
Integrated Analysis of Microbiome and Metabolome Reveals Disease-Specific Profiles in Inflammatory Bowel Diseases and Intestinal Behçet’s Disease
 , ,
, ,
Abstract
1. Introduction
2. Results
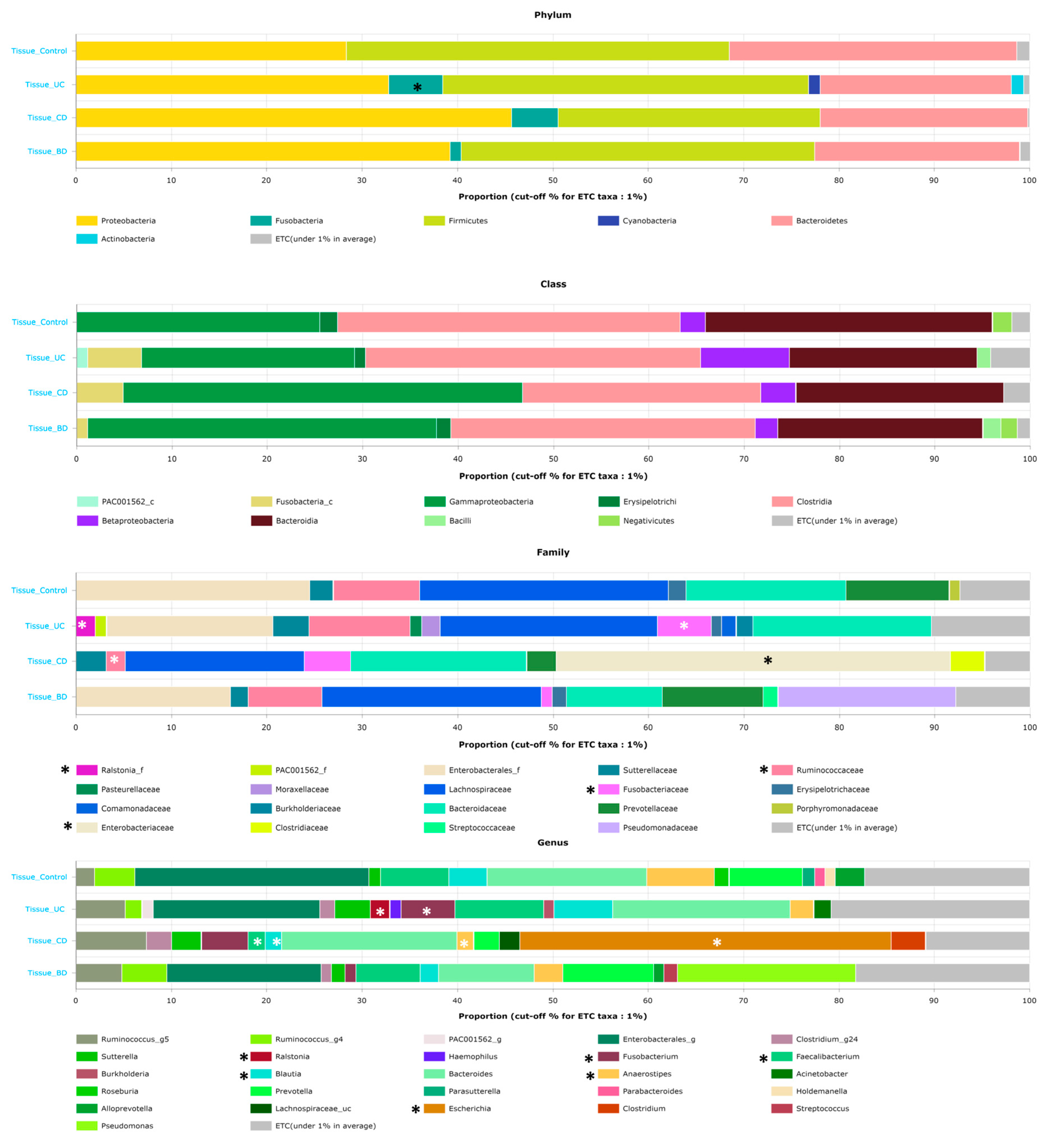
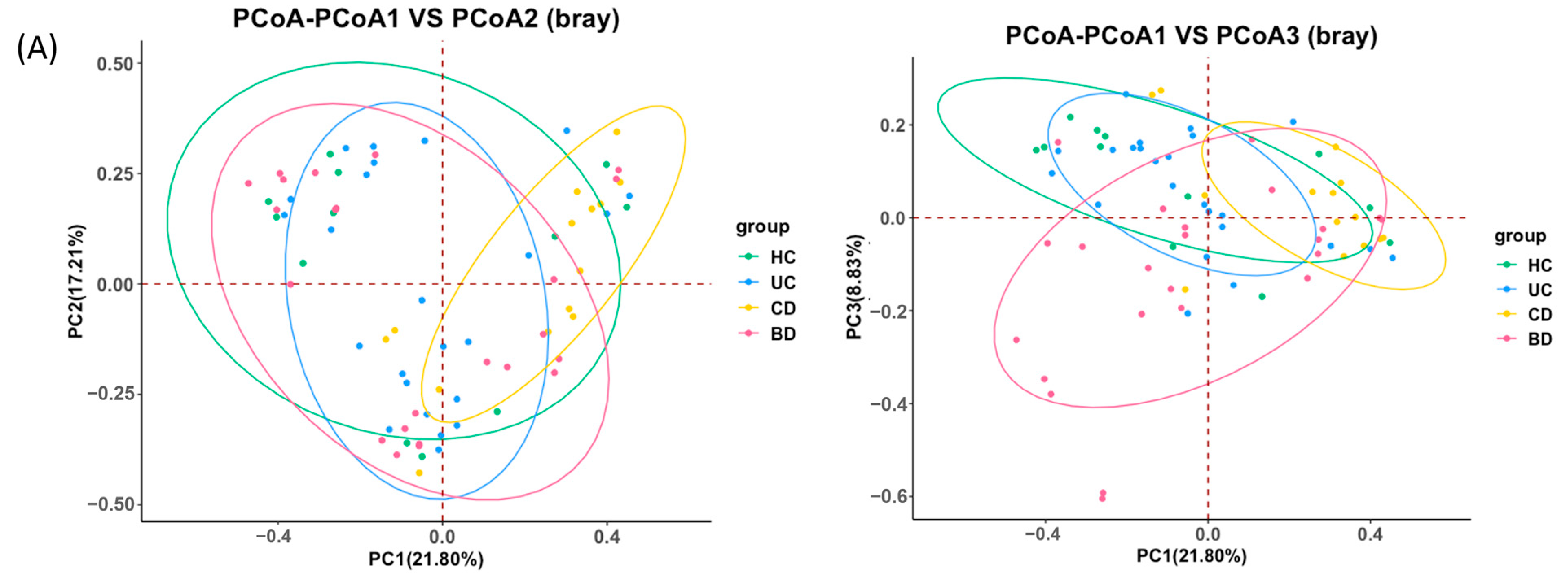
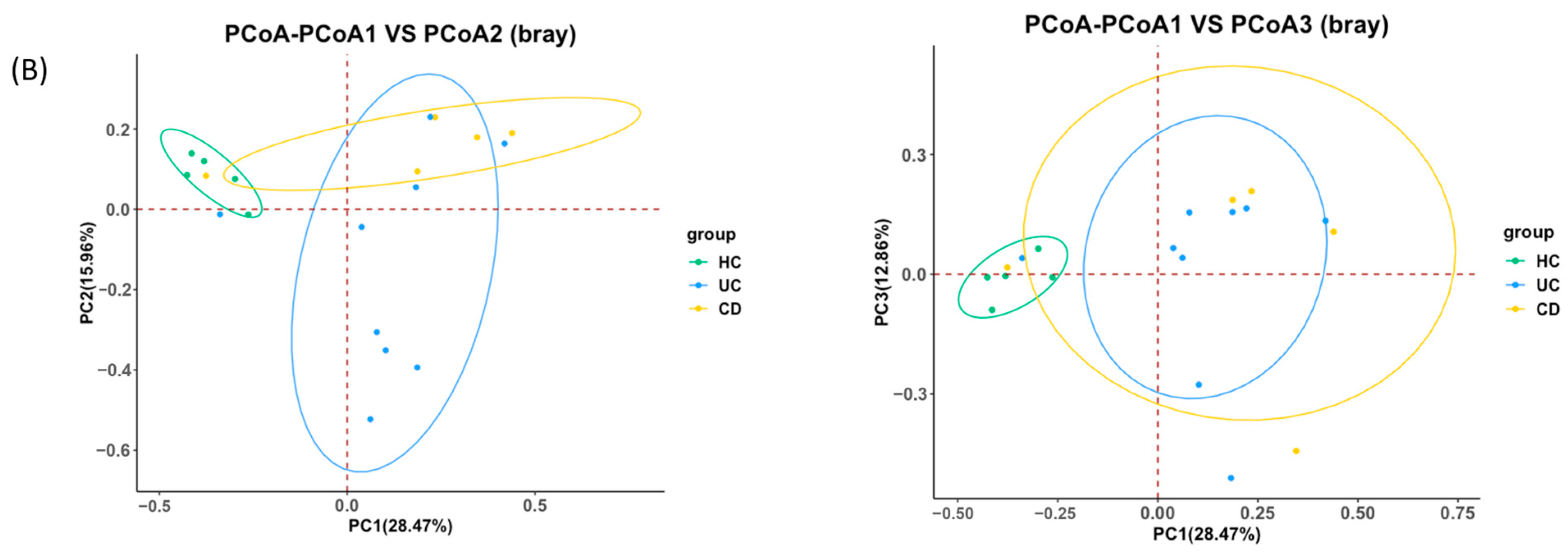
2.1. Ulcerative Colitis, Crohn’s Disease, and Intestinal Behçet’s Disease Display Distinct Tissue and Fecal Microbiota Compositions
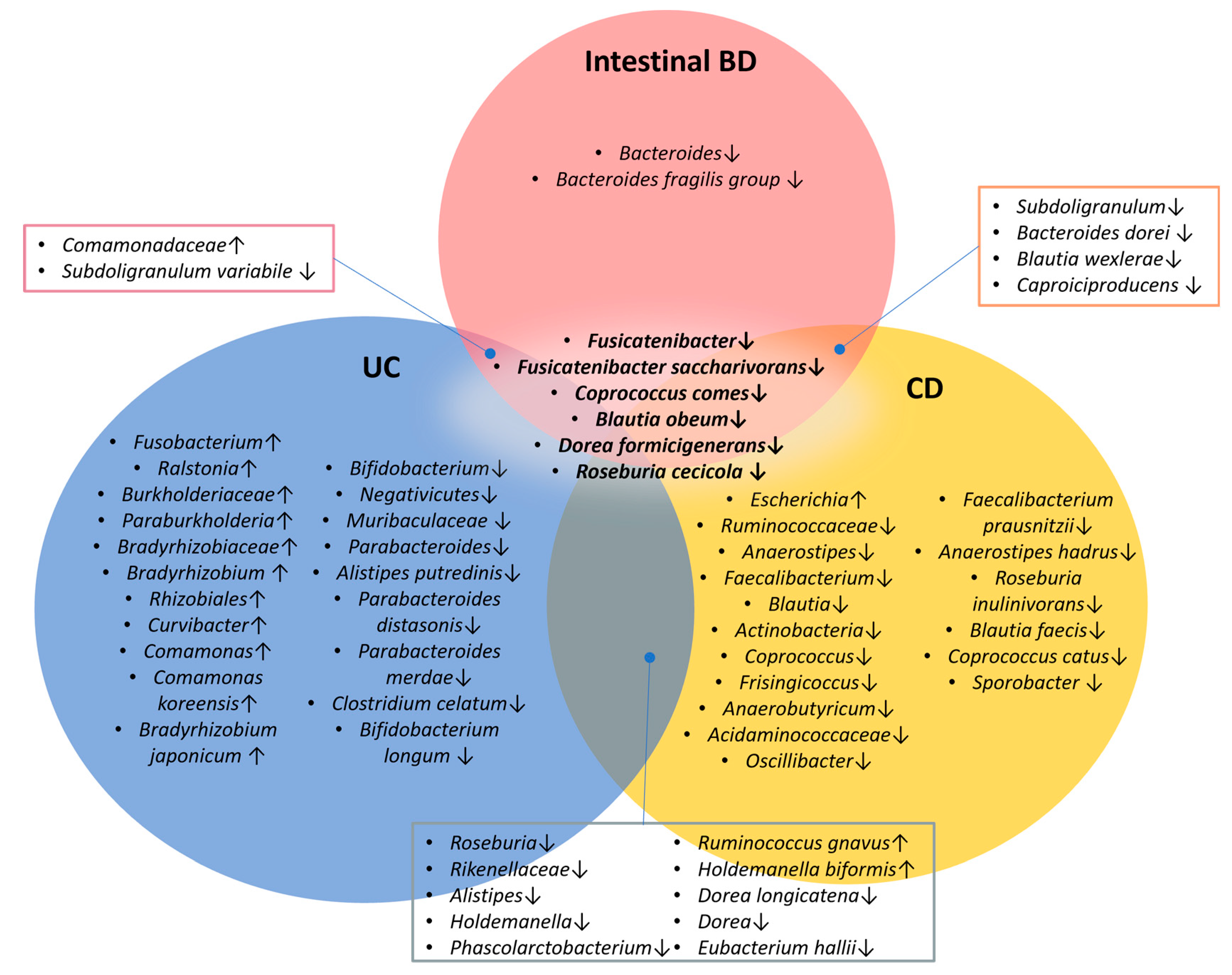
2.2. Intestinal BD Displays Distinctive Microbiota in Both Tissue and Feces, Unlike Other IBDs
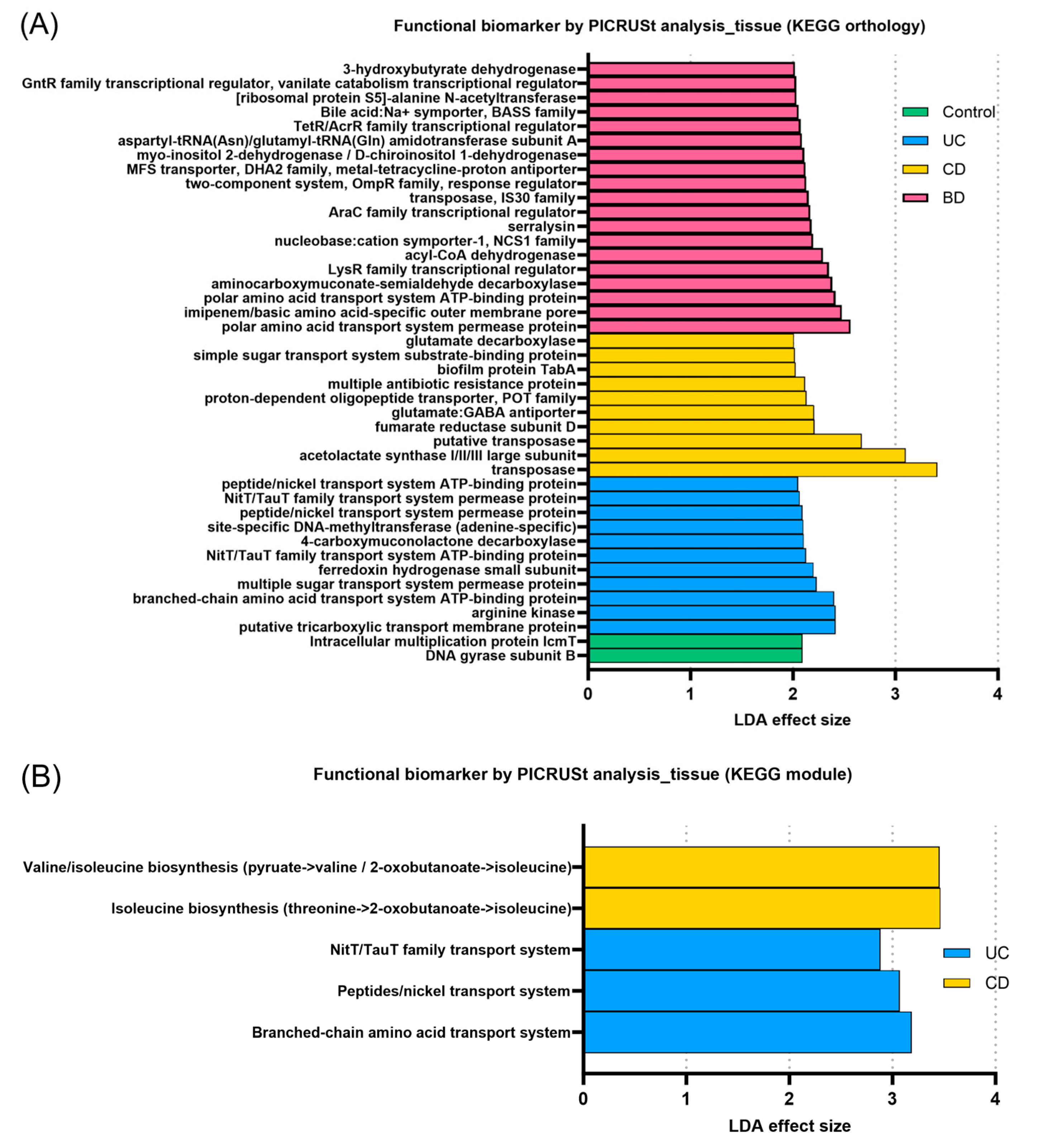
2.3. Inflammatory Bowel Diseases Exhibit Pronounced Functional Profiling of the Microbiome According to Subtype
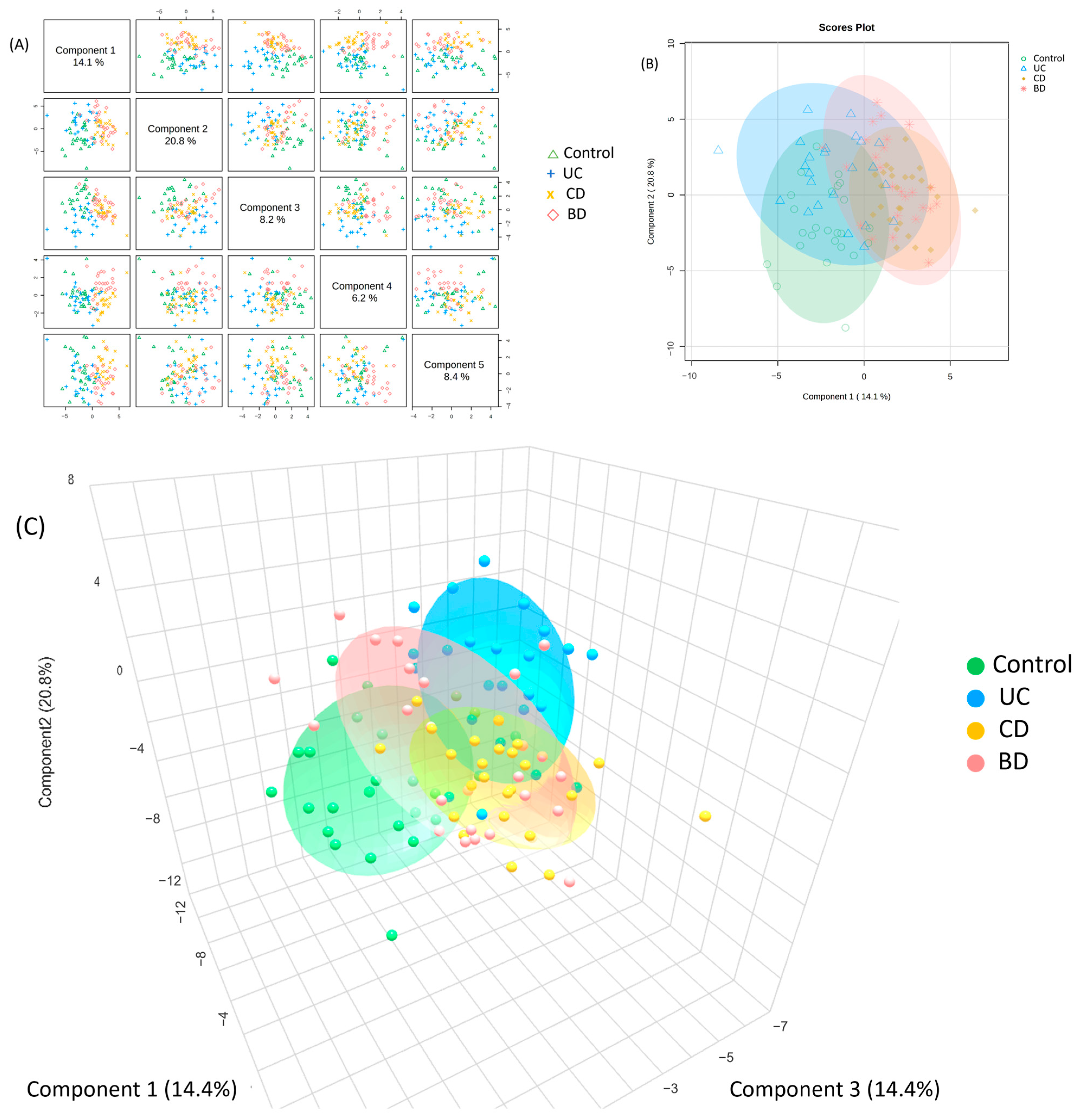
2.4. Intestinal Behçet’s Disease Metabolomic Profiles Share Similarities with Crohn’s Disease and Diverge from Ulcerative Colitis
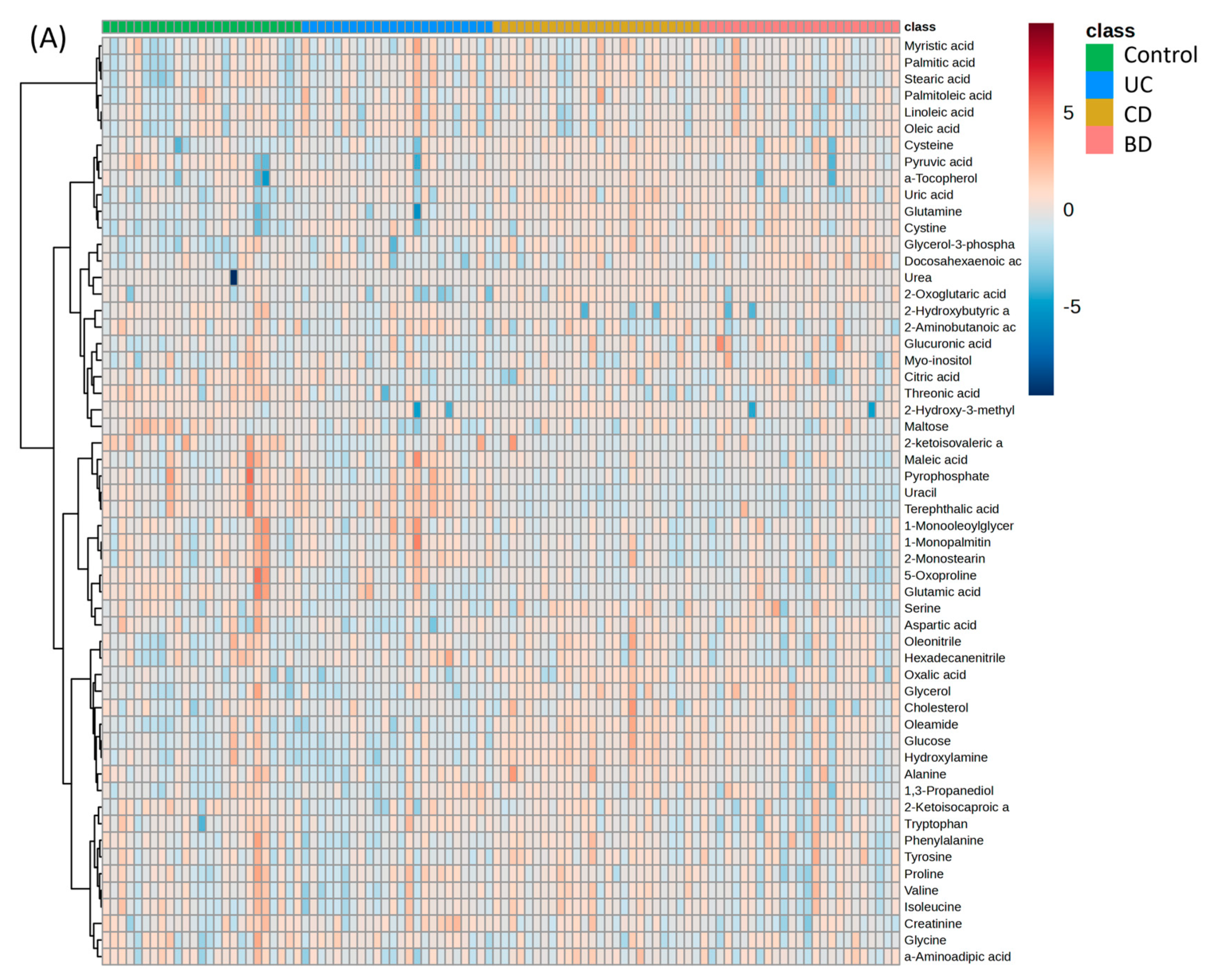
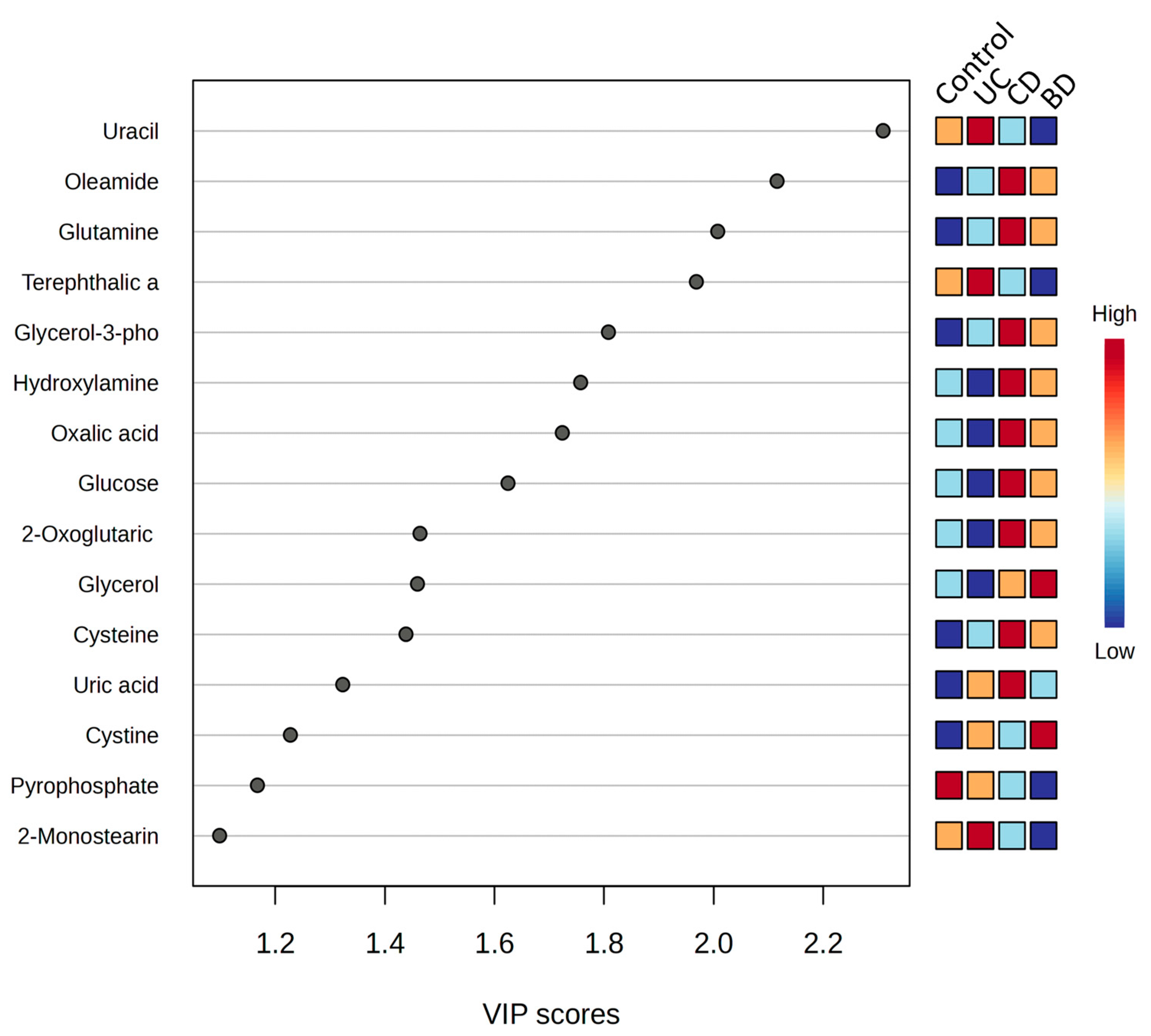
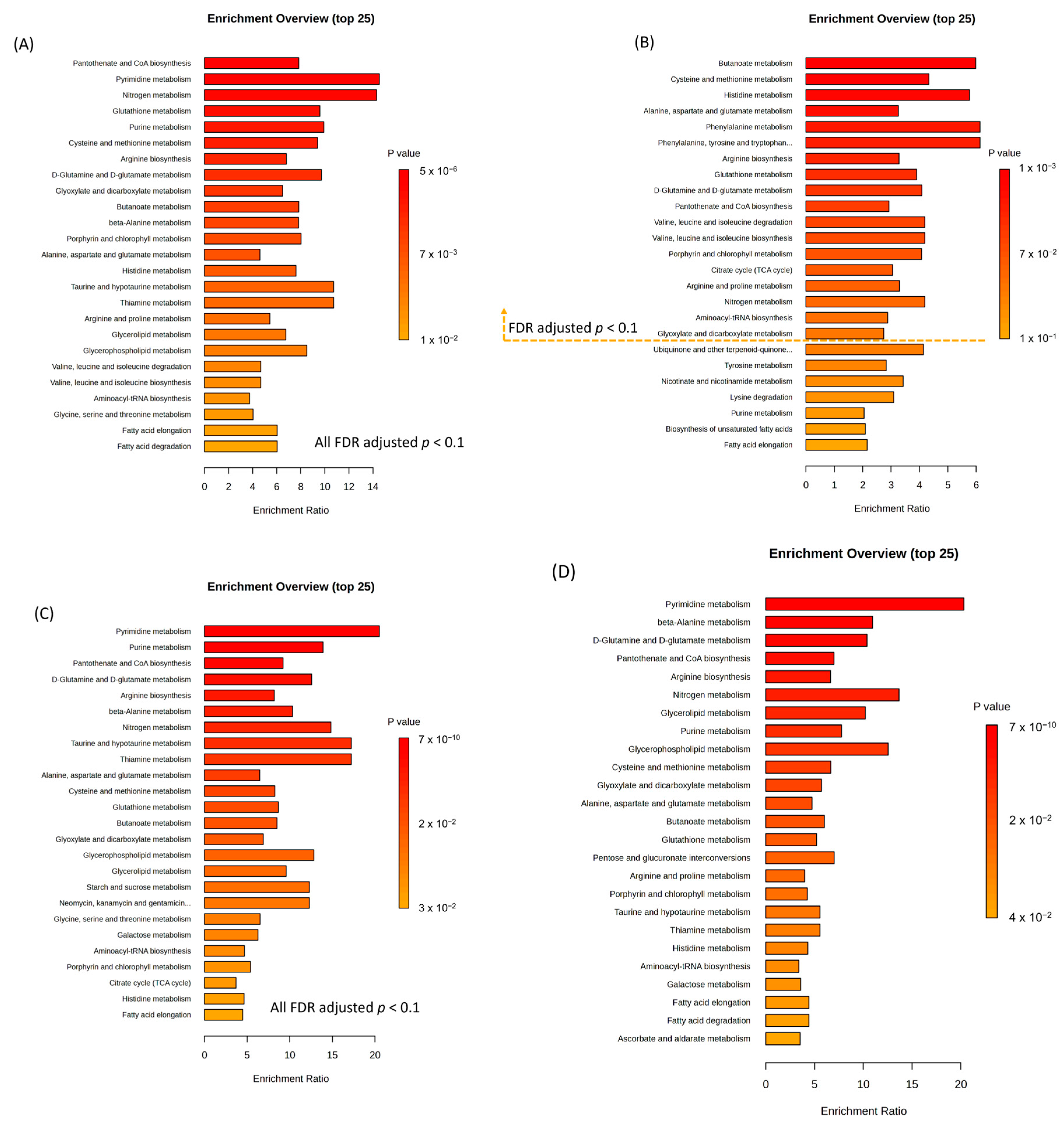
2.5. Functional Aspects of Metabolomic Biomarkers in Inflammatory Bowel Diseases and Intestinal Behçet’s Disease Differ from Those in Healthy Controls
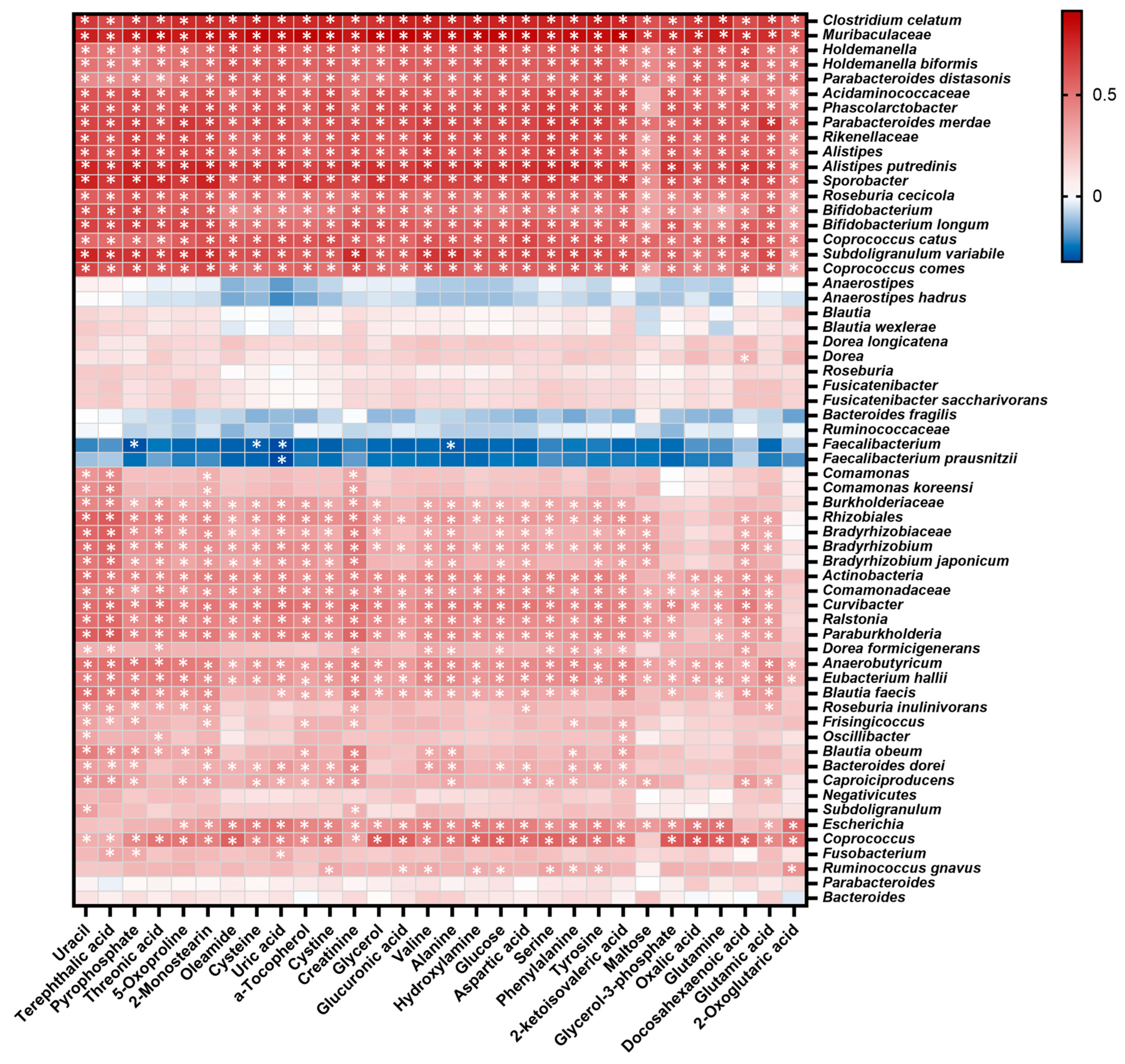
2.6. Integration of Microbial and Metabolomic Biomarkers Showed Correlations between Functional Aspects of Microbiota and Metabolites
3. Discussion
4. Materials and Methods
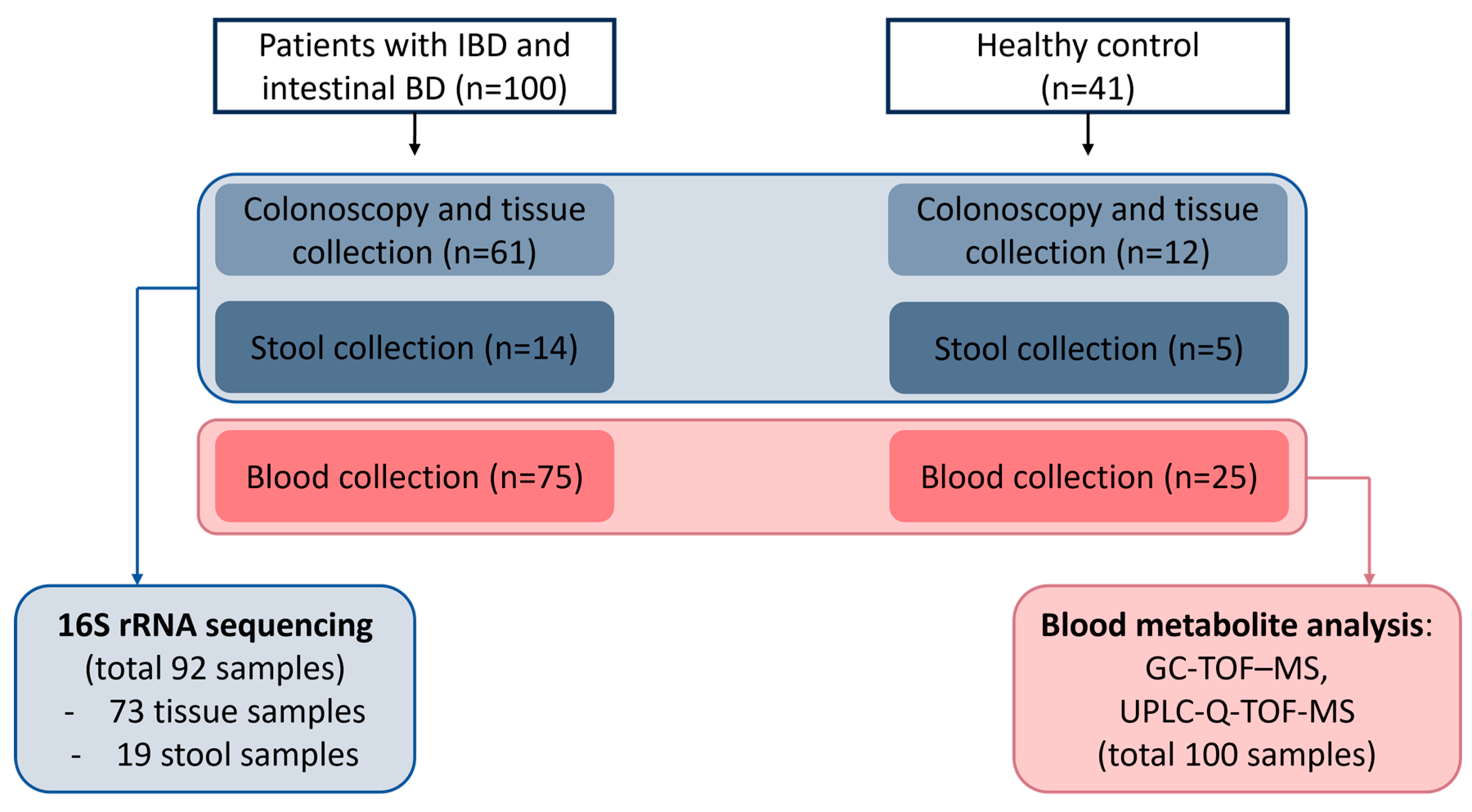
4.1. Study Subjects
4.2. Clinical Data Collection
4.3. Collecting Tissue, Blood, and Stool Samples
4.4. Microbiome Analysis
4.4.1. DNA Extraction
4.4.2. PCR Amplification and 16S rRNA Amplicon Sequencing
4.4.3. Microbiome Data Analysis
4.5. Metabolomic Analysis
4.5.1. Sample Preparation for Metabolomic Analysis
4.5.2. GC-TOF-MS Analysis
4.5.3. UPLC-Q-TOF-MS Analysis
4.5.4. Data Analysis
4.6. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Imhann, F.; Vich Vila, A.; Bonder, M.J.; Fu, J.; Gevers, D.; Visschedijk, M.C.; Spekhorst, L.M.; Alberts, R.; Franke, L.; van Dullemen, H.M.; et al. Interplay of host genetics and gut microbiota underlying the onset and clinical presentation of inflammatory bowel disease. Gut 2018, 67, 108–119. [Google Scholar] [CrossRef] [PubMed]
