramatic reduction in the incidence of ICC throughout India. The nature and role of a second factor in the causation of ICC remained unclear, although an inherited defect in copper metabolism was strongly suspected. ICC, however, was not considered to be a straightforward early onset of Wilson disease because ceruloplasmin was consistently normal and clinical and histologic recovery was maintained in the long term despite the withdrawal of D-penicillamine therapy. Descriptions of an ICC-like illness in the West suggested that different mechanisms including environmental and/or genetic factors can lead to the same end-stage liver disease: copper-associated childhood cirrhosis [111,113,114]. The early conclusion was reached that ICC probably represents a specific form of copper-associated childhood cirrhosis that requires high environmental copper ingestion for its full expression [109].
The initial concept of ICC [109] was challenged by subsequent studies [111,113]. While there is agreement that liver diseases of infancy and childhood are generally rare and within the spectrum of these disorders [109,113], only a few subtypes are related to abnormal hepatic copper accumulation [113]. In particular, idiopathic copper toxicosis has now been defined as such a subtype, characterized by distinct clinical and pathologic features; however, its exact etiology is still controversial [113]. Based on a review of the literature and observations of 138 cases endemic to western Austria, the hypothesis was presented that idiopathic copper toxicosis is caused by a constructive interaction of an autosomal-recessive inherited defect in copper metabolism and excess dietary copper [113], classified before as an ecogenetic disorder [112]. In line with these considerations is a case report from Germany on a female child of non-consanguineous, healthy German parents who fell ill at the age of 7 months with a progressive liver disease leading to irreversible hepatic failure 3 months later [111]. Histological examination revealed severe liver cell necrosis, excessive Mallory body formation and veno-occlusive-like changes associated with a massive storage of copper, similar to ICC. Chronic copper contamination of drinking water was the only detectable etiological factor. The conclusion is reached that ICC most probably is an environmental disease, also occurring outside the Indian subcontinent, and is likely to be underdiagnosed in the Western world [111].
This severe form of rapidly progressive cirrhosis associated with a marked increase in hepatic copper has been described in children from rural, middle class Hindu families in India [110,113]. Originally termed Indian childhood cirrhosis, similar clinical cases have been reported worldwide, and this disorder is now referred to as idiopathic childhood cirrhosis or idiopathic copper toxicosis [110,111,113,114,115]. Affected children are diagnosed by two years of age with hepatosplenomegaly, an elevation in the activity of serum aminotransferases, cirrhosis, and elevated liver copper [110]. Interestingly, the serum ceruloplasmin in these patients is normal or elevated, suggesting that the defect in biliary copper excretion is distal to the role of ATP7B in this process. Epidemiological investigations of idiopathic childhood cirrhosis indicate that both genetic and environmental factors may play a role in this disease [110]. Studies have revealed an increased copper content in the diet consumed by the affected children while an analysis of some families suggests autosomal recessive inheritance with incomplete penetrance [115]. In support of an underlying defect in hepatic copper excretion, D-penicillamine is effective in many cases and hepatic transplantation can be curative [110]. Overall, this disease, now called idiopathic childhood cirrhosis syn idiopathic copper toxicosis, is quite different from Wilson disease, traced back solely to genetic variability rather than to additional increased copper consumption through food or beverages.
3.5. Wilson Disease
3.5.1. Natural Course
As a copper-storing liver disease caused by inheritable malfunctioning or missing ATP7B, Wilson disease is characterized by a disturbed cellular homeostasis of copper handling primarily in the liver cell [106]. Released from the liver cells due to limited storage capacity, the toxic copper enters the circulation and arrives at other organs, causing local accumulation and cell injury. This explains why copper injures not only the liver but is also responsible for liver-unrelated symptoms and various clinical features.
3.5.2. Clinical Characteristics
Wilson disease is a multifaceted disorder, difficult to diagnose and often misdiagnosed [106], in part due to physicians’ limited knowledge of its clinical features and a low prevalence, ranging from 1:40,000 to 1:50,000 in the general population [116]. In 22.5% of patients, the diagnosis was delayed and was not achieved until three years after the initial symptoms were evident, and the diagnosis was established at a mean age of 20.4 years (SD 10.6, range 4–56) [117]. However, Wilson disease has been described even in infants with an age range from a few months up to 4 years [118].
Apart from the liver, the most implicated organs were the brain, kidneys, eyes, heart, muscles, and bones [106]. Depending on the organ involved and the clinical stage of the disease, patients with Wilson disease may be polysymptomatic, oligosymptomatic, or monosymptomatic, but some patients may present even as asymptomatic, especially in initial stages of the disease.
Details of clinical manifestations were perfectly and comprehensively listed in [106]: (1) findings related to the liver may include abnormal LTs, chronic active hepatitis, cirrhosis with portal hypertension, and acute liver failure; (2) psychiatric features comprise affect, cognition, and behavioral disorders as well as depression and psychosis; (3) neurologically, tremor, dysarthria, ataxia, nystagmus, writing problems, and dysphagia with pseudohypersalivation prevail; (4) renal tubular dysfunction; (5) Kayser–Fleischer corneal rings to be verified through split-beam examination by an eye specialist; and (6) various findings like cardiomyopathy, cardiac arrhythmias, rhabdomyolysis, osteoporosis, osteomalacia, arthritis, and arthralgia. In addition, Coombs-negative hemolytic anemia is a key feature of Wilson disease with undetectable serum haptoglobin, high serum activities of lactate dehydrogenase, and high reticulocyte counts [119,120].
3.5.3. Diagnostic Approach
In any patient with suspected Wilson disease, first and second-degree relatives need to be screened for Wilson disease [2,121]. Such family screening facilitated the early diagnosis of Wilson disease, and substantially increased the prevalence, as the mean age of patients was significantly lower compared with patients diagnosed at a symptomatic stage of the disease (15.5 vs. 20.4 years; p = 0.021) [117].
A mutation analysis of the coding region of ATP7B (except exons two, three, and twenty-one) performed in 150 patients with Wilson disease showed no detectable mutations in 15% of patients, and mutations causing disease were found in 57% of patients on both chromosomes and in 29% of patients on one chromosome [122]. There were no significant differences in the frequency of pathological laboratory test values between the two study cohorts with detectable mutations. Thus, a negative ATP7B does not rule out Wilson disease, and the conclusion may be drawn that Wilson disease can develop without an association with ATP7B. Nevertheless, the search for ATP7B mutation is obligatory for a complete assessment in each patient with suspected Wilson disease [106] but certainly restricted in countries lacking test availability or confronted with high costs.
There is not a single clinical feature viewed as key diagnostic sign that would help with the early recognition of Wilson disease [106,117]. It should be suspected preferentially in younger patients with jaundice as a sign of liver disease or hemolysis, or if psychiatric or neurologic symptoms are evident [106,117]. In infants before the age of three years Wilson disease is uncommon [60]. As expected, liver injury as one of the key features in Wilson disease received much attention in various publications [115,116,117,118,119,120,121,122]. Among these, liver manifestations were recently summarized [122]. Accordingly, patients may be asymptomatic, especially in the early disease stage, whereas symptoms may start with fatigue, reduced appetite, and eventually jaundice. With the development of jaundice, patients present to their practitioner, who may establish or even miss the diagnosis of Wilson disease. Provided hemolysis is excluded, jaundice is commonly due to acute flares of any hepatobiliary disease including Wilson disease. In Wilson disease, jaundice may reflect acute liver injury with a sharp elevation of LTs. Later stages may include hepatomegaly as well as fibrotic or cirrhotic changes of the liver, as evidenced by portal hypertension with splenomegaly, ascites or bleeding esophageal varices as signs of decompensated cirrhosis [122]. In addition, 3–5% of the Wilson patients present with acute liver failure requiring urgent liver transplantation [122].
Laboratory results of serum LTs are variable and not helpful to suspect the diagnosis of Wilson disease. Serum ALP activities were described with low levels for unknown reasons [2,123,124]. In Wilson disease serum activities of AST are often much higher as compared with ALT [119,123]. High serum ALT activities up to 800 U/L were commonly found only in younger children with an age range from 4 to 8 years, with low ALT activities in infants with an age <4 years partially due to hemolytic crisis, and higher ALT values in 3 other infants [118]. Consequently, a realistic R value to determine the liver injury pattern based on LT values is not feasible, as also seen in a recent study providing serum activities for ALT of 46.9 ± 33.8 U/L and for ALP of 158.0 ± 119.4 U/L but without giving ranges of normal [124].
Moreover, increased plasma levels of inflammatory cytokines and chemokines were found in patients with Wilson disease as compared with healthy controls, an interesting finding but not contributing as a diagnostic biomarker [125]. Serum autoimmune parameter including anti-nuclear antibodies (ANA) were rarely reported at the start of the therapy, with higher frequencies during treatment with D-penicillamine [126]. Other laboratory parameters in patients with Wilson disease were abnormal, including serum ceruloplasmin <0.2 g/L, non-ceruloplasmin-bound serum copper >25 µg/L, urinary copper excretion >1.6 µmol/24 h, and liver copper >250 µg/g dry wt [122].
Mainstream opinion describes Wilson disease as a disorder difficult to establish as a firm diagnosis due to variable clinical features and abundant laboratory results that differ in their validity [106,122]. In this context, artificial intelligence (AI) provides a forum whereby complicated processes including those prevalent in the diagnosis of human diseases are solved by intelligent diagnostic algorithms using relevant characteristics that summarize given individual scores, providing a final score with grades of probability [41]. Using such a scoring method, complex diagnoses of various diseases were firmly diagnosed, including DILI and HILI [32,33,41,42], autoimmune hepatitis [127], and Wilson disease, for which the Leipzig scoring system of 2003 [128] or the subsequently modified Leipzig soring method of 2019 was successfully applied [129]. Key items of Wilson disease received individual scores, and the sum of these scores classified the suspected Wilson disease as an established diagnosis (score ≥ 4), as possible (score 3), or as very unlikely (score ≤ 2) (Table 3).
Table 3.
Modified Leipzig scoring system for Wilson disease.
This modified Leipzig score was derived from a previous report [129], but for reasons of precision, the score was now revised: under the item of 24 h urinary copper, the previous term “acute hepatitis” was replaced by “chronic cholestatic liver disease”.
The note in the modified Leipzig scoring system as published [129] under the item 24 h urinary copper (in the absence of acute hepatitis) is not based on any evidence as plain acute hepatitis is not known for increased urinary copper excretion, as opposed to cholestatic autoimmune hepatitis (AIH) or any other liver disease presenting with cholestatic features like primary biliary cholangitis (PBC) or primary sclerosing cholangitis (PSC) [66,130], which may masquerade as Wilson disease due to high urinary copper excretion [130]. For all three cholestatic liver diseases (AIH, PBC, and PSC), a range of diagnostic parameters are available for exclusion purposes (Table 2). Thus, the term acute hepatitis has now been replaced by chronic cholestatic liver disease (Table 3).
Liver histopathology shows, in the initial stages of Wilson disease, mild unspecific lobular changes, and occasionally also shows singular apoptotic hepatocytes and spotty necrosis with surrounding lymphocytes, mild macrovesicular steatosis, and ballooned or glycogenated hepatocytes [131]. With progressing disease, inflammation increases, leading to fibrosis und ultimately to cirrhosis with the clinical end stage of acute liver failure. Increased copper accumulation in the hepatocytes may be visible by histochemical staining with rhodamine or rubeanic acid through direct binding to copper [131,132]. However, the absence of visible copper-binding protein does not exclude Wilson disease [131]. More specifically, at the time of diagnosis, liver histology obtained by liver biopsy in 78 patients showed chronic liver injury in 73% of patients, with fibrosis occurring in 36% and cirrhosis in 37% of patients, while steatosis was found in 54% of the evaluated liver specimens and a normal liver histology was described in three patients [122].
Ultrastructural analysis by an electron microscopy of the liver at the early Wilson disease stage of steatosis reveals specific mitochondrial abnormalities [133,134]. Typical findings include variability in size and shape, increased density of the matrix material, and numerous inclusions including lipid and fine granular material that may be copper. The most striking alteration is increased intracristal space with a dilatation of the tips of the cristae, creating a cystic appearance [134]. In the absence of cholestasis, these changes are considered to be pathognomonic of Wilson disease. At later stages of the disease, dense deposits within lysosomes are present. Ultrastructural analysis may be a useful adjunct for diagnosis [133].
3.5.4. Therapy
There are no prospective studies using RCTs available with a focus on the efficacy and risks of therapeutic approaches in patients with Wilson disease [66], partly attributable to the low disease frequency [129,135]. However, as for any new therapy approach, a control group would necessarily have any drug treatment withheld, but this would provide them with treatment too late and would be unethical. Instead, empirical therapy was applied early based on anecdotal reports, personal experience, and considering mechanisms leading to the liver injury [66,133]. To achieve a reduction in copper excess in the liver, two strategies have to be discussed, one refers to the uncommonly proposed prevention through a reduction in alimentary copper [52,57,66,135], and the other one focuses on increasing renal copper elimination as the better alternative [66,120].
Most of the commonly consumed foods contain trace amounts of copper [66,136], which makes dietary copper restriction less feasible, although foods with high copper content should be avoided to support therapy with chelators [66]. Foods with a high copper content include cocoa derived from Owena cocoa (Theobroma cacao L.) and contaminated with copper from copper-containing fungicides, dark chocolate from contaminated cocoa, nuts, raisins, shellfish, oysters, and butchery foodstuff from liver and kidneys derived from cattle grazing, possibly, on grounds with plants contaminated by copper [67,68,128,136].
Patients with Wilson disease are well treated first line with copper chelators, like D-penicillamine, that help remove circulating copper bound to albumin, which facilitates urinary copper excretion via the kidneys [66]. Based on previous considerations that drug treatment with D-penicillamine in patients with Wilson disease may interfere with pyridoxin, comedication with pyridoxal 5′phosphate, the biologically active form of vitamin B6 25–50 mg daily is commonly prescribed [106]. However, a recent preliminary study from France questioned the need for a routine comedication with vitamin B6 as its blood levels were in the normal range in Wilson disease patients treated with D-penicillamine in the absence of additional vitamin B6 supplementation [124]. In this context, it was mentioned that the usual diet contains sufficient amounts of vitamin B6, and vitamin B6 needs to be supplemented only if food deficient in vitamin B6 is consumed. There was also the note that optic neuropathy was observed in four patients in association with a D-penicillamine therapy [124]. However, a temporal association is not necessarily a causal association. Finally, it was argued that little published data on vitamin B6 supplementation for patients treated with D-penicillamine is available and no consensus recommendation exists [124].
D-penicillamine is initially given at a daily dose of 300 mg and increased weekly by 300 mg up to 1500 mg/d [66]. Remission is achieved with the normalization of free copper blood levels and urinary copper output, but lifelong maintenance treatment is mandatory with mostly 600–900 mg daily [128]. Similar to any other drug treatment for most diseases, the therapy with D-penicillamine is not without adverse drug reactions (ADRs). Initial allergic reactions can be scoped by intermittent drug cessation or concomitant use of corticosteroids [66,117,124]. More serious is an initial deterioration of neurological symptoms occurring in 14% of treated patients, hardly to be reversed and of unknown etiology [66,117]. Of concern is the development of autoimmunity due to D-penicillamine use, as evidenced by a new appearance of serum anti-nuclear antibodies (ANA) with variable titers [128] and to be differentiated from increased ANA titers already occasionally found before starting drug therapy for Wilson disease itself [126]. In the case of new autoimmunity, switching from D-penicillamine to alternative, second-line therapies of zinc is recommended [66].
For a long time, treatment with trientine has been recommended as a second-line therapy for patients with Wilson disease although no studies with head-to-head comparisons for initial treatment were available [66,133,137,138,139]. Respective previous recommendations are now outdated. Therapy with trientine 2HCl was restricted to patients intolerant to D-penicillamine and requires slowly increasing doses up to 1800 mg daily and reducing the doses down to 600–1000 mg daily, sufficient as maintenance doses for most patients [66,137]. Comedication with B6 was not required. Treatment efficacy was previously found to be similar for D-penicillamine and trientine, with fewer ADRs for trientine although neurologic features may worsen [66,133]. Also on the market are trientine 4HCl, which is stable at room temperature, and trientine 2HCl, which requires storage at 2–8 °C [138]. As copper-chelating agents, the trientine salts bind with excess copper, forming a stable complex which is excreted mainly in the urine [138]. It has been suggested that trientine salts may also decrease intestinal copper absorption [133,138,139].
The use of zinc as zinc salts or the more tolerable zinc–amino acid-bound medications, is associated with fewer ADRs except for severe abdominal discomfort that may lead to drug cessation [66]. The recommended dose for adults is 3 × 50 mg daily and for children or adolescents 3 × 25 mg daily [140]. Regarding the amelioration of liver injury, it was deemed inferior to chelators [66,135,137,141]. Zinc induces intestinal metallothionein, which blocks copper absorption, increases excretion in the stools, and results in an improvement in symptoms [141]. In addition, two meta-analyses and several large retrospective studies indicate that zinc is equally effective as a chelator for the treatment of Wilson disease, with the advantages of a very low level of toxicity and only the minor side effect of gastric disturbance. Thus, zinc may have a role as a first-line therapy in Wilson disease patients with neurological symptoms [133,141], is gaining acceptance for patients with hepatic presentations, and is universally recommended for lifelong therapy [141].
A short note on Bis-choline tetrathiomolybdate (TTM), also known as ALXN1840, is warranted as it is a drug in the pipeline of AstraZeneca for treating Wilson disease. TTM was recently cut at clinical phase III after results of additional phase II studies were disappointing, not reaching the study aims, and following consultations with regulatory agencies, communicated in spring 2023 by Pascal Soriot as the Chief Executive Officer (CEO) of AstraZeneca [142]. On theoretical grounds, treatment with TTM appeared promising due to its capacity to increase biliary copper excretion [66,142,143,144,145]. However, little attention was paid to the fact that copper may be reabsorbed from the intestine via the entero-hepatic circulation, counteracting hepatic copper depletion. Moreover, any new therapy approach should be more effective compared with previous treatment modalities like D-penicillamine, trientine, or zinc. Although initial studies with TTM were viewed as positive [66,143,144,145], some criticism was communicated regarding the low case numbers in the study cohort and quantitative copper measurement for assessing efficient copper removal from the blood by an old drug in a new design for Wilson disease, raising the question of whether TTM is good for the brain and liver [144]. It will be interesting to see the final information from AstraZeneca on why clinical phase III was cut [110].
Liver transplantation is an option as an ultima ratio in the end stage of Wilson disease, including in cirrhosis with untreatable complications or acute liver failure, providing a life-saving approach, curative treatment of the disease, restoration of the liver function, and mitigation of portal hypertension [146,147], with excellent survival rates of one and five years without disease recurrence [146]. In the case of deceased donor liver shortage, living related liver transplantation is an option [146]. Although promising data were obtained in animal studies, future RCT studies in humans are required to assess innovative approaches directed at hepatocyte transplantation [146,148], the transplantation of bone marrow cells [149], and gene therapy [106,146]. The indication for a liver transplantation has to be carefully considered in Wilson disease patients with severe psycho-neurological symptoms [146,147].
3.5.5. Prognosis
Prognosis is poor in patients with Wilson disease without the benefit of drug therapy, with the median life expectancy reduced to 40 years [66,106]. Instead, the prognosis is commonly good in Wilson disease, provided the disease is diagnosed early and patients receive an appropriate drug therapy, which might, rarely, include liver transplantation [66]. Under these conditions, the cumulative survival in 51 patients with Wilson disease was 95% at 33 years after diagnosis and identical with 95% of a control group matched for sex and age, considering that survival was only slightly reduced during the early observation period when liver transplantation was not available for acute liver failure.
During the observation period of 33 years and under the D-penicillamine treatment, all clinical signs improved with the exception of gynecomastia and esophageal varices, and treatment was associated with the improvement of all neurological symptoms and amelioration of all hematological laboratory test results [66,106].
3.5.6. Cascade of Molecular Events Leading to Copper Liver Injury
A broad range of proposals on how copper may cause molecular and mechanistic liver injury have been published [25,26,150], based on human or animal data [99]. Mechanistic events are similar in animals or humans exposed to high amounts of exogenous copper [92,99] compared to patients with Wilson disease, except for their genetic ATP7B mutations as the basis for this specific clinical liver injury [2,66] and transcuprein/alpha-2-macroglobulin of the blood following intestinal uptake, regulated by demand through intestinal transporters [151].
The second step represents the genetic impairment of biliary copper excretion [133]. This causes copper overload not only in the liver but also in other organs [66].
Third, the oxidized form of copper is the injurious metal that first attacks subcellular organelles of the hepatocyte like mitochondria, as evidenced by electron microscopy in patients with initial stages of Wilson disease [133,134]. These ultrastructural mitochondrial injuries are associated with functional disturbances of the mitochondrial respiratory chain, responsible for energy by ATP production and fatty acid oxidation [152,153], that cause steatosis, visible with light and electron microscopy [131,133,134]. Apart from liver mitochondria, other organelles of the liver cells can be damaged, because their membranes contain proteins and unsaturated fatty acids, which can easily be attacked by reactive oxygen species (ROS) [154] and generate lipid peroxides as evidenced by high plasma malondialdehyde (MDA), low glutathione (GSH) levels, and reduced total antioxidant capacity (TAC) [155,156]. Toxic intermediates are generated in the course of an interaction between the reduced form of copper and oxygen via the Fenton reaction [154], leading to superoxide anion, hydrogen peroxide, and hydroxyl radicals comprising ROS generated through oxidative stress, and capable of triggering, at least partially, liver injury [99,150]. In this context, cuproptosis is a newly described copper-dependent form of regulated cell death considered to be a contributory causative mechanism in Wilson disease [150]. Copper induces the aggregation of lipoylated dihydrolipoamide S-acetyltransferase (DLAT). This is associated with the mitochondrial tricarboxylic acid (TCA) cycle, resulting in proteotoxic stress in line with the proposed cuproptosis [150]. The copper-dependent, regulated cell death is distinct from known death mechanisms because of its dependency on the mitochondrial respiration. Notably, copper hepatotoxicity is not just oxidative stress by this heavy metal because zinc, by replacing copper, may be a major cofactor in various cellular processes of liver injury [157].
Fourth, recent evidence suggests a correlation between dysbiosis in gut microbiome and multiple diseases such as genetic disorders, including Wilson disease. As an example, 16S rRNA sequencing was performed on fecal samples from 14 patients with Wilson disease and was compared to the results from 16 healthy individuals [158]. The diversity and composition of the gut microbiome in the Wilson disease group were significantly lower than those in healthy individuals. The Wilson disease group presented a unique richness of Gemellaceae, Pseudomonadaceae, and Spirochaetaceae at the family level, which was hardly detected in healthy controls. This group had a markedly lower abundance of Actinobacteria, Firmicutes, and Verrucomicrobia, and a higher abundance of Bacteroidetes, Proteobacteria, Cyanobacteria, and Fusobacteria than healthy individuals. The Firmicutes to Bacteroidetes ratio in the Wilson disease group was significantly lower than that of healthy controls. In addition, the functional profile of the gut microbiome from Wilson disease patients showed a lower abundance of bacterial groups involved in the host immune- and metabolism-associated systems pathways such as transcription factors and ABC-type transporters, compared to healthy individuals. These results implied the dysbiosis of gut microbiota may be influenced by the host metabolic disorders of Wilson disease, which may provide a new understanding of the pathogenesis and even new possible therapeutic targets for Wilson disease. However, the impact of intestinal microbiota polymorphisms in Wilson disease have not been fully elaborated and need to be explored for seeking some microbiota benefit for Wilson disease patients [158].