- Davatchi, F.; Chams-Davatchi, C.; Shams, H.; Shahram, F.; Nadji, A.; Akhlaghi, M.; Faezi, T.; Ghodsi, Z.; Sadeghi Abdollahi, B.; Ashofteh, F.; et al. Behcet’s disease: Epidemiology, clinical manifestations, and diagnosis. Expert Rev. Clin. Immunol. 2017, 13, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Han, S.J.; Kang, E.A.; Park, J.; Park, S.J.; Park, J.J.; Kim, T.I.; Kim, W.H.; Cheon, J.H. Risk Factors for Surgery in Patients with Intestinal Behcet’s Disease During Anti-Tumor Necrosis Factor-Alpha Therapy. Yonsei Med. J. 2023, 64, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Leccese, P.; Alpsoy, E. Behçet’s Disease: An Overview of Etiopathogenesis. Front. Immunol. 2019, 10, 1067. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Cheon, J.H. Incidence and Prevalence of Inflammatory Bowel Disease across Asia. Yonsei Med. J. 2021, 62, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Pittayanon, R.; Lau, J.T.; Leontiadis, G.I.; Tse, F.; Yuan, Y.; Surette, M.; Moayyedi, P. Differences in Gut Microbiota in Patients with vs without Inflammatory Bowel Diseases: A Systematic Review. Gastroenterology 2020, 158, 930–946.e931. [Google Scholar] [CrossRef]
- Dovrolis, N.; Drygiannakis, I.; Filidou, E.; Kandilogiannakis, L.; Arvanitidis, K.; Tentes, I.; Kolios, G.; Valatas, V. Gut Microbial Signatures Underline Complicated Crohn’s Disease but Vary Between Cohorts; An In Silico Approach. Inflamm. Bowel Dis. 2019, 25, 217–225. [Google Scholar] [CrossRef]
- Gilbert, J.A.; Blaser, M.J.; Caporaso, J.G.; Jansson, J.K.; Lynch, S.V.; Knight, R. Current understanding of the human microbiome. Nat. Med. 2018, 24, 392–400. [Google Scholar] [CrossRef]
- Pascal, V.; Pozuelo, M.; Borruel, N.; Casellas, F.; Campos, D.; Santiago, A.; Martinez, X.; Varela, E.; Sarrabayrouse, G.; Machiels, K.; et al. A microbial signature for Crohn’s disease. Gut 2017, 66, 813–822. [Google Scholar] [CrossRef]
- Gevers, D.; Kugathasan, S.; Denson, L.A.; Vázquez-Baeza, Y.; Van Treuren, W.; Ren, B.; Schwager, E.; Knights, D.; Song, S.J.; Yassour, M.; et al. The treatment-naive microbiome in new-onset Crohn’s disease. Cell Host Microbe 2014, 15, 382–392. [Google Scholar] [CrossRef]
- Metwaly, A.; Dunkel, A.; Waldschmitt, N.; Raj, A.C.D.; Lagkouvardos, I.; Corraliza, A.M.; Mayorgas, A.; Martinez-Medina, M.; Reiter, S.; Schloter, M.; et al. Integrated microbiota and metabolite profiles link Crohn’s disease to sulfur metabolism. Nat. Commun. 2020, 11, 4322. [Google Scholar] [CrossRef] [PubMed]
- Vester-Andersen, M.K.; Mirsepasi-Lauridsen, H.C.; Prosberg, M.V.; Mortensen, C.O.; Träger, C.; Skovsen, K.; Thorkilgaard, T.; Nøjgaard, C.; Vind, I.; Krogfelt, K.A.; et al. Increased abundance of proteobacteria in aggressive Crohn’s disease seven years after diagnosis. Sci. Rep. 2019, 9, 13473. [Google Scholar] [CrossRef] [PubMed]
- Bamba, S.; Inatomi, O.; Nishida, A.; Ohno, M.; Imai, T.; Takahashi, K.; Naito, Y.; Iwamoto, J.; Honda, A.; Inohara, N.; et al. Relationship between the gut microbiota and bile acid composition in the ileal mucosa of Crohn’s disease. Intest. Res. 2022, 20, 370–380. [Google Scholar] [CrossRef]
- Shin, S.Y.; Kim, Y.; Kim, W.S.; Moon, J.M.; Lee, K.M.; Jung, S.A.; Park, H.; Huh, E.Y.; Kim, B.C.; Lee, S.C.; et al. Compositional changes in fecal microbiota associated with clinical phenotypes and prognosis in Korean patients with inflammatory bowel disease. Intest. Res. 2023, 21, 148–160. [Google Scholar] [CrossRef]
- Franzosa, E.A.; Sirota-Madi, A.; Avila-Pacheco, J.; Fornelos, N.; Haiser, H.J.; Reinker, S.; Vatanen, T.; Hall, A.B.; Mallick, H.; McIver, L.J.; et al. Gut microbiome structure and metabolic activity in inflammatory bowel disease. Nat. Microbiol. 2019, 4, 293–305. [Google Scholar] [CrossRef] [PubMed]
- Kolho, K.L.; Pessia, A.; Jaakkola, T.; de Vos, W.M.; Velagapudi, V. Faecal and Serum Metabolomics in Paediatric Inflammatory Bowel Disease. J. Crohn’s Colitis 2017, 11, 321–334. [Google Scholar] [CrossRef]
- Scoville, E.A.; Allaman, M.M.; Brown, C.T.; Motley, A.K.; Horst, S.N.; Williams, C.S.; Koyama, T.; Zhao, Z.; Adams, D.W.; Beaulieu, D.B.; et al. Alterations in Lipid, Amino Acid, and Energy Metabolism Distinguish Crohn’s Disease from Ulcerative Colitis and Control Subjects by Serum Metabolomic Profiling. Metabolomics 2018, 14, 17. [Google Scholar] [CrossRef]
- Wilmanski, T.; Rappaport, N.; Earls, J.C.; Magis, A.T.; Manor, O.; Lovejoy, J.; Omenn, G.S.; Hood, L.; Gibbons, S.M.; Price, N.D. Blood metabolome predicts gut microbiome alpha-diversity in humans. Nat. Biotechnol. 2019, 37, 1217–1228. [Google Scholar] [CrossRef]
- Consolandi, C.; Turroni, S.; Emmi, G.; Severgnini, M.; Fiori, J.; Peano, C.; Biagi, E.; Grassi, A.; Rampelli, S.; Silvestri, E.; et al. Behcet’s syndrome patients exhibit specific microbiome signature. Autoimmun. Rev. 2015, 14, 269–276. [Google Scholar] [CrossRef]
- Metwaly, A.; Reitmeier, S.; Haller, D. Microbiome risk profiles as biomarkers for inflammatory and metabolic disorders. Nat. Rev. Gastroenterol. Hepatol. 2022, 19, 383–397. [Google Scholar] [CrossRef]
- Qiu, P.; Ishimoto, T.; Fu, L.; Zhang, J.; Zhang, Z.; Liu, Y. The Gut Microbiota in Inflammatory Bowel Disease. Front. Cell. Infect. Microbiol. 2022, 12, 733992. [Google Scholar] [CrossRef] [PubMed]