As the fifth step in the further course of Wilson disease, hepatic immune cells become more involved, as evidenced by the detection of inflammatory mediators like cytokines in the plasma of patients with Wilson disease, changing a silent liver into an organ that provides information on processes occurring in the injured human liver to the blood of patients, easily available for further analysis by physicians [154]. The interaction of copper with immune cells producing mediators is well documented [159,160,161], explicitly shown by the reduced interleukin-2 (IL-2) production and IL-2 mRNA in human T lymphocytes caused by copper deficiency [161]. In this context and of importance regarding mechanistic steps in Wilson disease, there are results of an increased expression of cytokines and chemokines in the plasma of patients, indicating that the dysregulation of mediators plays a role in Wilson disease [154]. The study cohorts consisted of 99 patients with Wilson disease and 32 healthy controls. Compared with controls, in patients with Wilson disease, there was a significant increase in plasma of T helper (Th) 1 cells (IL-2, TNF-α, and TNF-β), Th2 cells (IL-5, IL-10, and IL-13), and Th17 (IL-23) (p < 0.05). Higher plasma Th 1 cells (IL-2, TNF-α, and TNF-β), Th 2 cells (IL-13), and Th 17 (TGF-β1, IL-23) levels were found in neurological patients compared with control groups (p < 0.01). In addition, Th 1 cells (TNF-α and TNF-β), Th 3 (TGF-β1), and Th 17 (IL-23) levels were significantly higher in hepatic and neurological patients (p < 0.05), whereby the higher Th1 cells (IL-2, TNF-α, and TNF-β), Th2 cells (IL-13), and Th17 (TGF-β1, IL-23) and the course of Wilson disease were associated with the severity of the neurological symptoms for Wilson disease patients [154]. Finally, a prolonged exposure of the liver to considerable amounts of copper will activate resident hepatic stellate cells to myofibroblasts, which secrete extracellular matrix proteins that generate collagen [162], leading to liver fibrosis and cirrhosis [106,122]. Despite some uncertainties, it remains to be established whether animal models [163,164], including the goldfish model [105] or the zebrafish model [165], can contribute to close the existing mechanistic gaps of excess copper liver injury.
Critical issues related to the liver injury due to copper have been discussed and quoted [99,150] and are now summarized (Figure 3).


Figure 3.
Issues connected to liver injury due to copper. Abbreviation: ROS, reactive oxygen species.
Figure 3.
Issues connected to liver injury due to copper. Abbreviation: ROS, reactive oxygen species.
4. Iron
Iron (Fe, from Latin: ferrum) is environmentally present [1,2,3,4,5] and became an indispensable element in humans for several vital biological processes [166] and in many other living organisms [167] including plants and microorganisms [168]. In a physiological range, iron is beneficial for human health, but lower or higher amounts are risky. Like other living organisms, humans cannot synthesize the metal but must acquire it by food from plants and the meat of cattle that consume plants rich in iron [166].
4.1. Physiology
As opposed to other minerals, iron levels in the human body are only controlled by absorption [166,169,170]. In line with experimental studies in rats [171,172], iron uptake in humans proceeds in the upper small intestine by an active, carrier-mediated mechanism involving a membrane iron-binding glycoprotein [170]. Intestinal absorption largely ensures body iron homeostasis in humans because iron lacks active excretory mechanisms [169,173].
Dietary iron is taken up by the divalent metal transporter 1 (DMT1) in enterocytes and moved to portal blood via ferroportin (FPN), where it is bound to transferrin and taken up by hepatocytes, macrophages and bone marrow cells via transferrin receptor 1 (TfR1) [167] with hepatic macrophages playing a crucial role for iron homeostasis [174]. As an unregulated process, iron excretion occurs through loss in sweat, menstruation, and the shedding of hair and skin cells, associated with rapid turnover and the excretion of enterocytes.
In the human body, iron exists mainly in erythrocytes as the heme compound hemoglobin with approximately 2 g of iron in men and 1.5 g in women, to a lesser extent in storage compounds like ferritin and hemosiderin, and in muscle cells as myoglobin [166]. Iron participates in ferroptosis [175,176] and DNA synthesis, electron transport, and oxygen transport [166]. The metal is also bound to proteins such as hemoproteins and non-heme enzymes [167] involved in oxidation–reduction reactions and the transfer of electrons (cytochromes and catalase) [166], with iron embedded in cytochrome P450 (CYP) [177] required for the metabolism of drugs and other exogenous substrates shown for the catalytic CYP cycle (Figure 4) [18].
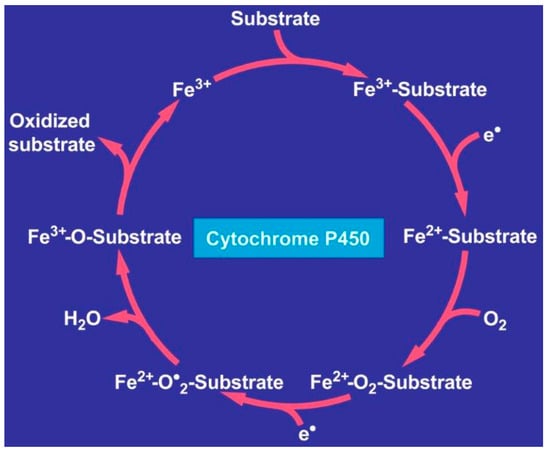
Figure 4.
Catalytic CYP cycle. Hepatic microsomal cytochrome P450 requires iron and releases it upon its degradation. Abbreviations: CYP, cytochrome P450; ROS, reactive oxygen species. The figure is derived from an open access report [18].
Under physiological conditions, iron in humans is commonly bound to hemoglobin in erythrocytes, while iron from senescent red blood cells is recycled by macrophages in the spleen, liver, and bone marrow [167]. Whereas most of the physiologically active iron is bound to hemoglobin, the major storage of iron occurs in the liver in a ferritin-bound fashion. In response to an increased iron load, hepatocytes secrete the peptide hormone hepcidin, which binds to and induces the internalization and degradation of the iron transporter ferroportin (FPN), thus controlling the amount of iron released from the cells into the blood.
In the blood, iron is commonly bound to transferrin as transferrin-bound iron (TBI). However, a small pool is also present as NTBI [167,178] as the major contributor to the iron loading of hepatocytes when transferrin is saturated [179]. In addition, liver hepatocytes ensures iron homeostasis by producing and releasing the 25-amino acid peptide hormone hepcidin [178]. Hepcidin is secreted into the bloodstream and inhibits the release of iron in several cells, such as duodenal enterocytes, macrophages, hepatocytes, and Kupffer cells [180]. By binding to FPN, hepcidin facilitates the ubiquitination, internalization, and degradation of FPN as well as directly blocking the channel for iron export out of the cell into the plasma [167,181]. The synthesis of the peptide hormone FPN is regulated at the transcriptional level, and controlled by serum iron concentrations [182]. When serum iron levels are increased, hepcidin expression is upregulated and results in the blocking of iron transport to the plasma via FPN, thus providing a negative feedback response preventing potential toxic iron accumulation in the body [167,182]. Low plasma levels of iron lead to low transferrin saturation [183], which in turn impairs the synthesis of hepcidin [182]. Therefore, iron concentration in biological fluids is tightly controlled to provide adequate intracellular and extracellular iron levels, thus preventing its toxic accumulation [183]. This is a key process, as any abnormalities in the distribution and content of iron in the body can have harmful effects on the physiological processes through ferroptosis, a new form of programmed cell death, which develops with iron dependence and occurs in patients with hemochromatosis [167,176].
The laboratory assessment of the iron status relies on serum parameters such as serum ferritin (SF), transferrin saturation, and soluble transferrin receptor (sTfR) [184]. These indicators present challenges for clinical practice and national nutrition surveys, and iron status interpretation is often based on the combination of several indicators. The diagnosis of iron deficiency (ID) through SF concentration, the most commonly used indicator, is complicated by concomitant inflammation since SF is also a diagnostic biomarker of acute phase inflammation [184]. On the other hand, sTfR concentration is an indicator of functional ID that is not an acute-phase reactant, but challenges in its interpretation arise because of a lack of assay standardization, common reference ranges, and common cutoffs [184]. It is unclear which indicators are best suited to assess excess iron status. The value of hepcidin, non-transferrin-bound iron, and reticulocyte indexes is being explored in research settings. Serum-based indicators are measured on fully automated clinical analyzers available in most hospitals. Although international reference materials have been available for years, the standardization of immunoassays is complicated by the heterogeneity of antibodies used and the absence of physicochemical reference methods to establish “true” concentrations. From 1988 to 2006, the assessment of iron status in NHANES was based on the multi-indicator ferritin model. However, the model did not indicate the severity of ID and produced categorical estimates. More recently, iron status assessment in the National Health and Nutrition Examination Survey (NHANES) has used the total body iron stores (TBI) model, in which the log ratio of sTfR to SF is assessed. Together, sTfR and SF concentrations cover the full range of iron status [184].
Serum parameters of normal and pathological iron status may differ among various laboratories depending on the analytical method used and definition of a cutoff, and are highly variable, influenced by age and gender ranges of normal [184,185]. While the determination of serum iron is of no clinical relevance due to its variability and lack of specificity, among the most used parameters in evaluating iron status are serum ferritin in combination with fasting transferrin saturation, although ferritin is also an acute phase protein that is increased in inflammatory conditions, confounding the interpretation of obtained values [185].
Iron as a trace metal in physiological amounts is beneficial [166,167] due to excellent measures that provide iron homeostasis, as comprehensively reviewed [167]. However, iron deficiency may be hazardous to human health due to anemia and various clinical manifestations [186], easily seen externally by pale skin and cheilitis [187,188]. Iron may also be injurious when present in excess due to acute and chronic intoxication by exogenous iron or in the course of genetic hemochromatosis with a hepatic overload of iron due to uncontrolled intestinal iron uptake [66,169,170,171,172], which is to be clinically differentiated from hemosiderosis with iron excess in the hepatic Kupffer cells due to a chronic release of iron from injured blood red cells as a consequence of genetic hemolytic disorders [189,190].
4.2. Iron as Pollutant and the Issue of Health Hazards
4.2.1. Sources of Iron as an Environmental Pollutant
Iron is the second most abundant metal in the earth’s crust, of which it accounts for about 5%, and is commonly found in nature in the form of its oxides [191]. The presence of iron was documented in different environmental matrices such as air, water, soil, food, plants, animals, and humans. This metal is one of the most common pollutants present in drinking water [191] but it was not found in plants of a municipal solid waste landfill [23]. Ethnomedical studies on the iron content of some medicinal herbs used in traditional medicine in Cote d’Ivoire for the treatment of anemia were reported [192], and iron as a suspected hepatotoxic pollutant was described in relation to medicinal herbs but studies on liver injury in patients using these herbal medicines are not published [24].
Environmental iron pollution is reported in the attic dust study, which used dust collected from the attic of old houses as an archive of historical air contamination by iron in the urban environment [71].
4.2.2. Elucidating Health Hazards of Environmental Iron
Several reports presented details and proposals for potential health hazards in connection with exposure to environmental iron. They were provided in condensed form and supplemented by critical comments if needed (Table 4) [183,193,194,195,196,197,198,199,200].
Table 4.
Iron sources from the natural environment and industry, with proposed impact on human health.
Overall, published work has revealed little if any convincing evidence of potential health hazards during and after exposure to iron derived from the environment (Table 4) [183,193,194,195,196,197,198,199,200]. Assessing the various key aspects is certainly a challenging approach as many confounding variables must be considered. They include different oxidation forms of the iron ion with different solubility characteristics. In principle, the environmental iron occurs in two stable oxidation states, preferentially as ferric (Fe3+) and ferrous (Fe2+), yet ferrous iron (Fe2+) is the form that is transported into enterocytes [201,202,203,204]. The oxidized or ferric (Fe3+) form is highly insoluble and must, therefore, be reduced to absorbable ferrous before transport through the intestinal mucosa. After entering the human gastrointestinal tract, the oxidized iron is poorly absorbed, conditions that help reduce the intestinal uptake of iron in amounts higher than needed for iron homeostasis. Many proposals were only based on theoretical considerations, like the hazard index, without proof that the iron actually entered the body of the exposed humans, because amounts of iron were not quantified [19,193,194,195,196,197,198,199,200]. There is also an information gap on how the highly insoluble ferric is absorbed by the pulmonary mucosa after exposure to the lungs by inhalation. Open for discussion is the assumption that neurogenerative diseases are causally related to the uptake of environmental iron (Table 4), especially in view of the commonly accepted view that demethylation in the basal ganglia is closely associated with age-related iron accumulation, though the pathophysiological mechanisms of the local iron accumulation have not yet been studied [205].
In essence, overall potential health risks following exposure to environmental iron lack the required level of evidence and seemingly are negligible, although some authors attributed a few disorders to the action of environmental iron (Table 4). However, healthy individuals not affected by gene mutation, which may disturb cellular iron homeostasis, are well prepared to manage increased amounts of iron entering the body through a reduction in intestinal iron uptake. Of course, this special approach of reduction does not work in patients with genetic hemochromatosis and in individuals exposed to iron in a setting of intoxication.
4.3. Acute human Liver Injury by Exogenous Iron
Acute intoxications with iron commonly occur through ingestion by intention or unintentionally [206,207,208,209,210,211,212,213,214,215]. There are reports on fatal iron toxicity in adults causing fulminant hepatic failure [208,212,213] with normal serum iron levels at 36 h after ingestion due to the redistribution of iron to the intracellular compartment [212]. In another patient, plasma iron levels were high on day 3 after intoxication with a further increase on day 6 and a concomitant decrease in unbound iron binding capacity, whereas plasma ferritin levels were high on day 3 [208]. In this patient, a radiodense content in the stomach was described on day 1, valuable information for future cases, and plasma activities were increased on day 2 with ALT and AST > 15,000 U/L, followed by a rapid decline to normal values. Despite repeated gastric and intestinal lavages, chelating therapy, and plasma exchange, the patient died of fulminant hepatic failure, and upon autopsy, liver histology showed the complete disappearance of hepatocytes [208]. There are no evidence-based guidelines on how best to treat patients with acute iron ingestion. Occasionally, a tightly packed collection of iron known as a bezoar is detected in the stomach if substantial amounts were ingested, for which endoscopic removal should first be approached before operative removal is considered [214].
Unintentional iron ingestion is one of the most common deadly poisonings among children through drugs containing iron prescribed to patients with anemia [209]. In this cohort, clinical features of acute iron ingestion are similar to those in adults [208]. Overall, details of cascade events leading to liver injury by acute iron intoxications in individuals lacking genetic hemochromatosis characteristics are, as expected, not available but likely are similar to those described below for hemochromatosis.
4.4. Chronic Human Liver Injury by Exogenous Iron
In a recent case report on prolonged iron use leading to hepatic overload, a postmenopausal patient consumed, for 30 years after menopause, 1–3 tablets daily, each containing 325 mg ferrous sulfate, because of a mistaken belief that it would benefit her general health [216]. Her serum transaminases and ALP activities were normal, but serum ferritin level was strikingly increased. In addition, magnetic resonance imaging (MRI) of the abdomen showed iron deposition in the liver and spleen compatible with overload by exogenous iron. In the liver histology, increased iron was found in Kupffer cells and hepatocytes with focal fibrosis but no cirrhosis. The issue of mechanistic steps involved in this type of prolonged human liver injury by iron attracted little scientific interest and led to no published data. However, respective pathogenetic events are likely similar to those of hemochromatosis with its characteristic features outlined further below. Both disease entities partially show a similar involvement of the liver, although the risk of liver injury is higher in the genetically based hemochromatosis. Studies on other similarities comparing both disease entities were not published.
Chronic exposure to iron oxide in steelworkers was reported in an early study but liver enzymes were not analyzed because the focus was on pulmonary aspects [217], confirmed by a later report with a focus on chronic obstructive pulmonary disease (COPD) [218]. A critical reappraisal report concluded that prolonged overload from exogenous iron rarely causes serious liver injury in humans and is only weakly fibrogenic in animal models, calling into question the concept that iron overload is a compelling cause of hepatotoxicity [219], in line with recent views [25,26].
4.5. Hemochromatosis
Hemochromatosis occurs in homozygotes with a mutation of the hemochromatosis gene (HFE) protein with a prevalence of 1:300 to 1:500 individuals [189]. A mutation in the HFE gene causes an increased absorption of iron despite a normal dietary iron intake. C282Y and H63D are the most common mutations of the HFE gene, present on the short arm of chromosome 6 (6p21.3) [189,220]. Different types of hemochromatosis are currently under discussion [189,221]; the most recent proposals are as follows [189]:
- Type 1 (HFE-related): This is the classic form of hemochromatosis that is inherited in an autosomal recessive fashion with worldwide prevalence [222,223].
- Type 2a (mutations of hemojuvelin gene) and type 2b (mutations of the hepcidin gene): Autosomal recessive disorder that is seen both in whites and non-whites. Its onset is usually at 15–20 years [189].
- Type 3 (mutations of transferrin receptor-2 gene): Autosomal recessive disorder that is seen both in whites and non-whites. Its onset is at 30–40 years [224].
- Type 4 (mutations of the ferroportin gene): Autosomal dominant disease that is seen both in whites and non-whites. Its onset is at 10–80 years [189].
Hemochromatosis, with its retained iron primarily deposited in the hepatocytes, is to be differentiated from hepatic iron overload syn hemosiderosis with its iron primarily deposited as hemosiderin in the reticuloendothelial cells of the liver [189]. This form of hepatic iron overload can occur in a variety of conditions like anemia with ineffective erythropoiesis due to thalassemia, sickle cell anemia, and hereditary spherocytosis, overall conditions often associated with multiple transfusions of red blood cells to treat anemia [190]. If iron overload is severe, chelation as a first-line therapy is indicated to avoid disease progress. Of course, venesection is not possible due to already existing anemia.
4.5.1. Natural Course
Hemochromatosis is a genetically heterogeneous disorder [189,221] that causes iron overload by an excess intestinal absorption of dietary iron, due to a decreased expression of intestinal hepcidin [225,226]. Excess amounts of iron are primarily found in the liver with its hepatocytes, which comprise around 80% of liver mass and are able to synthesize a high amount of the iron storage protein ferritin [167]. Iron is also stored in other parenchymal cells like pancreatic cells and cardiomyocytes [227] as well as joints, thyroid, skin, gonads, and pituitary [189].
4.5.2. Clinical Characteristics
Initial symptoms of hemochromatosis like asthenia, joint pain, arthritis, chondrocalcinosis, diabetes mellitus, hypopituitarism, hypogonadotropic hypogonadism, cardiopathy, heart failure, and cardiac arrhythmias are uncharacteristic [221]. This is why patients are often seen by non-hepatologists including general practitioners, orthopedists, diabetologists, cardiologists, endocrinologists, and dermatologists for skin pigmentation, rarely and after long disease only. In addition, koilonychia, a nail disease that causes thin, flat, or concave nails, affecting the thumb and index finger has been observed in 50% of patients while 25% of patients tend to have it in all nails [189]. This is more common in the male gender, with an odds ratio of 4.3 (95% CI 2.97–6.18) for liver disease [27], as pre-menopausal females lose iron through blood physiologically via menstruation. This results in delayed disease detection in females, often reported in their sixth decade as compared with males in their fifth decade [189].
In hemochromatosis as seen by light microscopy, liver injury may start with steatosis [228] or mild steatohepatitis [229,230]. If not treated, the injury proceeds to chronic liver disease, fibrosis, cirrhosis, and eventually hepatocellular carcinoma (HCC) [229,231]. These initial stages are clinically not easily detectable by conventional laboratory parameters. Similarly, even when cirrhosis develops it is often a silent cirrhosis lacking symptoms, before clinical features of decompensated cirrhosis develop with jaundice, ascites, bleeding esophageal varices, and hepatic encephalopathy [228]. End stages of the natural course are acute liver failure, hepatocellular carcinoma, and death [27,228].
Using electron microscopy for assessing the liver of patients with hemochromatosis, injurious changes to the mitochondria and endoplasmic reticulum were described in necrotic hepatocytes with the highest liver iron content, associated with large iron-laden lysosomes in the hepatocytes that were also encountered in the Kupffer cells [232] in the pre-cirrhotic stage of idiopathic hemochromatosis; the first evident ultrastructural changes are in the lysosomal compartment. These changes correlate well with the iron overload also in advanced stages of the disease, and are reversed after iron removal.
4.5.3. Diagnostic Approach
Initially, serum LTs like ALT and AST are normal or marginally increased up to two times the ULN [189,233], whereas later stages such as cirrhosis are clinically easier to recognize and show higher values of ALT, AST, and ALP [233]. Considering that mild abnormalities in the biochemical liver profile are common in hemochromatosis, patients with an unexplained abnormality in the liver profile should be screened for hemochromatosis with a serum ferritin and transferrin saturation [233]. Empirically, high values of serum ferritin and elevated transferrin saturation as well as genetic screening may help identify suspected hemochromatosis [221]. In general, a liver biopsy for histology examination is not required [231] and is no longer recommended [133]. Instead, the use of magnetic resonance imaging (MRI) allows for iron detection and quantification not only in the liver but also in other organs and has emerged as the reference standard imaging modality, as ultrasound is unable to detect iron overload and computed tomography findings are nonspecific and influenced by multiple confounding variables [231].
Despite many reports on the disease, there is no quantitative diagnostic algorithm available, which could provide information on the probability grades as to whether the diagnosis of hemochromatosis is likely, possible, or excluded [221]. Various diagnostic approaches were published, but none of the algorithms were based on evidence or quantitative results derived from scored key features [133,221,234].
4.5.4. Therapy
RCTs in patients with hemochromatosis are lacking because venesection syn phlebotomy was introduced early as a first-line of therapy based on theoretical considerations that this therapeutic approach is the best way to efficiently remove an excess of iron from the body [133,235,236,237,238], and further research is considered as unlikely to change the confidence in the estimate of the benefit and risk of venesection in this disease [133]. Consensus exists that venesection is a first-line therapy for patients with hemochromatosis with similar proposals on how best to proceed [133,239]. As an example of qualification for venesection, patients should have a serum ferritin level >300 μg/L in men and >200 μg/L in women in combination with an elevated fasting transferrin saturation (>45%) for both men and women. Weekly or fortnightly venesection aims to achieve an initial serum ferritin target of 50–100 μg/L [238]. Each venesection typically would remove 500 mL of blood corresponding to 250 mg iron. Venesection intervals were increased, or a lower volume of blood was removed if the patient did not tolerate phlebotomy because of weakness, hypotension, or the development of anemia [238]. Venesection ameliorates overall prognosis but must be conducted as a lifelong therapy [133,235,236,237].