- Schirmer, M.; Garner, A.; Vlamakis, H.; Xavier, R.J. Microbial genes and pathways in inflammatory bowel disease. Nat. Rev. Microbiol. 2019, 17, 497–511. [Google Scholar] [CrossRef] [PubMed]
- Vich Vila, A.; Imhann, F.; Collij, V.; Jankipersadsing, S.A.; Gurry, T.; Mujagic, Z.; Kurilshikov, A.; Bonder, M.J.; Jiang, X.; Tigchelaar, E.F.; et al. Gut microbiota composition and functional changes in inflammatory bowel disease and irritable bowel syndrome. Sci. Transl. Med. 2018, 10, eaap8914. [Google Scholar] [CrossRef] [PubMed]
- Takada, T.; Kurakawa, T.; Tsuji, H.; Nomoto, K. Fusicatenibacter saccharivorans gen. nov., sp. nov., isolated from human faeces. Int. J. Syst. Evol. Microbiol. 2013, 63, 3691–3696. [Google Scholar] [CrossRef] [PubMed]
- Takeshita, K.; Mizuno, S.; Mikami, Y.; Sujino, T.; Saigusa, K.; Matsuoka, K.; Naganuma, M.; Sato, T.; Takada, T.; Tsuji, H.; et al. A Single Species of Clostridium Subcluster XIVa Decreased in Ulcerative Colitis Patients. Inflamm. Bowel Dis. 2016, 22, 2802–2810. [Google Scholar] [CrossRef] [PubMed]
- Gryaznova, M.V.; Solodskikh, S.A.; Panevina, A.V.; Syromyatnikov, M.Y.; Dvoretskaya, Y.D.; Sviridova, T.N.; Popov, E.S.; Popov, V.N. Study of microbiome changes in patients with ulcerative colitis in the Central European part of Russia. Heliyon 2021, 7, e06432. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Lu, X.; Zhang, W.; Yang, L.; Wang, D.; Xu, J.; Jia, Y.; Wang, X.; Xie, H.; Li, S.; et al. Gut microbiota in the early stage of Crohn’s disease has unique characteristics. Gut Pathog. 2022, 14, 46. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.C.; Park, M.J.; Park, S.; Lee, E.S. Alteration of the Fecal but Not Salivary Microbiome in Patients with Behçet’s Disease According to Disease Activity Shift. Microorganisms 2021, 9, 1449. [Google Scholar] [CrossRef] [PubMed]
- Trosvik, P.; de Muinck, E.J. Ecology of bacteria in the human gastrointestinal tract--identification of keystone and foundation taxa. Microbiome 2015, 3, 44. [Google Scholar] [CrossRef]
- Ramakrishna, C.; Kujawski, M.; Chu, H.; Li, L.; Mazmanian, S.K.; Cantin, E.M. Bacteroides fragilis polysaccharide A induces IL-10 secreting B and T cells that prevent viral encephalitis. Nat. Commun. 2019, 10, 2153. [Google Scholar] [CrossRef]
- Zhang, W.; Zhou, Q.; Liu, H.; Xu, J.; Huang, R.; Shen, B.; Guo, Y.; Ai, X.; Xu, J.; Zhao, X.; et al. Bacteroides fragilis strain ZY-312 facilitates colonic mucosa regeneration in colitis via motivating STAT3 signaling pathway induced by IL-22 from ILC3 secretion. Front. Immunol. 2023, 14, 1156762. [Google Scholar] [CrossRef] [PubMed]
- Porter, N.T.; Luis, A.S.; Martens, E.C. Bacteroides thetaiotaomicron. Trends Microbiol. 2018, 26, 966–967. [Google Scholar] [CrossRef] [PubMed]
- Mazmanian, S.K.; Liu, C.H.; Tzianabos, A.O.; Kasper, D.L. An immunomodulatory molecule of symbiotic bacteria directs maturation of the host immune system. Cell 2005, 122, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.C.; Ching, Y.H.; Chiu, C.C.; Liu, J.Y.; Hung, S.W.; Huang, W.C.; Huang, Y.T.; Chuang, H.L. TLR2 and interleukin-10 are involved in Bacteroides fragilis-mediated prevention of DSS-induced colitis in gnotobiotic mice. PLoS ONE 2017, 12, e0180025. [Google Scholar] [CrossRef] [PubMed]
- Mazmanian, S.K.; Round, J.L.; Kasper, D.L. A microbial symbiosis factor prevents intestinal inflammatory disease. Nature 2008, 453, 620–625. [Google Scholar] [CrossRef]
- Studd, M.; McCance, D.J.; Lehner, T. Detection of HSV-1 DNA in patients with Behçet’s syndrome and in patients with recurrent oral ulcers by the polymerase chain reaction. J. Med. Microbiol. 1991, 34, 39–43. [Google Scholar] [CrossRef] [PubMed]
- Lloyd-Price, J.; Arze, C.; Ananthakrishnan, A.N.; Schirmer, M.; Avila-Pacheco, J.; Poon, T.W.; Andrews, E.; Ajami, N.J.; Bonham, K.S.; Brislawn, C.J.; et al. Multi-omics of the gut microbial ecosystem in inflammatory bowel diseases. Nature 2019, 569, 655–662. [Google Scholar] [CrossRef]
- Marchesi, J.R.; Holmes, E.; Khan, F.; Kochhar, S.; Scanlan, P.; Shanahan, F.; Wilson, I.D.; Wang, Y. Rapid and noninvasive metabonomic characterization of inflammatory bowel disease. J. Proteome Res. 2007, 6, 546–551. [Google Scholar] [CrossRef]
- Le Gall, G.; Noor, S.O.; Ridgway, K.; Scovell, L.; Jamieson, C.; Johnson, I.T.; Colquhoun, I.J.; Kemsley, E.K.; Narbad, A. Metabolomics of fecal extracts detects altered metabolic activity of gut microbiota in ulcerative colitis and irritable bowel syndrome. J. Proteome Res. 2011, 10, 4208–4218. [Google Scholar] [CrossRef]
- Jansson, J.; Willing, B.; Lucio, M.; Fekete, A.; Dicksved, J.; Halfvarson, J.; Tysk, C.; Schmitt-Kopplin, P. Metabolomics reveals metabolic biomarkers of Crohn’s disease. PLoS ONE 2009, 4, e6386. [Google Scholar] [CrossRef]