In a recent randomized crossover trial of erythrocytapheresis, which removes only red blood cells, versus phlebotomy, which removes blood with all cells, the conclusion was reached that erythrocytapheresis reduced the number of annual treatment procedures but at higher costs compared with venesection in the maintenance treatment of patients with HFE hemochromatosis [239]; erythrocytapheresis was subsequently classified as personalized erythrocytapheresis, representing the preferred treatment in selected cases [133].
As a second line of therapy in hemochromatosis for patients experiencing problems with venesection, iron chelation therapy using the once-daily, oral iron chelator deferasirox (Exjade) is available in selected patients [240,241] with contraindications for venesection such as anemia, severe heart disease, or poor venous access [240].
4.5.5. Prognosis
Based on a large cohort consisting of 163 patients with hemochromatosis, cumulative survival was 92% at 5 years after diagnosis, 76% at 10 years, 59% at 15 years, and 49% at 20 years [235,236,237]. More specifically, life expectancy was lower in patients with cirrhosis as compared with those without cirrhosis, in patients with diabetes mellitus as compared with those without diabetes, and in patients who could not be depleted of iron during the first 18 months of venesection therapy as compared with those who could be depleted, while prognosis was not influenced by sex. On the other hand, patients without cirrhosis had a life expectancy that was similar to that expected in an age- and sex-matched normal population. Compared with the normal population, causes of death analyzed in fifty-three patients showed that hepatocellular carcinoma was two hundred nineteen times more frequent among the patients (sixteen patients), cardiomyopathy was three hundred six times more frequent (three patients), cirrhosis was thirteen times more frequent (ten patients), and diabetes mellitus was seven times more frequent (three patients). Finally, death rates for other causes, including extrahepatic carcinomas (seven patients), were similar to the rates expected. The conclusion was reached that patients with hemochromatosis diagnosed in the pre-cirrhotic stage and treated by venesection have a normal life expectancy, whereas cirrhotic patients have a shortened life expectancy and a considerable risk of liver cancer even when complete iron depletion has been achieved [235,236,237].
4.5.6. Cascade of Molecular Events Leading to Iron Liver Injury
The pathogenesis of the iron liver injury was analyzed and discussed in previous reports [25,26,189,242,243]. The focus is on hemochromatosis, including intoxications by overdosed iron, in patients but rarely in animal studies.
The first pathogenetic step in hemochromatosis causing liver injury can be traced back to genetically triggered overwhelming iron absorption through the intestinal tract [169] specifically in the human upper small intestine microvillous membrane vesicles [170] and in the rat model [171,172]. This causes a toxic iron overload of the human organism including the liver [133,236,237,238,243,244,245,246]. Hemochromatosis is an autosomal recessive disorder caused by mutations in genes involved in iron metabolism, which results in increased intestinal iron absorption [246]. The abnormally high intestinal uptake of iron is likely due to mutations in several genes, including HFE, transferrin receptor 2 (TFR2), hepcidin, ferroportin (SLC40A1), and hemojuvelin (HFE2) [189,220,242,245], with a preference for the HFE gene that encodes the HFE protein [220,244]. This protein normally regulates the production of hepcidin responsible for iron homeostasis and especially, the control of iron uptake by cells through its interaction with the transferrin receptors, processes that do not function in patients with hemochromatosis [220]. This is shown by the downregulation of hepcidin synthesis, leading to increased intestinal iron absorption [244]. Thus, mutations in the HFE gene lead to excess iron absorption and iron overload in hemochromatosis [232].
The second mechanistic step represents the excessive iron uptake by the hepatocytes from blood, where it is transported by transferrin [228,246,247]. This follows the intestinal iron uptake as the first step of the cascade of events leading to liver injury [248].
The third step in hemochromatosis occurs in the liver itself with iron overloaded hepatocytes. It starts with injurious attacks of iron as divalent ferrous iron (Fe2+), a cation capable of reacting with hydrogen peroxide, generating one of the reactive oxygen species (ROS), the hydroxyl radical, while being oxidized to Fe3+ [228]. Radicals generated in the so-called Fenton–Haber–Weiss reaction are known as some of the most dominant oxidants found in the human body, attacking proteins, lipids, nucleic acids, and carbohydrates and leading to peroxidation and cell apoptosis [167,248,249,250]. ROS injure membranes of subcellular organelles, such as mitochondria or the endoplasmic reticulum, which contain structural proteins and phospholipids with polyunsaturated fatty acids (PUFAs) that are peroxidized, as evidenced by lipid peroxide markers found in patients with hemochromatosis or overloaded by exogenous iron [249,251,252]. As an example, in patients with hemochromatosis, liver biopsy specimens were immunostained for protein adducts with malondialdehyde and 4-hydroxynonenal, and both adducts were found to be more abundant as compared with controls, whereby the staining had a predominance in acinar zone 1 that followed the localization of iron [253]. Similarly, enhanced oxidative stress was described in patients with hemochromatosis, as evidenced by hepatic malondialdehyde (MDA)-protein adducts and by increased oxidatively modified serum proteins [251]. MDA-lysine epitopes and oxidatively modified serum proteins, as well as immunoglobulin G autoantibodies against MDA-lysine epitopes, were increased in untreated hemochromatosis patients compared with normal individuals, and these markers of ongoing oxidative stress decreased with phlebotomy treatment in hemochromatosis patients. In addition, TGF-beta1 colocalized with hepatic iron and MDA protein adducts in hepatocytes and sinusoidal cells of hepatic acinar zone 1 and normalized after iron removal. Iron overload increases both lipid peroxidation and TGF-beta1 expression, which together could promote hepatic injury and fibrogenesis leading to liver fibrosis and finally cirrhosis [251].
Ferroptosis, an iron-dependent form of regulated cell death [254,255,256,257], is closely related to mechanistic sequalae described in conditions of iron overload like hemochromatosis [176,256,257], and was discussed in detail more recently [257]. There is some evidence that ferroptosis is triggered by ferritinophagy, an autophagic process that specifically involves ferritin to release intracellular free iron [257]. At the morphological level, ferroptosis causes injury to mitochondria, with condensed, ruptured outer membranes, associated with initial iron accumulation, excessive ROS production, and excessive lipid peroxides [26,257]. Polyunsaturated fatty acids (PUFAs), as structural components of membrane phospholipids, are easily peroxidized, forming lipid peroxides triggered by ROS, generated through the Haber–Weiss reaction. Ferroptosis is also involved in alcoholic liver injury via liable iron accumulation and in the associated hepatic glutathione exhaustion [257].
The fourth step addresses the possible role of cytokines in hemochromatosis. The liver is commonly a secret-keeping organ providing few markers like cytokines or chemokines in the blood for a quick analysis of mechanistic processes within the liver in the context of liver injury. In patients with hemochromatosis, several cytokines were analyzed including IL1α, IL1β, IL2, IL4, IL6, IL8, IL10, IL12, IL17, IFNγ, TNFα, and Gm-CSF, which could help investigate the inflammatory status of the study population. As opposed to none of the individuals in the control group, serum IL8 was elevated in 42% of C282Y homozygotes and 46% of H63D patients, as homozygotes or in combination with C282Y [245]. The observation that several hemochromatosis patients had elevated levels of IL8 is difficult to explain but may be due to the fact that the C282Y HFE protein induces the transcription factor NF-κB, which consequently resulted in a marked increase in the protein production of IL8 along with an increased transcriptional activation of IL8 in C282Y HFE-expressing cells [245,258]. Elevated levels of IL8 were especially observed in patients who had recently been treated with phlebotomy, which might suggest a relation between disease severity and levels of IL8 [245]. Similar to other cytokines, IL8 as a chemokine is produced by macrophages and other cell types such as epithelial cells and airway smooth muscle cells [259], and can be significantly upregulated in patients harboring the H63D mutation; conceptually, however, the causes of neither the increased serum IL8 levels nor the unchanged levels of the other cytokines observed in hemochromatosis patients were thoroughly investigated, and these results currently do not add to our understanding regarding mechanistic steps involving mediators that are secreted by non-parenchymal cells within the liver and may interact among each other [167,245]. The failure to detect many cytokines [245] may be ascribed to the fact that the respective studies were carried out in patients with advanced stages of hemochromatosis already requiring phlebotomy therapy, rather than in those with the early phases of the disease. As an alternative, the immune cells commonly secreting cytokines under normal conditions may have so heavily been injured by the iron that they no longer produced and secreted cytokines.
Finally, the fifth mechanistic step has a focus on the gut microbiome, which is altered in hemochromatosis patients [238]. In this study cohort, systemic iron reduction by phlebotomy was associated with an alteration of the gut microbiome, with changes evident in those who experienced reduced fecal iron availability with venesection. For example, levels of Faecalibacterium prausnitzii, a bacterium associated with improved colonic health, were increased in response to fecal iron reduction. During iron depletion, iron absorption from the gastrointestinal tract increases to compensate for iron lost with treatment. Consequently, iron availability is limited in the gastrointestinal tract and is crucial to the growth and function of many gut bacteria. Moreover, increased colonic iron has been associated with colonic inflammation and oxidative stress. Similarly, metabolomic changes were seen in association with reduced fecal iron levels, with significant changes in microbial metabolites after treatment, when increases in pyruvate, tyrosine, methionine, glycine, and aspartate were observed. In these patients, there was a greater separation in the metabolome, where a shift was observed towards a more positive metabolomic profile with treatment compared with baseline [238].
The main critical issues related to liver injury caused by iron were discussed and quoted before [254,255,256,257] with a special focus on ferroptosis [257], and are summarized (Figure 5).

Figure 5.
Listing of key issues related to liver injury caused by iron. Abbreviation: ROS, reactive oxygen species.
5. Cadmium
Cadmium (Cd) (from ancient Greek καδμία kadmía or Latin cadmia) is environmentally present [2,260,261,262,263], and is encountered by humans [263].
5.1. Physiology
Cadmium in its ionic form of Cd2+ is a non-essential divalent metal and toxic in eukaryotic cells [264]. Consequently, cadmium is not required for any biological process involving, for instance, enzymes. Hence, there is no physiological downsizing of this metal once it enters the human body because mechanisms of ensuring cadmium homeostasis and that would control its uptake and release are missing. Actually, infants may already have cadmium in their body, provided by their mothers, since cadmium crosses the placental barrier and easily reaches the fetus [265]. In this context, cadmium was found in the cord blood, maternal blood, and placental tissue [266]. With advancing age and a continuous intake of cadmium via the lungs and the gastrointestinal tract [267], there is an increasing accumulation of cadmium in the body, where it may remain for 20 to 30 years as a non-biodegradable metal [263,267].
The presence of substantial amounts of cadmium accumulated in kidney, liver, bones, eyes, and other organs suggests an effective intestinal absorption of cadmium derived from ingested food contaminated with environmental cadmium and a functioning systemic transport of cadmium [268]. This is accomplished by employing multiple cellular transporters that are primarily used for the acquisition of the calcium, zinc, and manganese needed by the body.
5.2. Cadmium as Pollutant and the Issue of Health Hazards
5.2.1. Sources of Cadmium as an Environmental Pollutant
Cadmium is regularly found together with other heavy metals such as zinc, copper, and lead [269], detectable ubiquitously in the environment [23,25,26,30,270,271,272]. Overall environmental cadmium pollution is well documented using the excellent attic dust approach, whereby dust collected from the attic of old houses is examined, providing an archive of historical air contamination by cadmium in the urban environment [71].
Environmental cadmium concentrations continuously increase due to soil disruption, earth volcanic activity, and as byproduct of industrial processes [30,269,272] because it is used in alloys, pigments, stabilizers, and batteries [30]. Industrially, its use is best known for electroplating and in the production of nickel-cadmium batteries [269]. Consequently, cadmium is a contaminant of municipal solid waste landfills [23,270], electronic waste [273], soils, and plant parts such as leaves, stems, and roots [23]. Human exposure may occur via inhalation, including cigarette smoking, as well as by food [30], such as plants and vegetables contaminated [23] through the uptake of cadmium via roots or rhizome or via horizontal transfer (Figure 3), including potatoes, grains, seeds, or mushrooms, in addition to crustaceans, mollusks, shellfish, mussels, cocoa powder, and dried seaweed [30].
5.2.2. Elucidating Health Hazards of Environmental Cadmium
The widespread occurrence of the pollutant cadmium and the inability of humans exposed to this heavy metal to safely remove it from their body explains the broad interest of scientists and physicians in the health hazards possibly caused by cadmium (Table 5) [263,268,269,274,275,276,277,278,279,280,281].
Table 5.
Cadmium sources from the natural environment and industry, with proposed impact on human health.
Food, water, and air contaminated with environmental cadmium represents the major health hazard for exposed humans (Table 5) [263,268,269,274,275,276,277,278,279,280,281]. As cadmium is not biodegradable in the human body, it accumulates and exerts its toxic effects on a variety of organs, which leads to functional impairment including the liver with increased LTs in one study [280] but not in two others [269,279]. Under clinical aspects and much more seriously, however, prolonged exposure to cadmium is associated with increased mortality due to cancer and cardiovascular diseases [281]. Despite worldwide efforts to reduce cadmium emission in the environment, cadmium exposure to humans is largely unavoidable as measures to reduce cadmium intake are not really effective.
In essence, when finally entering the earth after being generated in the universe, cadmium is not an essential element in humans, who are not prepared to provide conditions for maintaining cellular cadmium homeostasis. Instead, cadmium is a typical pollutant responsible for many diseases due to unwanted continuous uptake and storage in the human body.
5.3. Acute Human Liver Injury by Cadmium
Acute cadmium intoxications in humans commonly occur by inhalation, resulting in lung injury, while liver injury has rarely been reported following the ingestion of cadmium salts [206,282,283,284], leaving liver values unchanged [284] or causing slightly increased LTs [283]. In one patient, the leading symptom was an initially unexplained hemolytic anemia with a complicated clinical course finally leading to death due to multiorgan failure. ALT activity was 86 U/L (normal range 5–45 U/L) thereby excluding a severe grade of liver injury, confirmed by an autopsy that showed by histology only diffuse centrilobular congestion and necrosis of the liver and by cadmium quantification low levels similar to the heart and brain, whereas extremely high cadmium levels were found in the kidneys, explaining the renal failure that required dialysis [283]. For the ingestion of cadmium, activated charcoal and gastric lavage are options in addition to mandatory monitoring for gastrointestinal injury as well as renal and hepatic dysfunction [269]. Chelators, though promising, have not been definitively proven to be useful in cadmium toxicity. However, it appears that if succimer is given early in the course, it has promising results. A comparison of the effectiveness of several chelators was published after a single experimental cadmium administration evaluating the toxicity, excretion, and distribution of cadmium [269,285].
5.4. Chronic Human Liver Injury by Cadmium
Serum LTs such as ALT, AST, and ALP in humans following prolonged cadmium exposure were commonly reported as having variable and mostly low values, often interpreted by the authors as signs of liver injury or liver disease [279,286,287,288,289,290,291,292]. However, published LT values did not commonly meet diagnostic criteria and thresholds of real liver injury as required [33], and occasionally normal ranges of the listed parameters were not provided. Low LT values in these patients are either due to low cadmium exposure or may reflect liver diseases that have nothing to do with cadmium, as causality assessment using RUCAM was not performed. Based on association studies analyzing serum cadmium levels, there is also the proposal that cadmium may cause hepatic steatosis and fibrosis [288], classified as non-alcoholic fatty liver disease (NAFLD) and non-alcoholic steatohepatitis (NASH) [289], now known as metabolic dysfunction-associated fatty liver disease (MAFLD) and metabolic dysfunction-associated steatohepatitis [292,293].
Chronic exposure to cadmium was described as related to positive associations with liver damage. Light microscopy of the liver specimens obtained from patients with chronic cadmium exposure showed significantly more fibrosis and hemosiderosis as compared with nonexposed controls [293]. Fibrosis was periportal, portal, perisinusoidal, or bridging. Additional features included inflammation, hepatitis, and macro- or microvesicular steatosis. These light microscopy changes in humans were grossly confirmed in corresponding animal models [294,295]. In other experimental microscopy studies, diffuse hepatocellular degeneration and necrosis were observed, also described as hemorrhagic lesions and coagulative necrosis, associated with high plasma ALT activity and high cadmium concentration in the liver [296]. The reported vacuolar degeneration was not further discussed but may represent vacuoles corresponding to empty fat cells following staining procedures with hematoxylin and eosin. In addition, other histopathological observations confirmed extensive parenchymal degeneration [297].
Transmission electron microscopy showed the presence of autophagosomes and dilated endoplasmic reticulum, which represents the microsomal fraction of the biochemists, and translucent vesicles and structural changes of the liver comprised the sinusoids being replaced by large gaps [298]. Additional ultrastructural studies focused on the proliferation of the endoplasmic reticulum and lysosomes and confirmed the presence of autophagosomes [296,298].
5.5. Cascade of Molecular Events Leading to Cadmium Liver Injury
The mechanism of liver injury caused by cadmium was discussed in several reports [25,26,264,279,299]. Most suggestions were derived from animal studies while respective data on patients were in the minority.
The first mechanistic step leading to liver injury after cadmium uptake by the human body and arrival in the circulation starts with with cadmium entering the liver; this is a complex approach because hepatocytes can only be targeted by Cd2+ if it hijacks physiological high-affinity entry pathways [264]. They help transport essential divalent metal ions in a process termed “ionic and molecular mimicry”. Hence, “free” Cd2+ ions and Cd2+ complexed with small organic molecules are transported across cellular membranes via ion channels, carriers, and ATP-hydrolyzing pumps, whereas receptor-mediated endocytosis internalizes Cd2+–protein complexes [264]. Within the liver cell, the ionic form of Cd2+ binds to the protein metallothionein (MT) to form the cadmium metallothionein (Cd–MT) complex, which allows for cadmium deposition in the liver and downgrades glutathione production [271].
The second step involves the covalent binding of Cd2+ to sulfhydryl groups on critical molecules such as hepatic mitochondria and cytosolic glutathione [271,299,300].
The third step is characterized by the reduced antioxidant function of the cytosolic glutathione and the overwhelming mitochondrial oxidative stress causing lipid peroxidation, which can no longer be mitigated by glutathione, contributing to the overall cellular production of ROS in hepatocytes detected by mass cytometry and fluorescence microscopy concomitantly with cadmium uptake [300]. An experimental exposure of hepatocytes to cadmium increased the concentration of malondialdehyde, a marker of lipid peroxidation, and decreased the activity of antioxidant enzymes, such as superoxide dismutase, catalase, glutathione reductase, and glutathione peroxidase [301]. Ferroptosis is involved in liver injury by cadmium, accompanied by the activation of the PERK-eIF2α-ATF4-CHOP signaling pathway, and ferroptosis in cadmium liver injury is dependent on endoplasmic reticulum stress [295]. The gut is also a target for experimental cadmium toxicity modulating the gut microbiome but studies in humans with the respective liver injury are not available, likely because of the small number of cases [302].
Fourth, experimental liver oxidative stress induced by cadmium was also ascribed to the activation of hepatic stellate cells [299].
The fifth step causing the injury may be triggered by cadmium causing the infiltration of polymorphonuclear neutrophils and Kupffer cells into the liver [303]. Cytokines and chemokines produced by activated Kupffer cells, such as tumor necrosis factor-alpha, interleukin-1, and interleukin-6, have been shown to be associated with inflammation and subsequent liver injury [299,300,304].
The last step of the liver injury focuses on the role of cadmium in modulating autophagy and tumorigenesis including hepatocellular carcinoma [271]. A recent meta-analysis proposed elevated serum and hair levels of cadmium as a risk factor for hepatocellular carcinoma [305].
The key critical issues related to liver injury caused by cadmium are summarized (Figure 6).

Figure 6.
Relevant challenges related to liver injury due to cadmium. Abbreviations: RCTs, randomized controlled trials; ROS, reactive oxygen species.
6. Arsenic
Arsenic (As, from Latin arsenicum) formed in the universe is environmentally present and potentially hazardous for human health [2,260,261].
6.1. Physiology
Arsenic is a non-essential element for human health because it is without a known nutritional or metabolic role and is not a constituent of any enzyme [306,307]. Similar to other non-essential metals like cadmium, arsenic, lead, nickel, barium, chromium, and mercury, it is harmful to health even at low concentrations [25,26,306]. Arsenic is commonly consumed together with food and affects intestinal homeostasis by disrupting barrier function and inducing inflammatory responses [308]. Based on experimental studies, processes involved in the transit of arsenic through the gastrointestinal tract can affect toxicity, as the more toxic of the ingested arsenic species may be formed in the intestinal lumen resulting from an interaction of arsenic with food components or gut microbiota, conditions that may modulate arsenic intestinal absorption and its adverse effects [308]. The experimental intestinal absorption of arsenic is variable and depends on the arsenic species used and the function of a possible paracellular transport [308]. Having no known cellular transporter system, arsenic is taken up into the cell by the phosphate carrier because arsenic is a phosphate analogue [307]. This process likely occurs in humans intoxicated with arsenic, which has been found in the liver and kidneys [309,310,311,312,313] with a maximum arsenic content of 6 mg/kg in the liver [310]. In this context, the normal human body contains 0.02–0.08 mg arsenic/kg body weight, which is concentrated in the liver, kidneys, lungs, bones, and hair [309].
6.2. Arsenic as Pollutant and the Issue of Health Hazards
Arsenic originating from the universe is frequently found in the environment of our earth [26,309,311,312,313] including the atmosphere originating from natural sources such as volcanic eruptions and industrial processes [313]. This metalloid is also a contaminant of soils [309,314], surface water [309,315], and ground water used as drinking water [310,313,316], and food [313,316]. In addition, arsenic was used in chemical warfare agents [309,313] and is a known component of some conventional or traditional medicines [24,309,311,313,317,318,319,320,321,322] including those used in Ayurveda medicine [24,320,323,324,325,326,327] and herbal Traditional Chinese Medicine (TCM) [24,328,329,330,331]. However, arsenic is not necessarily a common pollutant in municipal solid waste landfills [24]. Finally, environmental arsenic pollution is a longstanding phenomenon as evidenced by the attic dust approach, whereby dust collected from the attic of old houses is examined, providing an archive of historical air contamination by arsenic in the urban environment [71].
6.2.1. Sources of Arsenic as an Environmental Pollutant
Arsenic exists as a metalloid (As0) [311] and is found in the environment as the inorganic As(III), arsenite [311,332], which is more toxic than As(V), arsenate [332]. Other forms include organic arsenic and arsine (AsH3) [311]. Reviewing the abundant reports, arsenic shows variable degrees of toxicity with As2O3 as one of the most powerful poisons [306,307,308,309,310,311,312,313,314,315,316,317,318,319,320,321,322,323,324,325,326,327,328,329,330,331,332]. The liver is the primary target organ not only for toxicity but also for the metabolism of arsenicals with its major metabolic pathway being via methylation [311,332], which leads to the methylated intermediates of monomethylarsonic acid (MMA) and dimethylarsinic acid (DMA) [332].