- Bauset, C.; Gisbert-Ferrándiz, L.; Cosín-Roger, J. Metabolomics as a Promising Resource Identifying Potential Biomarkers for Inflammatory Bowel Disease. J. Clin. Med. 2021, 10, 622. [Google Scholar] [CrossRef]
- Gallagher, K.; Catesson, A.; Griffin, J.L.; Holmes, E.; Williams, H.R.T. Metabolomic Analysis in Inflammatory Bowel Disease: A Systematic Review. J. Crohn’s Colitis 2021, 15, 813–826. [Google Scholar] [CrossRef]
- Anand, S.; Kaur, H.; Mande, S.S. Comparative In silico Analysis of Butyrate Production Pathways in Gut Commensals and Pathogens. Front. Microbiol. 2016, 7, 1945. [Google Scholar] [CrossRef] [PubMed]
- Arp, D.J.; Stein, L.Y. Metabolism of inorganic N compounds by ammonia-oxidizing bacteria. Crit. Rev. Biochem. Mol. Biol. 2003, 38, 471–495. [Google Scholar] [CrossRef] [PubMed]
- Tong, S.; Zhang, P.; Cheng, Q.; Chen, M.; Chen, X.; Wang, Z.; Lu, X.; Wu, H. The role of gut microbiota in gout: Is gut microbiota a potential target for gout treatment. Front. Cell. Infect. Microbiol. 2022, 12, 1051682. [Google Scholar] [CrossRef]
- Zhu, F.; Feng, D.; Zhang, T.; Gu, L.; Zhu, W.; Guo, Z.; Li, Y.; Lu, N.; Gong, J.; Li, N. Altered uric acid metabolism in isolated colonic Crohn’s disease but not ulcerative colitis. J. Gastroenterol. Hepatol. 2019, 34, 154–161. [Google Scholar] [CrossRef]
- Parthasarathy, G.; Chen, J.; Chen, X.; Chia, N.; O’Connor, H.M.; Wolf, P.G.; Gaskins, H.R.; Bharucha, A.E. Relationship Between Microbiota of the Colonic Mucosa vs Feces and Symptoms, Colonic Transit, and Methane Production in Female Patients With Chronic Constipation. Gastroenterology 2016, 150, 367–379.e361. [Google Scholar] [CrossRef]
- Fukata, M.; Arditi, M. The role of pattern recognition receptors in intestinal inflammation. Mucosal Immunol. 2013, 6, 451–463. [Google Scholar] [CrossRef]
- Halfvarson, J.; Brislawn, C.J.; Lamendella, R.; Vázquez-Baeza, Y.; Walters, W.A.; Bramer, L.M.; D’Amato, M.; Bonfiglio, F.; McDonald, D.; Gonzalez, A.; et al. Dynamics of the human gut microbiome in inflammatory bowel disease. Nat. Microbiol. 2017, 2, 17004. [Google Scholar] [CrossRef]
- Gomollon, F.; Dignass, A.; Annese, V.; Tilg, H.; Van Assche, G.; Lindsay, J.O.; Peyrin-Biroulet, L.; Cullen, G.J.; Daperno, M.; Kucharzik, T.; et al. 3rd European Evidence-based Consensus on the Diagnosis and Management of Crohn’s Disease 2016: Part 1: Diagnosis and Medical Management. J. Crohn’s Colitis 2017, 11, 3–25. [Google Scholar] [CrossRef]
- Magro, F.; Gionchetti, P.; Eliakim, R.; Ardizzone, S.; Armuzzi, A.; Barreiro-de Acosta, M.; Burisch, J.; Gecse, K.B.; Hart, A.L.; Hindryckx, P.; et al. Third European Evidence-based Consensus on Diagnosis and Management of Ulcerative Colitis. Part 1: Definitions, Diagnosis, Extra-intestinal Manifestations, Pregnancy, Cancer Surveillance, Surgery, and Ileo-anal Pouch Disorders. J. Crohn’s Colitis 2017, 11, 649–670. [Google Scholar] [CrossRef] [PubMed]
- Cheon, J.H.; Kim, E.S.; Shin, S.J.; Kim, T.I.; Lee, K.M.; Kim, S.W.; Kim, J.S.; Kim, Y.S.; Choi, C.H.; Ye, B.D.; et al. Development and validation of novel diagnostic criteria for intestinal Behcet’s disease in Korean patients with ileocolonic ulcers. Am. J. Gastroenterol. 2009, 104, 2492–2499. [Google Scholar] [CrossRef] [PubMed]
- Cheon, J.H.; Han, D.S.; Park, J.Y.; Ye, B.D.; Jung, S.A.; Park, Y.S.; Kim, Y.S.; Kim, J.S.; Nam, C.M.; Kim, Y.N.; et al. Development, validation, and responsiveness of a novel disease activity index for intestinal Behcet’s disease. Inflamm. Bowel Dis. 2011, 17, 605–613. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.S.; Kim, J.N.; Yoon, S.H.; Chun, J.; Cerniglia, C.E. Impact of enrofloxacin on the human intestinal microbiota revealed by comparative molecular analysis. Anaerobe 2012, 18, 310–320. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.S.; Song, M.Y.; Kim, H. The anti-obesity effect of Ephedra sinica through modulation of gut microbiota in obese Korean women. J. Ethnopharmacol. 2014, 152, 532–539. [Google Scholar] [CrossRef] [PubMed]
- Yun, K.H.; Lee, H.S.; Nam, O.H.; Moon, C.Y.; Lee, J.H.; Choi, S.C. Analysis of bacterial community profiles of endodontically infected primary teeth using pyrosequencing. Int. J. Paediatr. Dent. 2017, 27, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.H.; Ha, S.M.; Kwon, S.; Lim, J.; Kim, Y.; Seo, H.; Chun, J. Introducing EzBioCloud: A taxonomically united database of 16S rRNA gene sequences and whole-genome assemblies. Int. J. Syst. Evol. Microbiol. 2017, 67, 1613–1617. [Google Scholar] [CrossRef] [PubMed]
- Jeon, Y.S.; Lee, K.; Park, S.C.; Kim, B.S.; Cho, Y.J.; Ha, S.M.; Chun, J. EzEditor: A versatile sequence alignment editor for both rRNA- and protein-coding genes. Int. J. Syst. Evol. Microbiol. 2014, 64, 689–691. [Google Scholar] [CrossRef] [PubMed]
- Langille, M.G.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Vega Thurber, R.L.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef]