6.2.2. Elucidating Health Hazards of Environmental Arsenic
Potential health risks have been reported in various publications, a selection of these are listed (Table 6) [310,333,334,335,336,337,338,339,340,341,342,343,344,345,346,347,348,349,350,351,352,353,354,355,356].
Table 6.
Arsenic sources from natural environments and industry, with proposed impact on human health issues.
Increased LTs due to a chronic intake of arsenic together with tap water or food have been published but the causal attribution of arsenic to real liver injury often remained unclear [310,332,357,358,359]. In general, increases were marginal with serum ALT values of 36.60 ± 11.07 U/L in exposed individuals vs. 31.80 ± 9.41 U/L in nonexposed ones [357] and much lower than those required for a significant liver injury [33]. Similarly, individuals were included with ALT values >1 times the ULN, again not representative for a significant liver injury [310]. These cohorts with marginally increased LTs [310,357] and partially associated clinical signs of hepatomegaly [310] should instead be interpreted as harmless liver adaptation syn liver tolerance because LTs commonly experience a spontaneous return to normal values even if the intake of the causative chemical is not stopped. There is also concern because efforts to exclude alternative causes unrelated to arsenic intake are limited. As individuals are exposed not only to arsenic alone but often also concomitantly to other heavy metals and toxins, causality attribution to arsenic may become a matter of debate. In an early study comprising 2248 patients with evidence of chronic arsenic toxicity, hepatomegaly was present in 190 of 248 patients (76.6%), non-cirrhotic portal fibrosis was the predominant lesion (91.3%) in liver histology, and the maximum arsenic content in the liver was 6 mg/kg, but it was undetected in six of twenty-nine samples studied [310]. Despite diagnostic shortcomings, sufficient evidence exists that a prolonged intake of environmental arsenic may cause liver injury (Table 6) [310,332,357,358,359].
Apart from targeting the liver, arsenic can jeopardize multiple organs (Table 6), a process that develops through various pathways, influenced by environmental bioprocesses [352]. In general, chronic exposure to arsenic may cause malignant, degenerative, and inflammatory diseases [339,345]. As a consequence, these arsenic-related disorders increased the disability-adjusted life years (DALYs) [350] and the mortality rates secondary to specific cancers [336].
6.3. Acute Human Liver Injury by Arsenic
Acute intoxication by a suicidal ingestion of around 8 g arsenic trioxide, which the patient found in the pharmacy of his grandmother, resulted in a fatal outcome and high arsenic amounts in the liver with 147 µg/g dry weight and As3+ as the predominant form, with a higher concentration of MMA compared with DMA, followed by the kidneys with 26.6 µg/g [360]. In other cases of acute arsenic intoxication, ante-mortem blood and urine concentrations ranged from 2.3 to 6.7 µg/mL [361].
Autopsy examinations in patients with acute arsenic poisoning revealed fat degeneration of the liver [362], or more specifically, microvesicular steatosis [363]. After a massive overdose of arsenic trioxide, LT were only marginally elevated with a serum AST activity of 51 U/L and ALT of 68 U/L [364].
Clinical symptoms of acute arsenic poisoning classically start with gastroenteritis involving diarrhea like “rice water” that may be bloody and is associated with severe hypotension as another hallmark viewed as secondary to dehydration and volume loss [312]. Other features may include colicky abdominal pain, arrhythmias, proteinuria, hematuria, and acute renal failure, as well as headache, delirium, encephalopathy, seizures, and a metallic taste occuring in the mouth with a slight odor of garlic in the breath [309]. Typical clinical signs of liver injury like jaundice or increased serum conjugated bilirubin levels are rarely observed [312] or not reported [309]. Acute arsenic exposure is confirmed at a level of 50 µg/L in spot urine or over 100 µg of total arsenic in a 24 h collection using a metal-free polyethylene container [312]. In acute exposure, 24 h urinary arsenic levels typically exceed several thousand micrograms. Spot urine levels commonly exceed 1000 µg/L. An abdominal radiograph may demonstrate intestinal radiopaque metallic flecks in arsenic ingestion [312].
Therapy using metal chelators must be quickly initiated at the first suspicion of an arsenic intoxication without waiting for urinary arsenic results [309,312]. Under consideration and thoroughly discussed are thiol chelators such as meso-2,3-dimercaptosuccinic acid, 2,3-dimercaptopropanol syn sodium 2,3-dimercapto-1-propanesulfonate known as the British-Lewisite antidote (BAL), penicillamine, and ethylenediaminetetraacetic acid, which provide the formation of inert chelator–metal complexes and help through the urinary excretion of arsenic from the body. Other measures include gastrointestinal lavage via mouth or large bowel lavage via rectum, based on anecdotal reports lacking evidence from RCTs due to small case numbers. This limitation also applies to any gastric bezoar suspected to consist of arsenic material upon radiology assessment that needs removal from the stomach by laparotomy or, currently preferred, by endoscopy. Lethality follows in the first few hours from shock or days later from acute renal or liver failure [365].
6.4. Chronic Human Liver Injury by Arsenic
Prolonged arsenic uptake by humans causes the toxicity of various organs including the liver [309,313,316,366] and leads to clinical features due to arsenic poisoning [38,309,313,317,318]. Abnormal LTs like serum activities of ALT, AST, and ALP have been published [272,318,319,321,332] with significantly higher values in patients exposed to substantial amounts of arsenic [332]. As an example, increased LTs were documented in two patients on a therapy including herbal, arsenic-containing Ayurvedic medicines: the first patient (68 years old, male) was on the arsenic-containing remedy for 32 days and showed increased LT values of ALT 473 U/L, AST 498 U/L, and ALP 188 U/L, whereas the second patient (48 years old, female) was on the arsenic medication for 10 days and showed LT increases of ALT 1122 U/L, AST 898 U/L, and ALP 364 U/L. Both cases were assessed for causality using the updated RUCAM [319] and were discussed regarding data interpretation [325,367].
Light microscopical liver histology in patients with prolonged arsenic uptake is characterized by liver fibrosis leading to clinical non-cirrhotic portal hypertension [368,369,370,371], whereby studies on the wedged hepatic vein pressure indicated that the obstruction to portal flow resided in the portal tracts [369]. Histology features of the liver include portal tract fibrosis [368,369,371] and an increase in the number of portal veins with thickening and hypertrophic walls in some reports [368,369]. A few inflammatory cells were described and mild fatty change was seen in the surrounding hepatocytes [368], a finding subsequently confirmed by electron microscopy [317].
6.5. Cascade of Molecular Events Leading to Arsenic Liver Injury
Pathogenetic sequalae involved in liver injury caused by arsenic were preferentially derived from animal studies and case reports, analyzed and reviewed in recent articles [25,26,309,311,312,372]. According to mainstream opinion, several steps are under discussion.
The first step depends on the active transport of the protein-bound arsenic from the blood into the liver. This is facilitated by anion exchange transporters in the plasma cell membrane of hepatocytes, in which arsenic replaces the phosphate [307]. As a non-essential element in humans, arsenic has no specific transporter system on its own.
The second step of the cascade of events is characterized by the binding of arsenic in its most toxic form of As3+ with sulfhydryl groups of enzymes and membrane proteins within the hepatocytes, conditions that lead to cross linkage [311,332,373,374]. Contributing factors to arsenic liver injury are oxidative DNA, acquired tolerance to apoptosis, enhanced cell proliferation, altered DNA methylation, and genomic instability [332,375]. Most of these alterations were confirmed in an experimental model using zebrafish (Danio rerio) intoxicated with arsenic and evaluated for metabolomic changes [376].
The third and final step considers, among others, the replacement of phosphate with arsenic in enzymes involved in the mitochondrial anion exchange transport system Na+/K+-ATPase, which impairs the function of the respiratory chain, as well as the binding of arsenic to the sulfhydryl groups of the ATPases [6,377,378].
Arsenic caused experimental liver injury via ROS, whereby specific radicals are generated [379,380,381], partially by mechanisms including CYP (Figure 3) [379]. Among the radicals were the nitric oxide NO•, singlet oxygen 1O2, hydrogen peroxide, and dimethylarsinic peroxyl radical (CH3)2AsOO•. The individual molecular steps leading to these radicals are difficult to firmly establish because the processes are fast and do not allow capture. However, ROS formation was experimentally ascertained because glutathione attenuated arsenic liver injury as evidenced by ameliorating the ROS parameters like malondialdehyde, superoxide dismutase, 8-hydroxy-2′-deoxyguanosin, and glutathione peroxidase [382].
In experimental studies, arsenic causes an imbalanced immune response as verified by the notable abnormal ratio of Th17 to Treg cells in peripheral blood as well as the secretion of inflammatory cytokines IL-17A, IL-6, TGF-β1, and IL-10 in the serum and liver [383]. In humans, a systematic review and meta-analysis revealed that inflammatory cytokines like interleukin (IL)-6, IL-8, and IL-12 levels were significantly higher in arsenic-exposed individuals compared with the control group, indicating that arsenic exposure can cause inflammatory responses [384].
The key critical issues related to liver injury caused by arsenic are presented in condensed form (Figure 7).

Figure 7.
Selected critical issues of liver injury due to arsenic. Abbreviations: CYP, cytochrome; ROS, reactive oxygen species.
7. Conclusions
Heavy metals share with other substances, like per- and polyfluoroalkyl substances (PFAS), the common characteristics of eternity, persistence, and issues of complete biodegradability that may be hazardous to human health. However, whereas both substance categories are derived from humankind and industry activities, heavy metals also have part of their origin in nature before portions thereof entered the industry segment. As shown in this analysis, heavy metals like copper, iron, cadmium, and arsenic may differently modify human health resulting in positive, negative, or mixed effects. More specifically, liver injury may become a key feature in individuals, if these heavy metals accumulate in the liver. This potentially dangerous accumulation is the result of limited mechanisms providing the required cellular heavy metal homeostasis through the control of uptake and excretion. For instance, genetic mutations cause copper accumulation in the liver of patients with Wilson disease or the same for iron in patients with hemochromatosis. As opposed to and unrelated to any genetic abnormality, cadmium and arsenic constantly accumulate during the whole lifespan in the liver, which is unable to provide the specific cellular homeostasis required. At the mechanistic level, any heavy metal may generate toxic radicals in excess amounts that are insufficiently detoxified through hepatic antioxidant capacities, which are exhausted in the meantime and thereby unable to prevent liver injury. Causality attribution for a single culprit heavy metal may be difficult as they are mostly encountered by humans not singly but in combination with other elements. In this context, the exclusion of alternative acute or chronic liver diseases is mandatory to clinically eliminate confounders. Humans occasionally tend to disqualify heavy metals globally, although essential heavy metals, with copper and iron as examples, among others, were used for human evolution and are still urgently needed for maintaining human health. Their benefits outweigh the risks by far, in addition to their role in ensuring the evolution of humankind, fauna, and flora. Without essential heavy metals, humankind would not exist. So, global criticism on these elements is not really warranted. Genetic disorders like Wilson disease or hemochromatosis can be treated well if recognized early, whereas health hazards due to cadmium or arsenic and other heavy metals are principally preventable diseases through avoiding or reducing their uptake but essentially require more regulatory efforts to help reduce their industrial emissions.
Funding
This research received no external funding.
Conflicts of Interest
The author declares that he has no conflicts of interest.
References
- Belford, R. The Origin of the Elements. Available online: https://chem.libretexts.org/Courses/University_of_Arkansas_Little_Rock/Chem_1403%3A_General_Chemistry_2/Text/21%3A_Nuclear_Chemistry/21.06%3A_The_Origin_of_the_Elements (accessed on 19 March 2024).
- Chaudhry, H.S.; Anilkumar, A.C. Wilson Disease. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2023. Available online: https://www.ncbi.nlm.nih.gov/books/NBK441990/ (accessed on 5 March 2024).
- Clery, D. Some of the Universe’s Heavier Elements are Created by Neutron Star Collisions. Available online: https://e.org/content/article/some-universe-s-heavier-elements-are-created-neutron-star-collisions (accessed on 19 March 2024).
- Frebel, A.; Beers, T.C. Some of the universe’s heavier elements are created by neutron star collisions. Phys. Today 2018, 71, 30–37. [Google Scholar] [CrossRef]
- Sheftel, A.D.; Mason, A.B.; Ponka, P. The long history of iron in the Universe and in health and disease. Biochim. Biophys. Acta 2012, 1820, 161–187. [Google Scholar] [CrossRef] [PubMed]
- Briffa, J.; Sinagra, E.; Blundell, R. Heavy metal pollution in the environment and their toxicological effects in humans. Heliyon 2020, 6, e04691. [Google Scholar] [CrossRef] [PubMed]
- Angon, P.B.; Islam, M.S.; Kc, S.; Das, A.; Anjum, N.; Poudel, A.; Suchi, S.A. Sources, effects and present perspectives of heavy metals contamination: Soil, plants and human food chain. Heliyon 2024, 10, e28357. [Google Scholar] [CrossRef] [PubMed]
- Trefts, E.; Gannon, M.; Wasserman, D.H. The liver. Curr. Biol. 2017, 27, R1147–R1151. [Google Scholar] [CrossRef] [PubMed]
- Kalra, A.; Yetiskul, E.; Wehrle, C.J.; Tuma, F. Physiology, Liver. 1 May 2023. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2024. Available online: https://www.ncbi.nlm.nih.gov/books/NBK535438/ (accessed on 18 March 2024).
- França, M.; Amorim, J.P. Liver increased iron deposition and storage diseases. In Imaging of the Liver and Intra-Hepatic Biliary Tract. Medical Radiology; Quaia, E., Ed.; Springer: Cham, Switzerland, 2021. [Google Scholar] [CrossRef]
- Vaja, R.; Rana, M. Drugs and the liver. Anaesth. Intensiv. Care Med. 2020, 21, 517–523. [Google Scholar] [CrossRef]
- Wen, Y.; Lambrecht, J.; Ju, C.; Tacke, F. Hepatic macrophages in liver homeostasis and diseases-diversity, plasticity and therapeutic opportunities. Cell Mol. Immunol. 2021, 18, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Judge, A.; Dodd, M.S. Metabolism. Essays Biochem. 2020, 64, 607–647. [Google Scholar] [CrossRef] [PubMed]
- Teschke, R.; Danan, G. Liver injury by drugs metabolized via cytochrome P450. J. Mod. Med. Chem. 2020, 8, 93–98. [Google Scholar] [CrossRef]
- Teschke, R.; Danan, G. Idiosyncratic drug induced liver injury, cytochrome P450, metabolic risk factors, and lipophilicity: Highlights and controversies. Int. J. Mol. Sci. 2021, 22, 3441. [Google Scholar] [CrossRef] [PubMed]
- Teschke, R.; Uetrecht, J. Mechanism of idiosyncratic drug induced liver injury (DILI): Unresolved basic issues. In special issue: Unresolved basic issues in hepatology, guest editors Ralf Weiskirchen and Wolfgang Stremmel. Ann. Transl. Med. 2021, 9, 730. [Google Scholar]
- Teschke, R. Alcoholic steatohepatitis (ASH) and acute alcoholic hepatitis (AH): Cascade of events, clinical features, and pharmacotherapy options. Exp. Opin. Pharmacother. 2018, 19, 779–793. [Google Scholar] [CrossRef] [PubMed]
- Teschke, R. Alcoholic liver disease: Alcohol metabolism, cascade of molecular mechanisms, cellular targets, and clinical aspects. Biomedicines 2018, 6, 106. [Google Scholar] [CrossRef] [PubMed]
- Teschke, R. Alcoholic liver disease: Current mechanistic aspects with focus on their clinical relevance. Biomedicines 2019, 7, 68. [Google Scholar]
- Teschke, R. Biochemical aspects of the hepatic microsomal ethanol-oxidizing system (MEOS). Resolved initial controversies and updated molecular views. Biochem. Pharmacol. 2019, 8, 267. [Google Scholar] [CrossRef]
- Teschke, R. Aliphatic halogenated hydrocarbons: Liver injury in 60 patients. J. Clin. Trans. Hepatol. 2018, 6, 1–12. [Google Scholar]
- Teschke, R. Liver injury by carbon tetrachloride intoxication in 16 patients treated with forced ventilation to accelerate toxin removal via the lungs: A clinical report. Toxics 2018, 6, 25. [Google Scholar] [CrossRef] [PubMed]
- Teschke, R. Intoxications by aliphatic halogenated hydrocarbons: Hepatotoxic risks for patients and clinical issues including role of CO2-induced hyperventilation as therapy option. J. Clin. Exp. Toxicol. 2018, 2, 25–29. [Google Scholar]
- Vongdala, N.; Tran, H.D.; Xuan, T.D.; Teschke, R.; Khanh, T.D. Heavy metal accumulation in water, soil, and plants of municipal solid waste landfill in Vientiane, Laos. Int. J. Environ. Res. Public. Health 2019, 16, 22. [Google Scholar] [CrossRef] [PubMed]
- Quan, N.V.; Xuan, T.D.; Teschke, R. Potential hepatotoxins found in herbal medicinal products: A systematic review. Int. J. Mol. Sci. 2020, 21, 5011. [Google Scholar] [CrossRef]
- Teschke, R.; Xuan, T.D. Heavy metals, halogenated hydrocarbons, phthalates, glyphosate, cordycepin, alcohol, drugs, and herbs, assessed for liver injury and mechanistic steps. Special Issue: Hepatotoxicity: Molecular Mechanisms and Pathophysiology. Front. Biosci. Landmark 2022, 27, 314. [Google Scholar] [CrossRef]
- Teschke, R. Aluminum, arsenic, beryllium, cadmium, chromium, cobalt, copper, iron, lead, mercury, molybdenum, nickel, platinum, thallium, titanium, vanadium, and zinc: Molecular aspects in experimental liver injury. Int. J. Mol. Sci. 2022, 23, 12213. [Google Scholar] [CrossRef] [PubMed]
- Adams, P.C.; Jeffrey, G.; Ryan, J. Haemochromatosis. Lancet 2023, 401, 10390. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.; Roberts, S.M.; Saab, I.N. Review of regulatory reference values and background levels for heavy metals in the human diet. Regul. Toxicol. Pharmacol. 2022, 130, 105122. [Google Scholar] [CrossRef] [PubMed]
- Tchounwou, P.B.; Yedjou, C.G.; Patlolla, A.K.; Sutton, D.J. Heavy metal toxicity and the environment. In: Luch, A. (eds) Molecular, Clinical and Environmental Toxicology. Exp. Suppl. 2012, 101, 133–164. [Google Scholar] [CrossRef] [PubMed]
- Aldridge, J.E.; Gibbons, J.A.; Flaherty, M.M.; Kreider, M.L.; Romano, J.A.; Levin, E.D. Heterogeneity of toxicant response: Sources of human variability. Toxicol. Sci. 2003, 76, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Danan, G.; Bénichou, C. Causality assessment of adverse reactions to drugs—I. A novel method based on the conclusions of international consensus meetings: Application to drug-induced liver injuries. J. Clin. Epidemiol. 1993, 46, 1323–1330. [Google Scholar] [CrossRef] [PubMed]
- Danan, G.; Teschke, R. RUCAM in drug and herb induced liver injury: The update. Int. J. Mol. Sci. 2016, 17, 14. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Guo, D.; Xu, Y.; Zhu, M.; Yao, C.; Chen, C.; Jia, W. Comparison of different liver test thresholds for drug-induced liver injury: Updated RUCAM versus other methods. Front. Pharmacol. 2019, 10, 816. [Google Scholar] [CrossRef] [PubMed]
- Allison, R.; Guraka, A.; Shawa, I.T.; Tripathi, G.; Moritz, W.; Kermanizadeh, A. Drug induced liver injury—A 2023 update. J. Toxicol. Environ. Health Part B 2023, 12, 875–883. [Google Scholar] [CrossRef] [PubMed]
- Hosack, T.; Damry, D.; Biswas, S. Drug-induced liver injury: A comprehensive review. Ther. Adv. Gastroenterol. 2023, 16, 17562848231163410. [Google Scholar]
- Kobayashi, T.; Iwaki, M.; Nogami, A.; Yoneda, M. Epidemiology and management of drug-induced liver injury: Importance of the updated RUCAM. J. Clin. Transl. Hepatol. 2023, 11, 1239–1245. [Google Scholar] [CrossRef] [PubMed]
- Kamal, S.; Abdelhakam, S.; Ghoraba, D.; Massoud, Y.; Aziz, K.A.; Hassan, H.; Hafez, T.; Abdel Sallam, A. The frequency, clinical course, and health related quality of life in adults with Gilbert’s syndrome: A longitudinal study. BMC Gastroenterol. 2019, 19, 22. [Google Scholar] [CrossRef] [PubMed]
- Teschke, R. Idiosyncratic DILI: Analysis of 46,266 cases assessed for causality by RUCAM and published from 2014 to early 2019. Special issue. Front. Pharmacol. 2019, 10, 730. [Google Scholar] [CrossRef] [PubMed]
- Ho, C.M.; Chen, C.L.; Chang, C.H.; Lee, M.R.; Wang, J.Y.; Hu, R.H.; Lee, P.H. Circulatory inflammatory mediators in the prediction of anti-tuberculous drug-induced liver injury using RUCAM for causality assessment. Biomedicines 2021, 9, 891. [Google Scholar] [CrossRef]
- Teschke, R. DILI, HILI, RUCAM algorithm, and AI, the Artificial Intelligence: Provocative issues, progress, and proposals. Arch. Gastroenterol. Res. 2020, 1, 4–11. [Google Scholar]
- Teschke, R.; Danan, G. Worldwide use of RUCAM for causality assessment in 81,856 idiosyncratic DILI and 14,029 HILI cases published 1993—Mid 2020: A comprehensive analysis. Medicines 2020, 7, 62. [Google Scholar] [CrossRef] [PubMed]
- Sivasailam, B.; Kumar, A.; Marciniak, E.; Deepak, J. Acute liver failure induced by Joss paper ingestion. Med. Case Rep. 2019, 5, 9. [Google Scholar]
- Witkowska, D.; Słowik, J.; Chilicka, K. Heavy metals and human health: Possible exposure pathways and the competition for protein binding sites. Molecules 2021, 26, 6060. [Google Scholar] [CrossRef] [PubMed]
- Teschke, R.; Vongdala, N.; Quan, N.V.; Quy, T.N.; Xuan, T.D. Metabolic toxification of 1,2-unsaturated pyrrolizidine alkaloids causes human hepatic sinusoidal obstruction syndrome: The update. Int. J. Mol. Sci. 2021, 22, 10419. [Google Scholar] [CrossRef] [PubMed]
- Nowak, M.; Wittke, C.; Lederer, I.; Klier, B.; Kleinwächter, M.; Selmar, D. Interspecific transfer of pyrrolizidine alkaloids: An unconsidered source of contaminations of phytopharmaceuticals and plant derived commodities. Food Chem. 2016, 213, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Selmar, D.; Wittke, C.; Beck von Wolffersdorff, I.; Klier, B.; Lewerenz, L.; Kleinwächter, M.; Nowak, M. Transfer of pyrrolizidine alkaloids between living plants: A disregarded source of contaminations. Environ. Pollut. 2019, 248, 456–461. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Lv, L.; Liu, Y.; Ji, M.; Zang, E.; Liu, Q.; Zhang, M.; Li, M. Applied analytical methods for detecting heavy metals in medicinal plants. Crit. Rev. Analyt. Chem. 2023, 53, 339–359. [Google Scholar] [CrossRef] [PubMed]
- Luo, L.; Wang, B.; Jiang, J.; Fitzgerald, M.; Huang, Q.; Yu, Z.; Li, H.; Zhang, J.; Wei, J.; Yang, C.; et al. Heavy metal contaminations in herbal medicines: Determination, comprehensive risk assessments, and solutions. Front. Pharmacol. 2021, 11, 595335. [Google Scholar] [CrossRef] [PubMed]
- Philips, A.B.; Ahamed, R.; Abduljaleel, J.K.; Rajesh, S.; Theruvath, A.H.; Raveendran, R.; Augustine, R. Ayurvedic treatment induced severe alcoholic hepatitis and non-cirrhotic portal hypertension in a 14-year-old girl. Oxf. Med. Case Rep. 2022, 2022, omac113. [Google Scholar] [CrossRef] [PubMed]
- Teschke, R. Molecular idiosyncratic toxicology of drugs in the human liver compared with animals: Basic considerations. Int. J. Mol. Sci. 2023, 24, 6663. [Google Scholar] [CrossRef] [PubMed]
- Collins, J.F. Copper nutrition and biochemistry and human (patho)physiology. Adv. Food Nutr. Res. 2021, 96, 311–364. [Google Scholar] [CrossRef] [PubMed]
- Tapiero, H.; Townsend, D.M.; Tew, K.D. Trace elements in human physiology and pathology. Copper. Biomed. Pharmacother. 2003, 57, 386–398. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Zheng, L.; Ma, S.R.; Li, J.; Yang, S. Cuproptosis: Emerging biomarkers and potential therapeutics in cancer. Front. Oncol. 2023, 13, 1288504. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Yang, Y.; Gao, Y.; He, J. Cuproptosis: Mechanisms and links with cancers. Mol. Cancer 2023, 22, 46. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Xiao, P.; Qiu, B.; Yu, H.F.; Teng, C.B. Copper chaperone antioxidant 1: Multiple roles and a potential therapeutic target. J. Mol. Med. 2023, 101, 527–542. [Google Scholar] [PubMed]
- Bost, M.; Houdart, S.; Oberli, M.; Kalonji, E.; Huneau, J.F.; Margaritis, I. Dietary copper and human health: Current evidence and unresolved issues. J. Trace Elem. Med. Biol. 2016, 35, 107–115. [Google Scholar] [CrossRef]
- Murray, L.; Arteche, A.; Fearon, P.; Halligan, S.; Goodyer, I.; Cooper, P. Maternal postnatal depression and the development of depression in offspring up to 16 years of age. J. Am. Acad. Child. Adolesc. Psychiatry 2011, 50, 460–470. [Google Scholar] [CrossRef] [PubMed]
- Roberts, E.A.; Sarkar, B. Liver as a key organ in the supply, storage, and excretion of copper. Am. J. Clin. Nutr. 2008, 88, 851S–854S. [Google Scholar] [CrossRef] [PubMed]
- Chanpong, A.; Dhawan, A. Wilson disease in children and young adults—State of the art. Saudi J. Gastroenterol. 2022, 28, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Gaetke, L.M.; Chow-Johnson, H.S.; Chow, C.K. Copper: Toxicological relevance and mechanisms. Arch. Toxicol. 2014, 88, 1929–1938. [Google Scholar] [CrossRef] [PubMed]
- Malinowska, E.; Jankowski, K. Copper and zinc concentrations in medicinal herbs and Soil surrounding ponds on agricultural land. Landsc. Ecol. Eng. 2017, 13, 183–188. [Google Scholar] [CrossRef]
- Nayak, N.C.; Chitale, A.R. Indian childhood cirrhosis (ICC) and ICC-like diseases: The changing scenario of facts versus notions. Indian. J. Med. Res. 2013, 137, 1029–1042. [Google Scholar] [PubMed]
- Yruela, I. Copper in plants: Acquisition, transport and interactions. Funct. Plant Biol. 2009, 36, 409–430. [Google Scholar] [CrossRef]
- Nawrot, N.; Wojciechowska, E.; Rezania, S.; Walkusz-Miotk, J.; Pazdro, K. The effects of urban vehicle traffic on heavy metal contamination in road sweeping waste and bottom sediments of retention tanks. Sci. Total Environ. 2020, 749, 141511. [Google Scholar] [CrossRef] [PubMed]
- Stremmel, W.; Weiskirchen, R. Therapeutic strategies in Wilson disease: Pathophysiology and mode of action. Ann. Transl. Med. 2021, 9, 732. [Google Scholar] [CrossRef] [PubMed]
- Weber, T.; Solioz, M. Evaluation of chocolate as a source of dietary copper. Eur. Food Technol. 2014, 238, 1063–1066. [Google Scholar] [CrossRef][Green Version]
- Adeyeye, E.I.; Arogundade, L.A.; Asaolu, S.S.; Olaofe, O. Fungicide-derived copper content in soil and vegetation component, Owena cocoa (Theobroma cacao L.) plantations in Nigeria. Bangladesh J. Sci. Ind. Res. 2006, 41, 129–140. [Google Scholar] [CrossRef]
- Flemming, C.A.; Trevors, J.T. Copper toxicity and chemistry in the environment: A review. Water Air Soil. Pollut. 1989, 44, 143–158. [Google Scholar] [CrossRef]
- Kovačič, G.R.; Lešnik, M.; Vršič, S. An overview of the copper situation and usage in viticulture. Bulg. J. Agric. Sci. 2013, 19, 50–59. [Google Scholar]
- Gaberšek, M.; Watts, M.J.; Gosar, M. Attic dust: An archive of historical air contamination of the urban environment and potential hazard to health? J. Hazard. Mat. 2022, 432, 128745. [Google Scholar] [CrossRef] [PubMed]
- Brüssow, H. What is health? Microb. Biotechnol. 2013, 6, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Bircher, J.; Kuruvilla, S. Defining health by addressing individual, social, and environmental determinants: New opportunities for health care and public health. J. Public Health Policy 2014, 35, 363–386. [Google Scholar] [CrossRef] [PubMed]
- Rehman, M.; Liu, L.; Wang, Q.; Saleem, M.H.; Bashir, S.; Ullah, S.; Peng, D. Copper environmental toxicology, recent advances, and future outlook: A review. Environ. Sci. Pollut. Res. Int. 2019, 26, 18003–18016. [Google Scholar] [CrossRef] [PubMed]
- Covre, W.P.; Ramos, S.J.; Pereira, W.V.D.S.; Souza, E.S.; Martins, G.C.; Teixeira, O.M.M.; Amarante, C.B.D.; Dias, Y.N.; Fernandes, A.R. Impact of copper mining wastes in the Amazon: Properties and risks to environment and human health. J. Hazard. Mater. 2022, 421, 126688. [Google Scholar] [CrossRef] [PubMed]
- Shabbir, Z.; Sardar, A.; Shabbir, A.; Abbas, G.; Shamshad, S.; Khalid, S.; Natasha Murtaza, G.; Dumat, C.; Shahid, M. Copper uptake, essentiality, toxicity, detoxification and risk assessment in soil-plant environment. Chemosphere 2020, 259, 127436. [Google Scholar] [CrossRef] [PubMed]
- Georgopoulos, P.G.; Roy, A.; Yonone-Lioy, M.J.; Opiekun, R.E.; Lioy, P.J. Environmental copper: Its dynamics and human exposure issues. J. Toxicol. Environ. Health B Crit. Rev. 2001, 4, 341–394. [Google Scholar] [CrossRef] [PubMed]
- Hsu, H.W.; Bondy, S.C.; Kitazawa, M. Environmental and dietary exposure to copper and its cellular mechanisms linking to Alzheimer’s disease. Toxicol. Sci. 2018, 163, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Pandita, S.; Singh Sidhu, G.P.; Sharma, A.; Khanna, K.; Kaur, P.; Bali, A.S.; Setia, R. Copper bioavailability, uptake, toxicity and tolerance in plants: A comprehensive review. Chemosphere 2021, 262, 127810. [Google Scholar] [CrossRef] [PubMed]
- Civardi, C.; Schwarze, F.W.; Wick, P. Micronized copper wood preservatives: An efficiency and potential health risk assessment for copper-based nanoparticles. Environ. Pollut. 2015, 200, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Yan, B.; Xu, D.M.; Chen, T.; Yan, Z.A.; Li, L.L.; Wang, M.H. Leachability characteristic of heavy metals and associated health risk study in typical copper mining-impacted sediments. Chemosphere 2020, 239, 124748. [Google Scholar] [CrossRef] [PubMed]
- Stern, B.R. Essentiality and toxicity in copper health risk assessment: Overview, update and regulatory considerations. J. Toxicol. Environ. Health A 2010, 73, 114–127. [Google Scholar] [CrossRef] [PubMed]
- Filimon, M.N.; Caraba, I.V.; Popescu, R.; Dumitrescu, G.; Verdes, D.; Petculescu Ciochina, L.; Sinitean, A. Potential ecological and human health risks of heavy metals in soils in selected copper mining areas-a case study: The Bor area. Int. J. Environ. Res. Public. Health 2021, 18, 1516. [Google Scholar] [CrossRef] [PubMed]
- Ameh, T.; Sayes, C.M. The potential exposure and hazards of copper nanoparticles: A review. Environ. Toxicol. Pharmacol. 2019, 71, 10322. [Google Scholar] [CrossRef]
- Chowdhury, R.; Ramond, A.; O′Keeffe, L.M.; Shahzad, S.; Kunutsor, S.K.; Muka, T.; Gregson, J.; Willeit, P.; Warnakula, S.; Khan, H.; et al. Environmental toxic metal contaminants and risk of cardiovascular disease: Systematic review and meta-analysis. BMJ 2018, 362, k3310. [Google Scholar] [CrossRef] [PubMed]
- Gujre, N.; Rangan, L.; Mitra, S. Occurrence, geochemical fraction, ecological and health risk assessment of cadmium, copper and nickel in soils contaminated with municipal solid wastes. Chemosphere 2021, 271, 129573. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A.A.; Tsuji, J.S.; Garry, M.R.; McArdle, M.E.; Goodfellow, W.L., Jr.; Adams, W.J.; Menzie, C.A. Critical review of exposure and effects: Implications for setting regulatory health criteria for ingested copper. Environ. Manag. 2020, 65, 131–159. [Google Scholar] [CrossRef] [PubMed]
- Nematollahi, M.J.; Keshavarzi, B.; Zaremoaiedi, F.; Rajabzadeh, M.A.; Moore, F. Ecological-health risk assessment and bioavailability of potentially toxic elements (PTEs) in soil and plant around a copper smelter. Environ. Monit. Assess. 2020, 192, 639. [Google Scholar] [CrossRef]
- Wang, M.; Lv, Y.; Lv, X.; Wang, Q.; Li, Y.; Lu, P.; Yu, H.; Wei, P.; Cao, Z.; An, T. Distribution, sources and health risks of heavy metals in indoor dust across China. Chemosphere 2023, 313, 137595. [Google Scholar] [CrossRef] [PubMed]
- Lerner, C.A.; Sundar, I.K.; Watson, R.M.; Elder, A.; Jones, R.; Done, D.; Kurtzman, R.; Ossip, D.J.; Robinson, R.; McIntosh, S.; et al. Environmental health hazards of e-cigarettes and their components: Oxidants and copper in e-cigarette aerosols. Environ. Pollut. 2015, 198, 100–107. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yang, Q. Association between serum copper levels and lung cancer risk: A meta-analysis. J. Int. Med. Res. 2018, 46, 4863–4873. [Google Scholar] [CrossRef] [PubMed]
- Royer, A.; Sharman, T. Copper Toxicity. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2023. Available online: https://www.ncbi.nlm.nih.gov/books/NBK557456/ (accessed on 18 March 2024).
- Carvalho, J.A.; Boavida, L.; Ferreira, R.; Favas, C.; Delgado Alves, J. Copper-induced haemolytic anaemia. Eur. J. Case Rep. Intern. Med. 2021, 8, 002785. [Google Scholar] [CrossRef]
- Gamakaranage, C.S.; Rodrigo, C.; Weerasinghe, S.; Gnanathasan, A.; Puvanaraj, V.; Fernando, H. Complications and management of acute copper sulphate poisoning; a case discussion. J. Occup. Med. Toxicol. 2011, 6, 34. [Google Scholar] [CrossRef]
- Jantsch, W.; Kulig, K.; Rumack, B.H. Massive copper sulfate ingestion resulting in hepatotoxicity. J. Toxicol. Clin. Toxicol. 1985, 22, 585–588. [Google Scholar] [CrossRef] [PubMed]
- Sinkovic, A.; Striding, A.; Svensek, F. Severe acute copper sulphate poisoning: A case report. Arh. Hig. Rada Toksikol. 2008, 59, 31–35. [Google Scholar] [CrossRef] [PubMed]
- Sonal Sekhar, M.; Rao, M. Clinical toxicology of copper: Source, toxidrome, mechanism of toxicity, and management. In Metal Toxicology Handbook; Bagchi, D., Bagchi, M., Eds.; CRC Press: Boca Raton, FL, USA, 2020; pp. 199–217. [Google Scholar]
- Berentsen, S.; Barcellini, W. Autoimmune hemolytic anemias. N. Engl. J. Med. 2021, 385, 1407–1419. [Google Scholar] [CrossRef] [PubMed]
- Lizaola-Mayo, B.C.; Dickson, R.C.; Lam-Himlin, D.M.; Chascsa, D.M. Exogenous copper exposure causing clinical Wilson disease in a patient with copper deficiency. BMC Gastroenterol. 2021, 21, 278. [Google Scholar] [CrossRef] [PubMed]
- Priya, S. Acute copper sulphate poisoning. Ind. J. Med. Spec. 2018, 9, 140–142. [Google Scholar] [CrossRef]
- Chugh, K.S.; Sharma, B.K.; Singhal, P.C.; Das, K.C.; Datta, B.N. Acute renal failure following copper sulphate intoxication. Postgrad. Med. J. 1977, 53, 18–23. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Araya, M.; Kelleher, S.L.; Arredondo, M.A.; Sierralta, W.; Vial, M.T.; Uauy, R.; Lönnerdal, B. Effects of chronic copper exposure during early life in rhesus monkeys. Am. J. Clin. Nutr. 2005, 81, 1065–1071. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Araya, M.; Núñez, H.; Pavez, L.; Arredondo, M.; Méndez, M.; Cisternas, F.; Pizarro, F.; Sierralta, W.; Uauy, R.; González, M. Administration of high doses of copper to Capuchin Monkeys does not cause liver damage but induces transcriptional activation of hepatic proliferative responses. J. Nutr. 2012, 142, 233–237. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Xu, M.; Luo, J.; Zhao, L.; Ye, G.; Shi, F.; Lv, C.; Chen, H.; Wang, Y.; Li, Y. Liver toxicity assessments in rats following sub-chronic oral exposure to copper nanoparticles. Environ. Sci. Eur. 2019, 31, 30. [Google Scholar] [CrossRef]
- Vogel, F.S. The deposition of exogenous copper under experimental conditions with observations on its neurotoxic and nephrotoxic properties in relation to Wilson’s disease. J. Exp. Med. 1959, 110, 801–810. [Google Scholar] [CrossRef]
- Stremmel, W.; Merle, U.; Weiskirchen, R. Clinical features of Wilson disease. Ann. Transl. Med. 2019, 7 (Suppl. S2), S61. [Google Scholar] [CrossRef] [PubMed]
- Garrido, I.; Marques, M.; Liberal, R.; Cardoso, H.; Lopes, S.; Macedo, G. Wilson disease in Northern Portugal: A long-term follow-up study. Orphanet J. Rare Dis. 2022, 17, 82. [Google Scholar] [CrossRef] [PubMed]
- Moini, M.; To, U.; Schilsky, M.K. Recent advances in Wilson disease. Trans. Gastroenterol. Hepatol. 2021, 6, 21. [Google Scholar] [CrossRef] [PubMed]
- Pandit, A.; Bhave, S. Present interpretation of the role of copper in Indian childhood cirrhosis. Am. J. Clin. Nutr. 1996, 63, 830S–835S. [Google Scholar] [CrossRef] [PubMed]
- Hamza, I.; Gitlin, J.D. Hepatic copper transport. In Madame Curie Bioscience Database; Landes Bioscience: Austin, TX, USA, 2000–2013. Available online: https://www.ncbi.nlm.nih.gov/books/NBK6381/ (accessed on 8 March 2024).
- Müller-Höcker, J.; Weiß, M.; Meyer, U.; Schramel, P.; Wiebecke, B.; Belohradsky, B.H.; Hübner, G. Fatal copper storage disease of the liver in a German infant resembling Indian childhood cirrhosis. Virchows Arch. A 1987, 411, 379–385. [Google Scholar] [CrossRef] [PubMed]
- Müller, T.; Feichtinger, H.; Berger, H.; Müller, W. Endemic Tyrolean infantile cirrhosis: An ecogenetic disorder. Lancet 1996, 347, 877–880. [Google Scholar] [CrossRef] [PubMed]
- Müller, T.; Müller, W.; Feichtinger, H. Idiopathic copper toxicosis. Am. J. Clin. Nutr. 1998, 67, 1082S–1086S. [Google Scholar] [CrossRef] [PubMed]
- Horslen, S.P.; Tanner, M.S.; Lyon, T.D.; Fell, G.S.; Lowry, M.F. Copper associated childhood cirrhosis. Gut 1994, 35, 1497–1500. [Google Scholar] [CrossRef] [PubMed]
- Scheinberg, I.H.; Sternlieb, I. Wilson disease and idiopathic copper toxicosis. Am. J. Clin. Nutr. 1996, 63, 842S–845S. [Google Scholar] [CrossRef]
- Sandahl, T.D.; Laursen, T.L.; Munk, D.E.; Vilstrup, H.; Weiss, K.H.; Ott, P. The prevalence of Wilson’s disease: An update. Hepatology 2020, 71, 722–732. [Google Scholar] [CrossRef] [PubMed]
- Merle, U.; Schaefer, M.; Ferenci, P.; Stremmel, W. Clinical presentation, diagnosis and long-term outcome of Wilson’s disease: A cohort study. Gut 2007, 56, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, H.; Watanabe, K.; Inui, A.; Kato, A.; Tatsumi, Y.; Okumura, A.; Fujisawa, T.; Kato, K. Alanine aminotransferase as the first test parameter for Wilson’s disease. J. Clin. Transl. Hepatol. 2019, 7, 293–296. [Google Scholar] [CrossRef] [PubMed]
- Hassoun, J.; Hammer, N.; Magini, G.; Ponte, B.; Ongaro, M.; Rougemont, A.L.; Goossens, N.; Frossard, J.L.; Spahr, L. Management of acute Wilsonian hepatitis with severe hemolysis: A successful combination of chelation and MARS dialysis. Case Rep. Hepatol. 2021, 2021, 5583654. [Google Scholar] [CrossRef] [PubMed]
- Kasztelan-Szczerbinska, B.; Cichoz-Lach, H. Wilson’s disease: An update on the diagnostic workup and management. J. Clin. Med. 2021, 10, 5097. [Google Scholar] [CrossRef] [PubMed]
- Mohr, I.; Weiss, K.H. Current anti-copper therapies in management of Wilson disease. Ann. Transl. Med. 2019, 7 (Suppl. S2), S69. [Google Scholar] [CrossRef]
- Alkhouri, N.; Gonzalez-Peralta, R.P.; Medici, V. Wilson disease: A summary of the updated AASLD Practice Guidance. Hepatol. Commun. 2023, 7, e0150. [Google Scholar] [CrossRef] [PubMed]
- Korman, J.D.; Volenberg, I.; Balko, J.; Webster, J.; Schiodt, F.V.; Squires, R.H., Jr.; Fontana, R.J.; Lee, W.M.; Schilsky, M.L.; Pediatric and Adult Acute Liver Failure Study Groups. Screening for Wilson disease in acute liver failure: A comparison of currently available diagnostic tests. Hepatology 2008, 48, 1167–1174. [Google Scholar] [CrossRef] [PubMed]
- Mbala, J.; Belmalih, A.; Guillaud, O.; Lachaux, A.; Couchonnal Bedoya, E. Evaluation of vitamin B6 supplementation in Wilson’s disease patients treated with D-penicillamine. BMJ Open Gastroenterol. 2023, 10, e001211. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Dong, J.; Cheng, N.; Yang, R.; Han, Y.; Han, Y. Inflammatory cytokines expression in Wilson’s disease. Neurol. Sci. 2019, 40, 1059–1066. [Google Scholar] [CrossRef] [PubMed]
- Antczak-Kowalska, M.; Członkowska, A.; Eyileten, C.; Palejko, A.; Cudna, A.; Wolska, M.; Piechal, A.; Litwin, T. Autoantibodies in Wilson disease: Impact on clinical course. JIMD Rep. 2022, 63, 508–517. [Google Scholar] [CrossRef] [PubMed]
- Hennes, E.M.; Zeniya, M.; Czaja, A.J.; Parés, A.; Dalekos, G.N.; Krawitt, E.L.; Bittencourt, P.L.; Porta, G.; Boberg, K.M.; Hofer, H.; et al. Simplified criteria for the diagnosis of autoimmune hepatitis. Hepatology 2008, 48, 169–176. [Google Scholar] [CrossRef]
- Ferenci, P.; Caca, K.; Loudianos, G.; Mieli-Vergani, G.; Tanner, S.; Sternlieb, I.; Schilsky, M.; Cox, D.; Berr, F. Diagnosis and phenotypic classification of Wilson disease. Liver Int. 2003, 23, 139–142. [Google Scholar] [CrossRef] [PubMed]
- Nagral, A.; Sarma, M.S.; Matthai, J.; Kukkle, P.L.; Devarbhavi, H.; Sinha, S.; Alam, S.; Bavdekar, A.; Dhiman, R.K.; Eapen, C.E.; et al. Wilson’s Disease: Clinical Practice Guidelines of the Indian National Association for Study of the Liver, the Indian Society of Pediatric Gastroenterology, Hepatology and Nutrition, and the Movement Disorders Society of India. J. Clin. Exp. Hepatol. 2019, 9, 74–98, Erratum in J. Clin. Exp. Hepatol. 2020, 10, 99. [Google Scholar] [CrossRef] [PubMed]
- Sood, V.; Rawat, D.; Khanna, R.; Alam, S. Cholestatic liver disease masquerading as Wilson disease. Indian J. Gastroenterol. 2015, 34, 174–177. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, S.M.; Matsukuma, K.E.; Medici, V. Wilson disease and the differential diagnosis of its hepatic manifestations: A narrative review of clinical, laboratory, and liver histological features. Ann. Transl. Med. 2021, 9, 1394. [Google Scholar] [CrossRef] [PubMed]
- Emanuele, P.; Goodman, Z.D. A simple and rapid stain for copper in liver tissue. Ann. Diagn. Pathol. 1998, 2, 125–126. [Google Scholar] [CrossRef]
- EASL. EASL clinical practice guidelines: Wilson’s disease. J. Hepatol. 2012, 56, 671–685. [Google Scholar] [CrossRef] [PubMed]
- Sternlieb, I. Mitochondrial and fatty changes in hepatocytes of patients with Wilson’s disease. Gastroenterology 1968, 55, 354–367. [Google Scholar] [CrossRef] [PubMed]
- Weiss, K.; Stremmel, W. Evolving perspectives in Wilson disease: Diagnosis, treatment and monitoring. Curr. Gastroenterol. Rep. 2012, 14, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Kolbaum, A.E.; Sarvan, I.; Bakhiya, N.; Spolders, M.; Pieper, R.; Schubert, J.; Jung, C.; Hackethal, C.; Sieke, C.; Grünewald, K.H.; et al. Long-term dietary exposure to copper in the population in Germany—Results from the BfR MEAL study. Food Chem. Toxicol. 2023, 176, 113759. [Google Scholar] [CrossRef] [PubMed]
- Ferenci, P.; Stremmel, W.; Członkowska, A.; Szalay, F.; Viveiros, A.; Stättermayer, A.F.; Bruha, R.; Houwen, R.; Pop, T.L.; Stauber, R.; et al. Age and sex but not ATP7B genotype effectively influence the clinical phenotype of Wilson disease. Hepatology 2019, 69, 1464–1476. [Google Scholar] [CrossRef] [PubMed]
- Woimant, F.; Debray, D.; Morvan, E.; Obadia, M.A.; Poujois, A. Efficacy and safety of two salts of trientine in the treatment of Wilson’s disease. J. Clin. Med. 2022, 11, 3975. [Google Scholar] [CrossRef] [PubMed]
- Siegemund, R.; Lössner, J.; Günther, K.; Kühn, H.J.; Bachmann, H. Mode of action of triethylenetetramine dihydrochloride on copper metabolism in Wilson’s disease. Acta Neurol. Scand. 1991, 83, 364–366. [Google Scholar] [CrossRef] [PubMed]
- Ranucci, G.; Di Dato, F.; Spagnuolo, M.I.; Vajro, P.; Iorio, R. Zinc monotherapy is effective in Wilson’s disease patients with mild liver disease diagnosed in childhood: A retrospective study. Orphanet J. Rare Dis. 2014, 9, 41. [Google Scholar] [CrossRef] [PubMed]
- Avan, A.; Członkowska, A.; Gaskin, S.; Granzotto, A.; Sensi, S.L.; Hoogenraad, T.U. The role of zinc in the treatment of Wilson’s disease. Int. J. Mol. Sci. 2022, 23, 9316. [Google Scholar] [CrossRef] [PubMed]
- Soriot, P. AstraZeneca Cuts Alexion’s PhIII Wilson Disease Drug, Takes $244M Write-Down. Available online: https://endpts.com/astrazeneca-cuts-alexions-phiii-wilson-disease-drug-takes-244m-writedown/ (accessed on 27 April 2023).
- Kim, P.; Zhang, C.C.; Thoröe-Boveleth, S.; Buhl, E.M.; Weiskirchen, S.; Stremmel, W.; Merle, U.; Weiskirchen, R. Analyzing the therapeutic efficacy of Bis-Choline-Tetrathiomolybdate in the Atp7b-/- copper overload mouse model. Biomedicines 2021, 9, 1861. [Google Scholar] [CrossRef] [PubMed]
- Stremmel, W. Bis-choline tetrathiomolybdate as old drug in a new design for Wilson’s disease: Good for brain and liver? Hepatology 2019, 69, 901–903. [Google Scholar] [CrossRef] [PubMed]
- Weiss, K.H.; Askari, F.K.; Czlonkowska, A.; Ferenci, P.; Bronstein, J.M.; Bega, D.; Ala, A.; Nicholl, D.; Flint, S.; Olsson, L.; et al. Bis-choline tetrathiomolybdate in patients with Wilson’s disease: An open-label, multicentre, phase 2 study. Lancet Gastroenterol. Hepatol. 2017, 2, 869–876. [Google Scholar] [CrossRef] [PubMed]
- Catana, A.M.; Medici, V. Liver transplantation for Wilson disease. World J. Hepatol. 2012, 4, 5–10. [Google Scholar] [CrossRef] [PubMed]
- Litwin, T.; Bembenek, J.P.; Antos, A.; Przybyłkowski, A.; Skowrońska, M.; Kurkowska-Jastrzębska, I.; Członkowska, A. Liver transplantation as a treatment for Wilson’s disease with neurological presentation: A systematic literature review. Acta Neurol. Belg. 2022, 122, 505–518. [Google Scholar] [CrossRef] [PubMed]
- Roy-Chowdhury, N.; Roy-Chowdhury, J. Hepatocyte Transplantation. Available online: https://www.uptodate.com/contents/hepatocyte-transplantation (accessed on 3 November 2023).
- Chen, X.; Xing, S.; Feng, Y.; Chen, S.; Pei, Z.; Wang, C.; Liang, X. Early stage transplantation of bone marrow cells markedly ameliorates copper metabolism and restores liver function in a mouse model of Wilson disease. BMC Gastroenterol. 2011, 11, 75. [Google Scholar] [CrossRef] [PubMed]
- Teschke, R.; Eickhoff, A. Wilson disease: Copper-mediated cuproptosis, iron-related ferroptosis, and clinical issues, with comprehensive and critical analysis update Special issue: Heavy metals. Int. J. Mol. Sci. 2024, 25, 4753. [Google Scholar] [CrossRef] [PubMed]
- Gioilli, B.D.; Kidane, T.Z.; Fieten, H.; Tellez, M.; Dalphin, M.; Nguyen, A.; Nguyen, K.; Linder, M.C. Secretion and uptake of copper via a small copper carrier in blood fluid. Metallomics 2022, 14, mfac006. [Google Scholar] [CrossRef] [PubMed]
- Bennett, C.F.; Latorre-Muro, P.; Puigserver, P. Mechanisms of mitochondrial respiratory adaptation. Nat. Rev. Mol. Cell Biol. 2022, 23, 817–835. [Google Scholar] [CrossRef] [PubMed]
- Morio, B.; Panthu, B.; Bassot, A.; Rieusset, J. Role of mitochondria in metabolic health and diseases. Cell Calcium 2021, 94, 102336. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Wang, J.; Pu, C.; Qiao, L.; Jiang, C. Wilson’s disease: A comprehensive review of the molecular mechanisms. Int. J. Mol. Sci. 2015, 16, 6419–6431. [Google Scholar] [CrossRef] [PubMed]
- Dalgiç, B.; Sönmez, N.; Biberoğlu, G.; Hasanoğlu, A.; Erbaş, D. Evaluation of oxidant stress in Wilson’s disease and non-Wilsonian chronic liver disease in childhood. Turk. J. Gastroenterol. 2005, 16, 7–11. [Google Scholar] [PubMed]
- Kalita, J.; Kumar, V.; Misra, U.K.; Kumar, S. Movement disorder in Wilson disease: Correlation with MRI and biomarkers of cell injury. J. Mol. Neurosci. 2021, 71, 338–346. [Google Scholar] [CrossRef] [PubMed]
- Barber, R.G.; Grenier, Z.A.; Burkhead, J.L. Copper toxicity is not just oxidative damage: Zinc systems and insight from Wilson disease. Biomedicines 2021, 9, 316. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Deng, L.; Ma, X.; Guo, Y.; Feng, Z.; Liu, M.; Guan, Y.; Huang, Y.; Deng, J.; Li, H.; et al. Altered diversity and composition of gut microbiota in Wilson’s disease. Sci. Rep. 2020, 10, 21825. [Google Scholar] [CrossRef] [PubMed]
- Kelley, D.S.; Daudu, P.A.; Taylor, P.C.; Mackey, B.E.; Turnlund, J.R. Effects of low-copper diets on human immune response. Am. J. Clin. Nutr. 1995, 62, 412–416. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, R.G.; Failla, M.L. Copper deficiency reduces interleukin-2 (IL-2) production and IL-2 mRNA in human T-lymphocytes. J. Nutr. 1997, 127, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, R.G.; Failla, M.L. Transcriptional regulation of interkeukin-2 gene expression is impaired by copper deficiency in Jurkat T lymphocytes. J. Nutr. 1999, 129, 596–601. [Google Scholar] [CrossRef] [PubMed]
- Kisseleva, T.; Brenner, D. Molecular and cellular mechanisms of liver fibrosis and its regression. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 151–166. [Google Scholar] [CrossRef] [PubMed]
- Fuentealba, I.C.; Aburto, E.M. Animal models of copper-associated liver disease. Comp. Hepatol. 2003, 2, 5. [Google Scholar] [CrossRef] [PubMed]
- Toyokuni, S.; Okada, S.; Hamazaki, S.; Fujioka, M.; Li, J.L.; Midorikawa, O. Cirrhosis of the liver induced by cupric nitrilotriacetate in Wistar rats. An experimental model of copper toxicosis. Am. J. Pathol. 1989, 134, 1263–1274. [Google Scholar]
- Zhao, L.; Xia, Z.; Wang, F. Zebrafish in the sea of mineral (iron, zinc, and copper) metabolism. Front. Pharmacol. 2014, 5, 33. [Google Scholar] [CrossRef] [PubMed]
- Ems, T.; St Lucia, K.; Huecker, M.R. Biochemistry, Iron Absorption. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2023. Available online: https://www.ncbi.nlm.nih.gov/books/NBK448204/ (accessed on 12 June 2024).
- Vogt, A.S.; Arsiwala, T.; Mohsen, M.; Vogel, M.; Manolova, V.; Bachmann, M.F. On iron metabolism and its regulation. Int. J. Mol. Sci. 2021, 22, 4591. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Xiong, Z.; Wu, W.; Ling, H.Q.; Kong, D. Iron in the symbiosis of plants and microorganisms. Plants 2023, 12, 1958. [Google Scholar] [CrossRef] [PubMed]
- McLaren, G.D.; Nathanson, M.H.; Jacobs, A.; Trevett, D.; Thomson, W. Regulation of intestinal iron absorption and mucosal iron kinetics in hereditary hemochromatosis. J. Lab. Clin. Med. 1991, 117, 390–401. [Google Scholar] [PubMed]
- Teichmann, R.; Stremmel, W. Iron uptake by human upper small intestine microvillous membrane vesicles. Indication for a facilitated transport mechanism mediated by a membrane iron-binding protein. J. Clin. Investig. 1990, 86, 2145–2153. [Google Scholar] [CrossRef] [PubMed]
- Stremmel, W.; Schneider, M.; Lotz, G.; Niederau, C.; Teschke, R.; Strohmeyer, G. Iron uptake by rat duodenal microvillous membrane vesicles. Gastroenterology 1985, 88, A1602. [Google Scholar]
- Stremmel, W.; Schneider, M.; Lotz, G.; Niederau, C.; Teschke, R.; Strohmeyer, G. Iron uptake by rat duodenal microvillous membrane vesicles: Evidence for a carrier-mediated process. Eur. J. Clin. Investig. 1987, 17, 136–145. [Google Scholar] [CrossRef]
- Piskin, E.; Cianciosi, D.; Gulec, S.; Tomas, M.; Capanoglu, E. Iron absorption: Factors, limitations, and Improvement methods. ACS Omega 2022, 7, 20441–20456. [Google Scholar] [CrossRef] [PubMed]
- Sukhbaatar, N.; Weichhart, T. Iron regulation: Macrophages in control. Pharmaceuticals 2018, 11, 13. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Stockwell, B.R.; Conrad, M. Ferroptosis: Mechanisms, biology and role in disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 266–282. [Google Scholar] [CrossRef]
- Li, J.; Cao, F.; Yin, H.L.; Huang, Z.J.; Lin, Z.T.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: Past, present and future. Cell Death Dis. 2020, 11, 88. [Google Scholar] [CrossRef]
- Gilardi, G.; Di Nardo, G. Heme iron centers in cytochrome P450: Structure and catalytic activity. Rend. Fis. Acc. Lincei 2017, 28, 159–167. [Google Scholar] [CrossRef]
- Rishi, G.; Subramaniam, V.N. The liver in regulation of iron homeostasis. Am. J. Physiol. Gastrointest. Liver Physiol. 2017, 313, G157–G165. [Google Scholar] [CrossRef] [PubMed]
- Fleming, R.E.; Ponka, P. Iron overload in human disease. N. Engl. J. Med. 2012, 366, 348–359, Erratum in N. Engl. J. Med. 2012, 366, 771. [Google Scholar] [CrossRef] [PubMed]
- Anderson, G.J.; Frazer, D.M. Hepatic iron metabolism. Semin. Liver Dis. 2005, 25, 420–432. [Google Scholar] [CrossRef] [PubMed]
- Qiao, B.; Sugianto, P.; Fung, E.; Del-Castillo-Rueda, A.; Moran-Jimenez, M.J.; Ganz, T.; Nemeth, E. Hepcidin-induced endocytosis of ferroportin is dependent on ferroportin ubiquitination. Cell Metab. 2012, 15, 918–924. [Google Scholar] [CrossRef] [PubMed]
- Daher, R.; Karim, Z. Iron metabolism: State of the art. Transfus. Clin. Biol. 2017, 24, 115–119. [Google Scholar] [CrossRef] [PubMed]
- Abbaspour, N.; Hurrell, R.; Kelishadi, R. Review on iron and its importance for human health. J. Res. Med. Sci. 2014, 19, 164–174. [Google Scholar] [PubMed]
- Pfeiffer, C.M.; Looker, A.C. Laboratory methodologies for indicators of iron status: Strengths, limitations, and analytical challenges. Am. J. Clin. Nutr. 2017, 106 (Suppl. S6), 1606S–1614S. [Google Scholar] [CrossRef] [PubMed]
- Daru, J.; Colman, K.; Stanworth, S.J.; De La Salle, B.; Wood, E.M.; Pasricha, S.R. Serum ferritin as an indicator of iron status: What do we need to know? Am. J. Clin. Nutr. 2017, 106 (Suppl. S6), 1634S–1639S. [Google Scholar] [CrossRef] [PubMed]
- Leung, A.K.C.; Lam, J.M.; Wong, A.H.C.; Hon, K.L.; Li, X. Iron Deficiency Anemia: An Updated Review. Curr. Pediatr. Rev. 2024, 20, 339–356. [Google Scholar] [CrossRef] [PubMed]
- Johnson-Wimbley, T.D.; Graham, D.Y. Diagnosis and management of iron deficiency anemia in the 21st century. Therap Adv. Gastroenterol. 2011, 4, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Lugović-Mihić, L.; Pilipović, K.; Crnarić, I.; Šitum, M.; Duvančić, T. Differential diagnosis of cheilitis—How to classify cheilitis? Acta Clin. Croat. 2018, 57, 342–351. [Google Scholar] [CrossRef]
- Porter, J.L.; Rawla, P. Hemochromatosis. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2023. Available online: https://www.ncbi.nlm.nih.gov/books/NBK430862/ (accessed on 12 June 2024).
- Salomao, M.A. Pathology of hepatic iron overload. Clin. Liver Dis. 2021, 17, 232–237. [Google Scholar] [CrossRef] [PubMed]
- Kamble, R.K.; Thakare, M.G.; Ingle, A.B. Iron in the environment. Indian J. Environ. Prot. 2013, 33, 881–888. [Google Scholar]
- Koné, W.M.; Koffi, A.G.; Bomisso, E.L.; Tra Bi, F.H. Ethnomedical study and iron content of some medicinal herbs used in traditional medicine in Cote d‘Ivoire for the treatment of anaemia. Afr. J. Tradit. Complement. Altern. Med. 2011, 9, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Zhang, J.; Li, W.; Xu, M.; Liu, S. Disruption of iron homeostasis and resultant health effects upon exposure to various environmental pollutants: A critical review. J. Environ. Sci. 2015, 34, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Schreinemachers, D.M.; Ghio, A.J. Effects of environmental pollutants on cellular iron homeostasis and ultimate links to human disease. Environ. Health Insights 2016, 10, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, G.C.; Khan, M.J.H.; Chakraborty, T.K.; Zaman, S.; Kabir, A.H.M.E.; Tanaka, H. Human health Risk assessment of elevated and variable iron and manganese intake with arsenic-safe groundwater in Jashore, Bangladesh. Sci. Rep. 2020, 10, 5206. [Google Scholar] [CrossRef] [PubMed]
- Lal, A. Iron in health and disease: An update. Indian J. Pediatr. 2020, 87, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Schildroth, S.; Kordas, K.; White, R.F.; Friedman, A.; Placidi, D.; Smith, D.; Lucchini, R.G.; Wright, R.O.; Horton, M.; Claus Henn, B. An industry-relevant metal mixture, iron status, and reported attention-related behaviors in Italian adolescents. Environ. Health Perspect. 2024, 132, 27008. [Google Scholar] [CrossRef] [PubMed]
- Maher, B.A.; González-Maciel, A.; Reynoso-Robles, R.; Torres-Jardón, R.; Calderón-Garcidueñas, L. Iron-rich air pollution nanoparticles: An unrecognised environmental risk factor for myocardial mitochondrial dysfunction and cardiac oxidative stress. Environ. Res. 2020, 188, 109816. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, E.D. The hazards of iron loading. Metallomics 2010, 2, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Diami, S.M.; Kusin, F.M.; Madzin, Z. Potential ecological and human health risks of heavy metals in surface soils associated with iron ore mining in Pahang, Malaysia. Environ. Sci. Pollut. Res. Int. 2016, 23, 21086–21097. [Google Scholar] [CrossRef] [PubMed]
- de Mello Gabriel, G.V.; Pitombo, L.M.; Rosa, L.M.T.; Navarrete, A.A.; Botero, W.G.; do Carmo, J.B.; de Oliveira, L.C. The environmental importance of iron speciation in soils: Evaluation of classic methodologies. Environ. Monit. Assess. 2021, 193, 63. [Google Scholar] [CrossRef]
- Roemhild, K.; von Maltzahn, F.; Weiskirchen, R.; Knüchel, R.; von Stillfried, S.; Lammers, T. Iron metabolism: Pathophysiology and pharmacology. Trends Pharmacol. Sci. 2021, 42, 640–656. [Google Scholar] [CrossRef]
- Gulec, S.; Anderson, G.J.; Collins, J.F. Mechanistic and regulatory aspects of intestinal iron absorption. Am. J. Physiol. Gastrointest. Liver Physiol. 2014, 307, G397–G409. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Jones, A.; Waite, T.D.; Chen, Y.; Huang, X.; Rosso, K.M.; Kappler, A.; Mansor, M.; Tratnyek, P.G.; Zhang, H. Fe(II) redox chemistry in the environment. Chem. Rev. 2021, 121, 8161–8233. [Google Scholar] [CrossRef] [PubMed]
- Biel, D.; Steiger, T.K.; Bunzeck, N. Age-related iron accumulation and demyelination in the basal ganglia are closely related to verbal memory and executive functioning. Sci. Rep. 2021, 11, 9438. [Google Scholar] [CrossRef] [PubMed]
- Bateman, D.N.; Eagling, V.; Sandilands, E.A.; Jackson, G.; Crawford, C.; Hawkins, L.; Cheung, T.; Cooper, G.; Bradberry, S.M.; Thompson, J.P.; et al. Iron overdose epidemiology, clinical features and iron concentration-effect relationships: The UK experience 2008–2017. Clin. Toxicol. 2018, 56, 1098–1106. [Google Scholar] [CrossRef] [PubMed]
- Verma, S. Long-term follow-up of overdose of iron tablets in adult successfully managed without liver transplant: A case report. J. Gstro. Hepato. 2022, V9, 1–3. [Google Scholar]
- Nishikawa, Y.; Matsuo, Y.; Watanabe, R.; Miyazato, M.; Matsuo, M.; Nagahama, Y.; Tanaka, H.; Ooshio, T.; Goto, M.; Okada, Y.; et al. Hepatocyte-specific damage in acute toxicity of sodium ferrous citrate: Presentation of a human autopsy case and experimental results in mice. Toxicol. Rep. 2023, 10, 669–679. [Google Scholar] [CrossRef] [PubMed]
- Yuen, H.W.; Becker, W. Iron Toxicity. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2024. Available online: https://www.ncbi.nlm.nih.gov/books/NBK459224/ (accessed on 12 June 2024).
- Eça, R.; Ferreira, S.; Gandara, J.; Pessegueiro, H.; Daniel, J. Liver transplantation for acute hepatic failure following intentional iron overdose. Cureus 2023, 15, e48392. [Google Scholar] [CrossRef] [PubMed]
- Morales-Cruz, M.J.; MejiasMorales, D.; Hunsaker, P.; Butler, E.; Tassone, M. Ingestion of toxic iron dose with benign outcome. Cureus 2023, 15, e40103. [Google Scholar] [CrossRef] [PubMed]
- Abhilash, K.P.; Arul, J.J.; Bala, D. Fatal overdose of iron tablets in adults. Indian J. Crit. Care Med. 2013, 17, 311–313. [Google Scholar] [CrossRef] [PubMed]
- Sane, M.R.; Malukani, K.; Kulkarni, R.; Varun, A. Fatal iron toxicity in an adult: Clinical profile and review. Indian J. Crit. Care Med. 2018, 22, 801–803. [Google Scholar] [CrossRef] [PubMed]
- Ng, H.W.; Tse, M.L.; Lau, F.L.; Chu, W. Endoscopic removal of iron bezoar following acute overdose. Clin. Toxicol. 2008, 46, 913–915. [Google Scholar] [CrossRef] [PubMed]
- Majdanik, S.; Potocka-Banas, B.; Glowinski, S.; Borowiak, K. Suicide by intoxication with iron (III) chloride. Forensic Toxicol. 2012, 39, 513–517. [Google Scholar] [CrossRef]
- Lands, R.; Isang, E. Secondary hemochromatosis due to chronic oral iron supplementation. Case Rep. Hematol. 2017, 2017, 2494167. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.G.; Warner, C.G. Chronic exposure to iron oxide, chromium oxide, and nickel oxide fumes of metal dressers in a steelworks. Br. J. Ind. Med. 1972, 29, 169–177. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bala, S.; Tabaku, A. Chronic obstructive pulmonary disease in iron-steel and ferrochrome industry workers. Cent. Eur. J. Public Health 2010, 18, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Bloomer, S.A.; Brown, K.E. Iron-induced liver injury: A critical reappraisal. Int. J. Mol. Sci. 2019, 20, 2132. [Google Scholar] [CrossRef] [PubMed]
- Feder, J.N.; Gnirke, A.; Thomas, W.; Tsuchihashi, Z.; Ruddy, D.A.; Basava, A.; Dormishian, F.; Domingo, R.; Ellis, M.C.; Fullan, A.; et al. A novel MHC class I-like gene is mutated in patients with hereditary haemochromatosis. Nat. Genet. 1996, 13, 399–408. [Google Scholar] [CrossRef]
- Girelli, D.; Busti, F.; Brissot, P.; Cabantchik, I.; Muckenthaler, M.U.; Porto G; on behalf of the Nomenclature Committee of the International Society for the Study of Iron in Biology and Medicine (BIOIRON Society). Hemochromatosis classification: Update and recommendations by the BIOIRON Society. Blood 2022, 139, 3018–3029. [Google Scholar] [CrossRef] [PubMed]
- Bardou-Jacquet, E.; Brissot, P. Diagnostic evaluation of hereditary hemochromatosis (HFE and non-HFE). Hematol. Oncol. Clin. N. Am. 2014, 28, 625–635. [Google Scholar] [CrossRef] [PubMed]
- Yun, S.; Vincelette, N.D. Update on iron metabolism and molecular perspective of common genetic and acquired disorder, hemochromatosis. Crit. Rev. Oncol. Hematol. 2015, 95, 12–25. [Google Scholar] [CrossRef] [PubMed]
- Joshi, R.; Shvartsman, M.; Morán, E.; Lois, S.; Aranda, J.; Barqué, A.; de la Cruz, X.; Bruguera, M.; Vagace, J.M.; Gervasini, G.; et al. Functional consequences of transferrin receptor-2 mutations causing hereditary hemochromatosis type 3. Mol. Genet. Genom. Med. 2015, 3, 221–232. [Google Scholar] [CrossRef] [PubMed]
- Tatsumi, Y.; Yano, M.; Wakusawa, S.; Miyajima, H.; Ishikawa, T.; Imashuku, S.; Takano, A.; Nihei, W.; Kato, A.; Kato, K.; et al. A Revised classification of primary iron overload syndromes. J. Clin. Transl. Hepatol. 2024, 12, 346–356. [Google Scholar] [CrossRef] [PubMed]
- Milman, N.T. Managing Genetic Hemochromatosis: An overview of dietary measures, which may reduce intestinal iron absorption in persons with iron overload. Gastroenterol. Res. 2021, 14, 66–80. [Google Scholar] [CrossRef] [PubMed]
- Brissot, P.; Thibault Cavey, T.; Martine Ropert, M.; Pascal Guggenbuhl, P.; Loréal, O. Clinical management of hemochromatosis: Current perspectives. Int. J. Clin. Transfus. Med. 2017, 5, 1–7. [Google Scholar] [CrossRef]
- Barton, J.C.; McLaren, C.E.; Chen, W.P.; Ramm, G.A.; Anderson, G.J.; Powell, L.W.; Subramaniam, V.N.; Adams, P.C.; Phatak, P.D.; Gurrin, L.C.; et al. Cirrhosis in hemochromatosis: Independent risk factors in 368 HFE p.C282Y homozygotes. Ann. Hepatol. 2018, 17, 871–879. [Google Scholar] [CrossRef] [PubMed]
- Martinelli, A.L.; Filho, R.; Cruz, S.; Franco, R.; Tavella, M.; Secaf, M.; Ramalho, L.; Zucoloto, S.; Rodrigues, S.; Zago, M. Hereditary hemochromatosis in a Brazilian university hospital in São Paulo State (1990–2000). Genet. Mol. Res. 2005, 4, 31–38. [Google Scholar] [PubMed]
- Nash, S.; Marconi, S.; Sikorska, K.; Naeem, R.; Nash, G. Role of liver biopsy in the diagnosis of hepatic iron overload in the era of genetic testing. Am. J. Clin. Pathol. 2002, 118, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Golfeyz, S.; Lewis, S.; Weisberg, I.S. Hemochromatosis: Pathophysiology, evaluation, and management of hepatic iron overload with a focus on MRI. Expert. Rev. Gastroenterol. Hepatol. 2018, 12, 767–778. [Google Scholar] [CrossRef] [PubMed]
- Stål, P.; Glaumann, H.; Hultcrantz, R. Liver cell damage and lysosomal iron storage in patients with idiopathic hemochromatosis. A light and electron microscopic study. J. Hepatol. 1990, 11, 172–180. [Google Scholar] [CrossRef] [PubMed]
- Lin, E.; Adams, P.C. Biochemical liver profile in hemochromatosis. A survey of 100 patients. J. Clin. Gastroenterol. 1991, 13, 316–320. [Google Scholar] [CrossRef]
- Tavill, A.S.; Adams, P.C. A diagnostic approach to hemochromatosis. Can. J. Gastroenterol. 2006, 20, 535–540. [Google Scholar] [CrossRef] [PubMed]
- Strohmeyer, G.; Niederau, C.; Stremmel, W. Survival and causes of death in hemochromatosis. Observations in 163 patients. Ann. N. Y. Acad. Sci. 1988, 526, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Niederau, C.; Fischer, R.; Sonnenberg, A.; Stremmel, W.; Trampisch, H.J.; Strohmeyer, G. Survival and causes of death in cirrhotic and in noncirrhotic patients with primary hemochromatosis. N. Engl. J. Med. 1985, 313, 1256–1262. [Google Scholar] [CrossRef] [PubMed]
- Niederau, C.; Fischer, R.; Pürschel, A.; Stremmel, W.; Häussinger, D.; Strohmeyer, G. Long-term survival in patients with hereditary hemochromatosis. Gastroenterology 1996, 110, 1107–1119. [Google Scholar] [CrossRef] [PubMed]
- Parmanand, B.; Watson, M.; Boland, K.J.; Ramamurthy, N.; Wharton, V.; Morovat, A.; Lund, E.K.; Collier, J.; Le Gall, G.; Kellingray, L.; et al. Systemic iron reduction by venesection alters the gut microbiome in patients with haemochromatosis. JHEP Rep. 2020, 2, 100154. [Google Scholar] [CrossRef] [PubMed]
- Rombout-Sestrienkova, E.; Winkens, B.; Essers, B.A.; Nieman, F.H.; Noord, P.A.; Janssen, M.C.; van Deursen, C.T.; Boonen, A.; Reuser-Kaasenbrood, E.P.; Heeremans, J.; et al. Erythrocytapheresis versus phlebotomy in the maintenance treatment of HFE hemochromatosis patients: Results from a randomized crossover trial. Transfusion 2016, 56, 261–270. [Google Scholar] [CrossRef] [PubMed]
- Phatak, P.; Brissot, P.; Wurster, M.; Adams, P.C.; Bonkovsky, H.L.; Gross, J.; Malfertheiner, P.; McLaren, G.D.; Niederau, C.; Piperno, A.; et al. A phase I-II dose-escalation trial of deferasirox for the treatment of iron overload in HFE-related hereditary hemochromatosis. Hepatology 2010, 52, 1671–1679. [Google Scholar] [CrossRef] [PubMed]
- Cançado, R.; Melo, M.R.; de Moraves Bastos, R.; Santos, P.C.; Guerra-Shinohara, E.M.; Chiattone, C.; Ballas, S.K. Deferasirox in patients with iron overload secondary to hereditary hemochromatosis: Results of a 1-yr Phase 2 study. Eur. J. Haematol. 2015, 95, 545–550. [Google Scholar] [CrossRef] [PubMed]
- Babitt, J.L.; Lin, H.Y. The molecular pathogenesis of hereditary hemochromatosis. Semin. Liver Dis. 2011, 31, 280–292. [Google Scholar] [CrossRef] [PubMed]
- Niederau, C.; Strohmeyer, G. Strategies for early diagnosis of haemochromatosis. Eur. J. Gastroenterol. Hepatol. 2002, 14, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Pantopoulos, K. Inherited disorders of iron overload. Front. Nutr. 2018, 5, 103. [Google Scholar] [CrossRef] [PubMed]
- Grønlien, H.K.; Christoffersen, T.E.; Nystrand, C.F.; Garabet, L.; Syvertsen, T.; Moe, M.K.; Olstad, O.K.; Jonassen, C.M. Cytokine and gene expression profiling in patients with HFE-associated hereditary hemochromatosis according to genetic profile. Acta Haematol. 2021, 144, 446–457. [Google Scholar] [CrossRef] [PubMed]
- Schmidtke, J. Twenty-Five years of contemplating genotype-based hereditary hemochromatosis population screening. Genes 2022, 13, 1622. [Google Scholar] [CrossRef] [PubMed]
- Faruqi, A.; Mukkamalla, S.K.R. Iron binding capacity. [Updated 2 January 2023]. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar] [PubMed]
- Ayala, A.; Muñoz, M.F.; Argüelles, S. Lipid peroxidation: Production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid. Med. Cell Longev. 2014, 2014, 360438. [Google Scholar] [CrossRef] [PubMed]
- Mehta, K.J.; Farnaud, S.J.; Sharp, P.A. Iron and liver fibrosis: Mechanistic and clinical aspects. World J. Gastroenterol. 2019, 25, 521–538. [Google Scholar] [CrossRef] [PubMed]
- Winterbourn, C.C. Toxicity of iron and hydrogen peroxide: The Fenton reaction. Toxicol. Lett. 1995, 82–83, 969–974. [Google Scholar] [CrossRef] [PubMed]
- Houglum, K.; Ramm, G.A.; Crawford, D.H.; Witztum, J.L.; Powell, L.W.; Chojkier, M. Excess iron induces hepatic oxidative stress and transforming growth factor beta1 in genetic hemochromatosis. Hepatology 1997, 26, 605–610. [Google Scholar] [CrossRef] [PubMed]
- Shizukuda, Y.; Tripodi, D.J.; Rosing, D.R. Iron Overload or Oxidative Stress? Insight into a mechanism of early cardiac manifestations of asymptomatic hereditary hemochromatosis subjects with C282Y homozygosity. J. Cardiovasc. Transl. Res. 2016, 9, 400–401. [Google Scholar] [CrossRef] [PubMed]
- Niemelä, O.; Parkkila, S.; Britton, R.S.; Brunt, E.; Janney, C.; Bacon, B. Hepatic lipid peroxidation in hereditary hemochromatosis and alcoholic liver injury. J. Lab. Clin. Med. 1999, 133, 451–460. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Chung, F.L. Oxidative stress and hepatocarcinogenesis. Hepatoma Res. 2018, 4, 39. [Google Scholar] [CrossRef] [PubMed]
- Macías-Rodríguez, R.U.; Inzaugarat, M.E.; Ruiz-Margáin, A.; Nelson, L.J.; Trautwein, C.; Cubero, F.J. Reclassifying hepatic cell death during liver damage: Ferroptosis—A novel form of non-apoptotic cell death? Int. J. Mol. Sci. 2020, 21, 1651. [Google Scholar] [CrossRef] [PubMed]
- Mao, L.; Zhao, T.; Song, Y.; Lin, L.; Fan, X.; Cui, B.; Feng, H.; Wang, X.; Yu, Q.; Zhang, J.; et al. The emerging role of ferroptosis in non-cancer liver diseases: Hype or increasing hope? Cell Death Dis. 2020, 11, 518. [Google Scholar] [CrossRef] [PubMed]
- Teschke, R. Hemochromatosis: Ferroptosis, ROS, gut microbiome, and clinical challenges with alcohol as variable. Int. J. Mol. Sci. 2024, 25, 2668. [Google Scholar] [CrossRef] [PubMed]
- Lawless, M.W.; Mankan, A.K.; White, M.; O′Dwyer, M.J.; Norris, S. Expression of hereditary hemochromatosis C282Y HFE protein in HEK293 cells activates specific endoplasmic reticulum stress responses. BMC Cell Biol. 2007, 8, 30. [Google Scholar] [CrossRef] [PubMed]
- Hedges, J.C.; Singer, C.A.; Gerthoffer, W.T. Mitogen-activated protein kinases regulate cytokine gene expression in human airway myocytes. Am. J. Respir. Cell Mol. Biol. 2000, 23, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Arcones, A.; Thielemann, F.K. Origin of the elements. Astron. Astrophys. Rev. 2023, 31, 1. [Google Scholar] [CrossRef]
- Johnson, J.A.; Fields, B.D.; Thompson, T.A. The origin of the elements: A century of progress. Phil. Trans. R. Soc. 2020, 378, 2019030120190301. [Google Scholar] [CrossRef] [PubMed]
- Roederer, I.U.; Karakas, A.I.; Pignatari, M.; Herwig, F. The diverse origins of neutron-capture elements in the metal-poor Star HD 94028: Possible detection of products of I-process nucleosynthesis. Astrophys. J 2016, 821, 37. [Google Scholar] [CrossRef]
- Genchi, G.; Sinicropi, M.S.; Lauria, G.; Carocci, A.; Catalano, A. The effects of cadmium toxicity. Int. J. Environ. Res. Public Health 2020, 17, 3782. [Google Scholar] [CrossRef] [PubMed]
- Thévenod, F.; Fels, J.; Lee, W.K.; Zarbock, R. Channels, transporters and receptors for cadmium and cadmium complexes in eukaryotic cells: Myths and facts. Biometals 2019, 32, 469–489. [Google Scholar] [CrossRef]
- Chandravanshi, L.; Shiv, K.; Kumar, S. Developmental toxicity of cadmium in infants and children: A review. Environ. Anal. Health Toxicol. 2021, 36, e2021003. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.; Li, H.; Zhang, B.; Zheng, T.; Li, Y.; Zhou, A.; Du, X.; Pan, X.; Yang, J.; Wu, C.; et al. Prenatal cadmium exposure and preterm low birth weight in China. J. Exp. Sci. Environ. Epidemiol. 2017, 27, 491–496. [Google Scholar] [CrossRef] [PubMed]
- Branca, J.J.V.; Morucci, G.; Pacini, A. Cadmium-induced neurotoxicity: Still much ado. Neural Regen. Res. 2018, 13, 1879–1882. [Google Scholar] [CrossRef] [PubMed]
- Satarug, S.; Garrett, S.H.; Sens, M.A.; Sens, D.A. Cadmium, environmental exposure, and health outcomes. Cien Saude Colet. 2011, 16, 2587–2602. [Google Scholar] [CrossRef] [PubMed]
- Koons, A.L.; Rajasurya, V. Cadmium Toxicity. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2023. Available online: https://www.ncbi.nlm.nih.gov/books/NBK536966/ (accessed on 19 March 2024).
- Adnan, M.; Xiao, B.; Xiao, P.; Zhao, P.; Bibi, S. Heavy metal, waste, COVID-19, and rapid industrialization in this modern era—Fit for sustainable future. Sustainability 2022, 14, 4746. [Google Scholar] [CrossRef]
- Niture, S.; Lin, M.; Qi, Q.; Moore, J.T.; Levine, K.E.; Fernando, R.A.; Kumar, D. Role of autophagy in cadmium-induced hepatotoxicity and liver diseases. J. Toxicol. 2021, 2021, 9564297. [Google Scholar] [CrossRef] [PubMed]
- Renu, K.; Chakraborty, R.; Myakala, H.; Koti, R.; Famurewa, A.C.; Madhyastha, H.; Vellingiri, B.; George, A.; Valsala Gopalakrishnan, A. Molecular mechanism of heavy metals (lead, chromium, arsenic, mercury, nickel and cadmium)—Induced hepatotoxicity—A review. Chemosphere 2021, 271, 129735. [Google Scholar] [CrossRef] [PubMed]
- Parvez, S.M.; Jahan, F.; Brune, M.N.; Gorman, J.F.; Rahman, M.J.; Carpenter, D.; Islam, Z.; Rahman, M.; Aich, N.; Knibbs, L.D.; et al. Health consequences of exposure to e-waste: An updated systematic review. Lancet Planet. Health 2021, 5, e905–e920. [Google Scholar] [CrossRef] [PubMed]
- WHO. Preventing Disease through Healthy Environments. Exposure to Cadmium: A Major Public Health Concern. Available online: https://iris.who.int/handle/10665/329480 (accessed on 22 March 2024).
- Charkiewicz, A.E.; Omeljaniuk, W.J.; Nowak, K.; Garley, M.; Nikliński, J. Cadmium toxicity and health effects—A brief summary. Molecules 2023, 28, 6620. [Google Scholar] [CrossRef] [PubMed]
- Godt, J.; Scheidig, F.; Grosse-Siestrup, C.; Esche, V.; Brandenburg, P.; Reich, A.; Groneberg, D.A. The toxicity of cadmium and resulting hazards for human health. J. Occup. Med. Toxicol. 2006, 1, 22. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, M.; Kortenkamp, A. Cadmium exposures and deteriorations of cognitive abilities: Estimation of a reference dose for mixture risk assessments based on a systematic review and confidence rating. Environ. Health 2022, 21, 6. [Google Scholar] [CrossRef] [PubMed]
- Rafati Rahimzadeh, M.; Rafati Rahimzadeh, M.; Kazemi, S.; Moghadamnia, A.A. Cadmium toxicity and treatment: An update. Caspian J. Intern. Med. 2017, 8, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Hong, D.; Min, J.Y.; Min, K.B. Association between cadmium exposure and liver function in adults in the United States: A cross-sectional study. J. Prev. Med. Public. Health 2021, 54, 471–480. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, M.; Watanabe, T.; Zhang, Z.W.; Moon, C.S.; Shimbo, S. The integrity of the liver among people environmentally exposed to cadmium at various levels. Int. Arch. Occup. Environ. Health 1997, 69, 379–385. [Google Scholar] [CrossRef]
- Moon, S.; Lee, J.; Yu, J.M.; Choi, H.; Choi, S.; Park, J.; Choi, K.; Kim, E.; Kim, H.; Kim, M.J.; et al. Association between environmental cadmium exposure and increased mortality in the U.S. National Health and Nutrition Examination Survey (1999–2018). J. Exp. Sci. Environ. Epidemiol. 2023, 33, 874–882. [Google Scholar] [CrossRef] [PubMed]
- Ohshiro, H.; Nose, T.; Sugiyama, K.; Meshitsuka, S.; Kurozawa, Y.; Kuranobu, M.; Funakawa, K.; Yamasaki, H.; Kinosita, K. [A case study of acute cadmium poisoning by welding work]. Sangyo Igaku 1988, 30, 210–211. (In Japanese) [Google Scholar] [CrossRef][Green Version]
- Raval, G.; Straughen, J.E.; McMillin, G.A.; Bornhorst, J.A. Unexplained hemolytic anemia with multiorgan failure. Clin. Chem. 2011, 57, 1485–1488. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Townshend, R.H. Acute cadmium pneumonitis: A 17-year follow-up. Br. J. Ind. Med. 1982, 39, 411–412. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Cantilena, L.R., Jr.; Klaassen, C.D. Comparison of the effectiveness of several chelators after single administration on the toxicity, excretion, and distribution of cadmium. Toxicol. Appl. Pharmacol. 1981, 58, 452–460. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Xu, X.; Zeng, Z.; Lin, X.; Qin, Q.; Huo, X. Blood lead and cadmium levels associated with hematological and hepatic functions in patients from an e-waste-polluted area. Chemosphere 2019, 220, 531–538. [Google Scholar] [CrossRef] [PubMed]
- Eslam, M.; Newsome, P.N.; Sarin, S.K.; Anstee, Q.M.; Targher, G.; Romero-Gomez, M.; Zelber-Sagi, S.; Wai-Sun Wong, V.; Dufour, J.F.; Schattenberg, J.M.; et al. A new definition for metabolic dysfunction-associated fatty liver disease: An international expert consensus statement. J. Hepatol. 2020, 73, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Sung, G.; Lee, S.; Han, K.J.; Han, H. Serum cadmium is associated with hepatic steatosis and fibrosis. Medicine 2022, 101, e28559. [Google Scholar] [CrossRef] [PubMed]
- Hyder, O.; Chung, M.; Cosgrove, D.; Herman, J.M.; Li, Z.; Firoozmand, A.; Gurakar, A.; Koteish, A.; Pawlik, T.M. Cadmium exposure and liver disease among US adults. J. Gastrointest. Surg. 2013, 17, 1265–1273. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.W.; Ock, J.; Moon, K.W.; Park, C.H. Association between Pb, Cd, and Hg exposure and liver injury among Korean adults. Int. J. Environ. Res. Publ. Health 2021, 18, 6783. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.C.; Lian, I.B.; Kor, C.T.; Chang, C.C.; Su, P.Y.; Chang, W.T.; Liang, Y.F.; Su, W.W.; Soon, M.S. Association between soil heavy metals and fatty liver disease in men in Taiwan: A cross sectional study. BMJ Open 2017, 7, e014215. [Google Scholar] [CrossRef] [PubMed]
- Méndez-Sánchez, N.; Bugianesi, E.; Gish, R.G.; Lammert, F.; Tilg, H.; Nguyen, M.H.; Sarin, S.K.; Fabrellas, N.; Zelber-Sagi, S.; Fan, J.G.; et al. Global multi-stakeholder endorsement of the MAFLD definition. Lancet Gastroenterol. Hepatol. 2022, 7, 388–390. [Google Scholar] [CrossRef] [PubMed]
- Baba, H.; Tsuneyama, K.; Yazaki, M.; Nagata, K.; Minamisaka, T.; Tsuda, T.; Nomoto, K.; Hayashi, S.; Miwa, S.; Nakajima, T.; et al. The liver in itai-itai disease (chronic cadmium poisoning): Pathological features and metallothionein expression. Mod. Pathol. 2013, 26, 1228–1234. [Google Scholar] [CrossRef] [PubMed]
- Go, Y.M.; Sutliff, R.L.; Chandler, J.D.; Khalidur, R.; Kang, B.Y.; Anania, F.A.; Orr, M.; Hao, L.; Fowler, B.A.; Jones, D.P. Low-dose cadmium causes metabolic and genetic dysregulation associated with fatty liver disease in mice. Toxicol. Sci. 2015, 147, 524–534. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Gao, J.; Hou, H.; Qi, Z.; Chen, H.; Zhang, X. Inhibition of mitochondrial fatty acid oxidation contributes to development of nonalcoholic fatty liver disease induced by environmental cadmium exposure. Environ. Sci. Technol. 2019, 53, 13992–14000. [Google Scholar] [CrossRef] [PubMed]
- Zou, H.; Sun, J.; Wu, B.; Yuan, Y.; Gu, J.; Bian, J.; Liu, X.; Liu, Z. Effects of cadmium and/or lead on autophagy and liver injury in rats. Biol. Trace Elem. Res. 2020, 198, 206–215. [Google Scholar] [CrossRef] [PubMed]
- Rana, K.; Verma, Y.; Rana, S.V.S. Possible mechanisms of liver injury induced by cadmium sulfide nanoparticles in rat. Biol. Trace Elem. Res. 2021, 199, 216–226. [Google Scholar] [CrossRef] [PubMed]
- Kuester, R.K.; Waalkes, M.P.; Goering, P.L.; Fisher, B.L.; McCuskey, R.S.; Sipes, I.G. Differential Hepatotoxicity Induced by Cadmium in Fischer 344 and Sprague-Dawley Rats. Toxicol. Sci. 2002, 65, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Zhu, Y.; Lu, Z.; Guo, W.; Tumen, B.; He, Y.; Chen, C.; Hu, S.; Xu, K.; Wang, Y.; et al. Cadmium Induces acute liver injury by inhibiting Nrf2 and the role of NF-κB, NLRP3, and MAPKs signaling pathway. Int. J. Environ. Res. Public Health 2020, 17, 138. [Google Scholar] [CrossRef] [PubMed]
- Rikans, L.E.; Yamano, T. Mechanisms of cadmium-mediated acute hepatotoxicity. J. Biochem. Mol. Toxicol. 2000, 14, 110–117. [Google Scholar] [CrossRef]
- Ikediobi, C.O.; Badisa, V.L.; Ayuk-Takem, L.T.; Latinwo, L.M.; West, J. Response of antioxidant enzymes and redox metabolites to cadmium-induced oxidative stress in CRL-1439 normal rat liver cells. Int. J. Mol. Med. 2004, 14, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Tinkov, A.A.; Gritsenko, V.A.; Skalnaya, M.G.; Cherkasov, S.V.; Aaseth J Skalny, A.V. Gut as a target for cadmium toxicity. Environ. Pollut. 2018, 235, 429–434. [Google Scholar] [CrossRef] [PubMed]
- Souza-Arroyo, V.; Fabián, J.J.; Bucio-Ortiz, L.; Miranda-Labra, R.U.; Gomez-Quiroz, L.E.; Gutiérrez-Ruiz, M.C. The mechanism of the cadmium-induced toxicity and cellular response in the liver. Toxicology 2022, 480, 153339. [Google Scholar] [CrossRef]
- Yamano, T.; DeCicco, L.A.; Rikans, L.E. Attenuation of cadmium-induced liver injury in senescent male fischer 344 rats: Role of Kupffer cells and inflammatory cytokines. Toxicol. Appl. Pharmacol. 2000, 162, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wang, Y.; Li, T.; Zhuo, W.; Zhu, Y. Elevated serum and hair levels of cadmium as a risk factor for liver carcinoma: A meta-analysis. Nutr. Cancer 2023, 75, 1438–1447. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Pan, Z.; Zhu, M.; Gao, R.; Wang, Y.; Cheng, Y.; Zhang, N. Exposure to essential and non-essential trace elements and risks of congenital heart defects: A narrative review. Front. Nutr. 2023, 10, 1121826. [Google Scholar] [CrossRef] [PubMed]
- Rosen, B.P.; Liu, Z. Transport pathways for arsenic and selenium: A minireview. Environ. Int. 2009, 35, 512–515. [Google Scholar] [CrossRef] [PubMed]
- Calatayud, M.; Laparra Llopis, J.M. Arsenic through the gastrointestinal tract. In Handbook of Arsenic Toxicology; Academic Press: Cambridge, MA, USA, 2015; pp. 281–299. [Google Scholar] [CrossRef]
- Bjørklund, G.; Oliinyk, P.; Lysiuk, R.; Rahaman, M.S.; Antonyak, H.; Lozynska, I.; Lenchyk, L.; Peana, M. Arsenic intoxication: General aspects and chelating agents. Arch. Toxicol. 2020, 94, 1879–1897. [Google Scholar] [CrossRef] [PubMed]
- Guha Mazumder, D.N. Arsenic and liver disease. J. Indian. Med. Assoc. 2001, 311, 314–315. [Google Scholar]
- Balali-Mood, M.; Naseri, K.; Tahergorabi, Z.; Khazdair, M.R.; Sadeghi, M. Toxic mechanisms of five heavy metals: Mercury, Lead, Chromium, Cadmium, and Arsenic. Front. Pharmacol. 2021, 12, 643972. [Google Scholar] [PubMed]
- Kuivenhoven, M.; Mason, K. Arsenic Toxicity. [Updated 12 June 2023]. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2023. Available online: https://www.ncbi.nlm.nih.gov/books/NBK541125/ (accessed on 18 March 2024).
- Muzaffar, S.; Khan, J.; Srivastava, R.; Gorbatyuk, M.S.; Athar, M. Mechanistic understanding of the toxic effects of arsenic and warfare arsenicals on human health and environment. Cell Biol. Toxicol. 2023, 39, 85–110. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Jiménez, E.; Esteban, E.; Peñalosa, J.M. The fate of arsenic in soil-plant systems. Rev. Environ. Contam. Toxicol. 2012, 215, 1–37. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi, M.; Naghibi, S.A.; Motevalli, A.; Hashemi, H. Human-induced arsenic pollution modeling in surface waters—An integrated approach using machine learning algorithms and environmental factors. J. Environ. Manag. 2022, 305, 114347. [Google Scholar] [CrossRef] [PubMed]
- Guha Mazumder, D.N.; Chakraborty, A.K.; Ghose, A.; Gupta, J.D.; Chakraborty, D.P.; Dey, S.B.; Chattopadhyay, N. Chronic arsenic toxicity from drinking tubewell water in rural West Bengal. Bull. World Health Organ. 1988, 66, 499–506. [Google Scholar] [PubMed]
- Gorby, M.S. Arsenic poisoning. West. J. Med. 1988, 149, 308–315. [Google Scholar] [PubMed]
- LiverTox. Arsenic. 2023. Available online: https://www.ncbi.nlm.nih.gov/books/NBK548490/?report=reader (accessed on 18 March 2024).
- Theruvath, A.H.; Raveendran, R.; Philips, C.A. Dangerous placebo during the COVID-19 pandemic: A series of homoeopathic arsenicum album-induced liver injury. Cureus 2022, 14, e26062. [Google Scholar] [CrossRef] [PubMed]
- Theruvath, A.H.; Raveendran, R.; Philips, C.A.; Ahamed, R.; Abduljaleel, J.K.; Tharakan, A.; Rajesh, S.; Augustine, P. A series of homeopathic remedies-related severe drug-induced liver injury from South India. Hepatol. Commun. 2023, 7, e0064. [Google Scholar] [CrossRef] [PubMed]
- Upshaw, C.B.; Claiborne, T.S. Medicinal arsenic poisoning: 27-year follow-up. South. Med. J. 1995, 88, 892–893. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Xi, S.; Liu, Z.; Yang, Y.; Zheng, Q.; Wang, F.; Xu, Y.; Wang, Y.; Zheng, Y.; Sun, G. Arsenic methylation metabolism and liver injury of acute promyelocytic leukemia patients undergoing arsenic trioxide treatment. Environ. Toxicol. 2013, 28, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Devarbhavi, H. Ayurvedic and herbal medicine-induced liver injury: It is time to wake up and take notice. Indian. J. Gastroenterol. 2018, 37, 5–7. [Google Scholar] [CrossRef] [PubMed]
- Hardin, J.; Seltzer, J.; Suhandynata, R.; Spiegel, B.; Silver, R.; Thomas, D.; Galust, H.; Friedman, N.; Clark, R.; Momper, J. Severe arsenic poisoning due to Ayurvedic supplements. Clin. Case Rep. 2023, 11, e7733. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, A.; Chara, M.K. Letter to the Editor: Homeopathic drug-induced liver injury—An example of biases pertaining to Roussel Uclaf causality assessment method. Hepatol. Commun. 2023, 7, e00177. [Google Scholar] [CrossRef] [PubMed]
- Philips, C.A.; Paramaguru, R.; Joy, A.K.; Antony, K.L.; Augustine, P. Clinical outcomes, histopathologic patterns and chemical analysis of ayurveda and herbal medicine associated with severe liver injury—A single center experience from South India. Indian J. Gastroenterol. 2018, 37, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Pinto, B.; Goyal, P.; Flora, S.J.; Gill, K.D.; Singh, S. Chronic arsenic poisoning following ayurvedic medication. J. Med. Toxicol. 2014, 10, 395–398. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Zhang, Y.; Yun, Z.; He, B.; Zhang, Q.; Hu, L.; Jiang, G. Speciation and bioaccessibility of arsenic in traditional Chinese medicines and assessment of its potential health risk. Sci. Total Environ. 2018, 619–620, 1088–1097. [Google Scholar] [CrossRef] [PubMed]
- Inada, I.; Kiuchi, F.; Urushihara, H. Comparison of regulations for arsenic and heavy metals in herbal medicines using pharmacopoeias of nine counties/regions. Ther. Innov. Regul. Sci. 2023, 57, 963–974. [Google Scholar] [CrossRef] [PubMed]
- Spilchuk, V.; Thompson, A. Chronic arsenic poisoning from traditional Chinese medicine. CMAJ 2019, 191, E424. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Yan, R.; Guan, R.; Du, Y.; Liu, Y.; Wu, S.; Zhu, S.; Song, M.; Hang, T. Arsenic-related health risk assessment of realgar-containing NiuHuangJieDu tablets in healthy volunteers po administration. Front. Pharmacol. 2022, 12, 761801. [Google Scholar] [CrossRef] [PubMed]
- Islam, K.; Haque, A.; Karim, R.; Fajol, A.; Hossain, E.; Salam, K.A.; Ali, N.; Saud, Z.A.; Rahman, M.; Rahman, M.; et al. Dose-response relationship between arsenic exposure and the serum enzymes for liver function tests in the individuals exposed to arsenic: A cross sectional study in Bangladesh. Environ. Health 2011, 10, 64. [Google Scholar] [CrossRef] [PubMed]
- Abernathy, C.O.; Liu, Y.P.; Longfellow, D.; Aposhian, H.V.; Beck, B.; Fowler, B.; Goyer, R.; Menzer, R.; Rossman, T.; Thompson, C.; et al. Arsenic: Health effects, mechanisms of actions, and research issues. Environ. Health Perspect. 1999, 107, 593–597. [Google Scholar] [CrossRef] [PubMed]
- Abernathy, C.O.; Thomas, D.J.; Calderon, R.L. Health effects and risk assessment of arsenic. J. Nutr. 2003, 133, 1536S–1538S. [Google Scholar] [CrossRef] [PubMed]
- Ng, J.C.; Wang, J.; Shraim, A. A global health problem caused by arsenic from natural sources. Chemosphere 2003, 52, 1353–1359. [Google Scholar] [CrossRef] [PubMed]
- Tchounwou, P.B.; Patlolla, A.K.; Centeno, J.A. Carcinogenic and systemic health effects associated with arsenic exposure--a critical review. Toxicol. Pathol. 2003, 31, 575–588. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.C.; Saha, K.C.; Pati, S.; Dutta, R.N.; Rahman, M.M.; Sengupta, M.K.; Ahamed, S.; Lodh, D.; Das, B.; Hossain, M.A.; et al. Murshidabad—One of the nine groundwater arsenic-affected districts of West Bengal, India. Part II: Dermatological, neurological, and obstetric findings. Clin. Toxicol. 2005, 43, 835–848. [Google Scholar] [CrossRef] [PubMed]
- Kapaj, S.; Peterson, H.; Liber, K.; Bhattacharya, P. Human health effects from chronic arsenic poisoning—A review. J. Environ. Sci. Health A Tox. Hazard. Subst. Environ. Eng. 2006, 41, 2399–2428. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Kumar, D.; Sahu, A.P. Arsenic in the environment: Effects on human health and possible prevention. J. Environ. Biol. 2007, 28, 359–365. [Google Scholar]
- Vahter, M. Health effects of early life exposure to arsenic. Basic. Clin. Pharmacol. Toxicol. 2008, 102, 204–211. [Google Scholar] [CrossRef] [PubMed]
- Fatmi, Z.; Abbasi, I.N.; Ahmed, M.; Kazi, A.; Kayama, F. Burden of skin lesions of arsenicosis at higher exposure through groundwater of taluka Gambat district Khairpur, Pakistan: A cross-sectional survey. Environ. Geochem. Health 2013, 35, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Naujokas, M.F.; Anderson, B.; Ahsan, H.; Aposhian, H.V.; Graziano, J.H.; Thompson, C.; Suk, W.A. The broad scope of health effects from chronic arsenic exposure: Update on a worldwide public health problem. Environ. Health Perspect. 2013, 121, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.J. Health hazards and mitigation of chronic poisoning from arsenic in drinking water: Taiwan experiences. Rev. Environ. Health 2014, 29, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Palma-Lara, I.; Martínez-Castillo, M.; Quintana-Pérez, J.C.; Arellano-Mendoza, M.G.; Tamay-Cach, F.; Valenzuela-Limón, O.L.; García-Montalvo, E.A.; Hernández-Zavala, A. Arsenic exposure: A public health problem leading to several cancers. Regul. Toxicol. Pharmacol. 2020, 110, 104539. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.I.; Ahmad, M.F.; Ahmad, I.; Ashfaq, F.; Wahab, S.; Alsayegh, A.A.; Kumar, S.; Hakeem, K.R. Arsenic exposure through dietary intake and associated health hazards in the Middle East. Nutrients 2022, 14, 2136. [Google Scholar] [CrossRef] [PubMed]
- Ozturk, M.; Metin, M.; Altay, V.; Bhat, R.A.; Ejaz, M.; Gul, A.; Unal, B.T.; Hasanuzzaman, M.; Nibir, L.; Nahar, K.; et al. Arsenic and human health: Genotoxicity, epigenomic effects, and cancer signaling. Biol. Trace Elem. Res. 2022, 200, 988–1001. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Ali, M.; Raj, V.; Kumari, A.; Rachamalla, M.; Niyogi, S.; Kumar, D.; Sharma, A.; Saxena, A.; Panjawani, G.; et al. Arsenic causing gallbladder cancer disease in Bihar. Sci. Rep. 2023, 13, 4259. [Google Scholar] [CrossRef] [PubMed]
- Aryan, Y.; Pon, T.; Panneerselvam, B.; Dikshit, A.K. A comprehensive review of human health risks of arsenic and fluoride contamination of groundwater in the South Asia region. J. Water Health 2024, 22, 235–267. [Google Scholar] [CrossRef] [PubMed]
- Demissie, S.; Mekonen, S.; Awoke, T.; Teshome, B.; Mengistie, B. Examining carcinogenic and noncarcinogenic health risks related to arsenic exposure in Ethiopia: A longitudinal study. Toxicol. Rep. 2024, 12, 100–110. [Google Scholar] [CrossRef] [PubMed]
- Dou, D.C.; Qi, R.; Xiao, S.M.; Su, G.X.; Guo, Y.X. Distribution Characteristics of Arsenic in Drinking Water in China and Its Health Risk Based on Disability-adjusted Life Years. Huan Jing Ke Xue 2024, 45, 131–139. (In Chinese) [Google Scholar] [CrossRef]
- EFSA Panel on Contaminants in the Food Chain (CONTAM); Schrenk, D.; Bignami, M.; Bodin, L.; Chipman, J.K.; Del Mazo, J.; Grasl-Kraupp, B.; Hogstrand, C.; Hoogenboom, L.R.; Leblanc, J.C.; et al. Update of the risk assessment of inorganic arsenic in food. EFSA J. 2024, 22, e8488. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Gao, T.; Li, X.; Wāng, Y. Didactical approaches and insights into environmental processes and cardiovascular hazards of arsenic contaminants. Chemosphere 2024, 352, 141381. [Google Scholar] [CrossRef] [PubMed]
- Issanov, A.; Adewusi, B.; Saint-Jacques, N.; Dummer, T.J.B. Arsenic in drinking water and lung cancer: A systematic review of 35 years of evidence. Toxicol. Appl. Pharmacol. 2024, 483, 116808. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.J.; Cao, Y.R.; Lee, D.Y. Assessment of health risks associated with prediction of vegetable inorganic arsenic concentrations given different soil properties. Environ. Geochem. Health 2024, 46, 71. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Li, W.; Zhang, J.; Yan, Y.; Zhou, Q.; Liu, Q.; Guan, Y.; Zhao, Z.; An, J.; Cheng, X.; et al. Associations of arsenic exposure and arsenic metabolism with the risk of non-alcoholic fatty liver disease. Int. J. Hyg. Environ. Health 2024, 257, 114342. [Google Scholar] [CrossRef] [PubMed]
- Sevak, P.; Pushkar, B. Arsenic pollution cycle, toxicity and sustainable remediation technologies: A comprehensive review and bibliometric analysis. J. Environ. Manag. 2024, 349, 119504. [Google Scholar] [CrossRef] [PubMed]
- Das, N.; Paul, S.; Chatterjee, D.; Banerjee, N.; Majumder, N.S.; Sarma, N.; Sau, T.J.; Basu, S.; Banerjee, S.; Majumder, P.; et al. Arsenic exposure through drinking water increases the risk of liver and cardiovascular diseases in the population of West Bengal, India. BMC Public Health 2012, 12, 639. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Jiang, X.; Qian, H.; Li, X.; Su, J.; Zhang, G.; Li, X. Associations of arsenic exposure with liver injury in US adults: NHANES 2003–2018. Environ. Sci. Pollut. Res. Int. 2023, 30, 48260–48269. [Google Scholar] [CrossRef] [PubMed]
- Yao, M.; Zeng, Q.; Luo, P.; Sun, B.; Liang, B.; Wei, S.; Xu, Y.; Wang, Q.; Liu, Q.; Zhang, A. Assessing the risk of coal-burning arsenic-induced liver damage: A population-based study on hair arsenic and cumulative arsenic. Environ. Sci. Pollut. Res. Int. 2021, 28, 50489–50499. [Google Scholar] [CrossRef] [PubMed]
- Benramdane, L.; Accominotti, M.; Fanton, L.; Malicier, D.; Vallon, J.J. Arsenic speciation in human organs following fatal arsenic trioxide poisoning—A case report. Clin. Chem. 1999, 45, 301–306. [Google Scholar] [CrossRef] [PubMed]
- Lech, T.; Trela, F. Massive acute arsenic poisonings. Forensic Sci. Int. 2005, 151, 273–277. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.; Ma, J.Q.; Li, F.; Xu, G.H.; Guo, W.; Zhou, H.M. A Fatal case of acute arsenic poisoning. J. Forensic Sci. 2019, 64, 1271–1273. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Tong, X.; Zhao, S.; Yu, Z.; Zhang, J.; Ma, L.; Shi, Q.; Zhou, Y. Four cases of fatal acute arsenic poisoning: Histopathology, toxicology, and new trends. Forensic. Sci. Med. Pathol. 2023; in press. [Google Scholar] [CrossRef]
- Kim, L.H.; Abel, S.J. Survival after a massive overdose of arsenic trioxide. Crit. Care Resusc. 2009, 11, 42–45. [Google Scholar] [CrossRef] [PubMed]
- Fowler, B.A.; Selene, C.H.; Chou, R.J.; Jones, D.L.; Sullivan, W., Jr.; Chen, C.J. Chapter 28—Arsenic. In Handbook on the Toxicology of Metals, 4th ed.; Nordberg, G.F., Fowler, B.A., Nordberg, M., Eds.; Academic Press: San Diego, CA, USA, 2015; pp. 581–624. [Google Scholar]
- Guha Mazumder, D.N. Effect of chronic intake of arsenic-contaminated water on liver. Toxicol. Appl. Pharmacol. 2005, 206, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Philips, C.A.; Theruvath, A.H. Homeopathic remedies and liver injury: Authors’ reply to central council for research in homeopathy. Hepatol. Commun. 2023, 7, e0170. [Google Scholar] [CrossRef] [PubMed]
- Datta, D.V.; Mitra, S.K.; Chhuttani, P.N.; Chakravarti, R.N. Chronic oral arsenic intoxication as a possible aetiological factor in idiopathic portal hypertension (non-cirrhotic portal fibrosis) in India. Gut 1979, 20, 378–384. [Google Scholar] [CrossRef] [PubMed]
- Morris, J.S.; Schmid, M.; Newman, S.; Scheuer, P.J.; Sherlock, S. Arsenic and noncirrhotic portal hypertension. Gastroenterology 1974, 66, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Neale, G.; Azzopardi, G. Chronic arsenical poisoning and non-cirrhotic portal hypertension. A case for diagnosis. Br. Med. J. 1971, 4, 725–730. [Google Scholar]
- Villeneuve, J.P.; Huet, P.M.; Joly, J.G.; Lafortune, M.; Lavoie, P.; Viallet, A. Idiopathic portal hypertension. Am. J. Med. 1976, 61, 459–464. [Google Scholar] [CrossRef] [PubMed]
- Goering, P.L.; Barber, D.S. Hepatotoxicity of copper, Iron, cadmium, and arsenic. Compr. Toxicol. 2010, 9, 501–526. [Google Scholar] [CrossRef]
- Akhand, A.A.; Du, J.; Liu, W.; Hossain, K.; Miyata, T.; Nagase, F.; Kato, M.; Suzuki, H.; Nakashima, I. Redox-linked cell surface-oriented signaling for T-cell death. Antiox. Redox Signal. 2002, 4, 445–454. [Google Scholar] [CrossRef] [PubMed]
- Hossain, K.; Akhand, A.A.; Kato, M.; Du, J.; Takeda, K.; Wu, J.; Takeuchi, K.; Liu, W.; Suzuki, H.; Nakashima, I. Arsenite induces apoptosis of murine T lymphocytes through membrane Raft-linked signaling for activation of c-Jun amino-terminal kinase. J. Immunol. 2000, 165, 4290–4297. [Google Scholar] [CrossRef] [PubMed]
- Rossman, T. Mechanism of arsenic carcinogenesis: An integrated approach. Mut. Res. 2003, 533, 37–65. [Google Scholar] [CrossRef]
- Li, C.; Li, P.; Tan, Y.M.; Lam, S.H.; Chan, E.C.Y.; Gong, Z. Metabolomic characterization of liver injury caused by acute arsenic toxicity in Zebrafish. PLoS ONE 2016, 11, e0151225. [Google Scholar] [CrossRef] [PubMed]
- Hussain, A.; Raveendran, V.A.; Kundu, S.; Samanta, T.; Shunmugam, R.; Pal, D.; Sarma, J.D. Mechanisms of Arsenic-Induced Toxicity with Special Emphasis on Arsenic-Binding Proteins; InTech: London, UK, 2018. [Google Scholar] [CrossRef]
- Miltonprabu, S.; Sumedha, N.C. Arsenic-induced hepatic mitochondrial toxicity in rats and its amelioration by diallyl trisulfide. Toxicol. Mech. Methods 2014, 24, 124–135. [Google Scholar] [CrossRef]
- Hrycay, E.G.; Bandiera, S.M. Involvement of cytochrome P450 in reactive oxygen species formation and cancer. Adv. Pharmacol. 2015, 74, 35–84. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Li, J.; Lou, B.; Wu, R.; Wang, G.; Lu, C.; Wang, H.; Pi, J.; Xu, Y. The role of reactive oxygen species in arsenic toxicity. Biomolecules 2020, 10, 240. [Google Scholar] [CrossRef]
- Shi, H.; Shi, X.; Liu, K.J. Oxidative mechanism of arsenic toxicity and carcinogenesis. Mol. Cell Biochem. 2004, 255, 67–78. [Google Scholar] [CrossRef]
- Zhang, H.; Jin, B.; Liu, L.; Li, H.; Zheng, X.; Li, M.; He, R.; Wang, K. Glutathione might attenuate arsenic-induced liver injury by modulating the Foxa2-XIAP Axis to reduce oxidative stress and mitochondrial apoptosis. Biol. Trace Elem. Res. 2023, 20, 5201–5212. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.; Liu, Y.; Wang, D.; Zhu, K.; Zou, Z.; Zhang, A. Imbalanced inflammatory response in subchronic arsenic-induced liver injury and the protective effects of Ginkgo biloba extract in rats: Potential role of cytokines mediated cell-cell interactions. Environ. Toxicol. 2021, 36, 2073–2092. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Pi, R.; Luo, J.; Liu, J.; Zhang, A.; Sun, B. Association between arsenic exposure and inflammatory cytokines and C-reaction protein: A systematic review and meta-analysis. Medicine 2022, 101, e32352. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).