- Pavoine, S.; Dufour, A.B.; Chessel, D. From dissimilarities among species to dissimilarities among communities: A double principal coordinate analysis. J. Theor. Biol. 2004, 228, 523–537. [Google Scholar] [CrossRef]
- Ramette, A. Multivariate analyses in microbial ecology. FEMS Microbiol. Ecol. 2007, 62, 142–160. [Google Scholar] [CrossRef] [PubMed]
- Kelly, B.J.; Gross, R.; Bittinger, K.; Sherrill-Mix, S.; Lewis, J.D.; Collman, R.G.; Bushman, F.D.; Li, H. Power and sample-size estimation for microbiome studies using pairwise distances and PERMANOVA. Bioinformatics 2015, 31, 2461–2468. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.Y.; Jung, E.S.; Suh, D.H.; Jeong, S.J.; Kim, K.; Chon, S.; Yu, S.Y.; Woo, J.T.; Lee, C.H. Plasma amino acids and oxylipins as potential multi-biomarkers for predicting diabetic macular edema. Sci. Rep. 2021, 11, 9727. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.Y.; Jung, E.S.; Park, H.M.; Jeong, S.J.; Kim, K.; Chon, S.; Yu, S.Y.; Woo, J.T.; Lee, C.H. Plasma glutamine and glutamic acid are potential biomarkers for predicting diabetic retinopathy. Metabolomics 2018, 14, 89. [Google Scholar] [CrossRef] [PubMed]
- Park, S.A.; Son, S.Y.; Lee, A.Y.; Park, H.G.; Lee, W.L.; Lee, C.H. Metabolite Profiling Revealed That a Gardening Activity Program Improves Cognitive Ability Correlated with BDNF Levels and Serotonin Metabolism in the Elderly. Int. J. Environ. Res. Public Health 2020, 17, 541. [Google Scholar] [CrossRef] [PubMed]
- Pang, Z.; Zhou, G.; Ewald, J.; Chang, L.; Hacariz, O.; Basu, N.; Xia, J. Using MetaboAnalyst 5.0 for LC-HRMS spectra processing, multi-omics integration and covariate adjustment of global metabolomics data. Nat. Protoc. 2022, 17, 1735–1761. [Google Scholar] [CrossRef] [PubMed]
- Goeman, J.J.; van de Geer, S.A.; de Kort, F.; van Houwelingen, H.C. A global test for groups of genes: Testing association with a clinical outcome. Bioinformatics 2004, 20, 93–99. [Google Scholar] [CrossRef]
- Glickman, M.E.; Rao, S.R.; Schultz, M.R. False discovery rate control is a recommended alternative to Bonferroni-type adjustments in health studies. J. Clin. Epidemiol. 2014, 67, 850–857. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tissue Microbiome Analysis (n = 73) | p | Stool Microbiome Analysis (n = 19) | p | Plasma Metabolite Analysis (n = 100) | p | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Characteristics | Control (n = 12) | UC (n = 24) | CD (n = 14) | BD (n = 23) | Control (n = 5) | UC (n = 9) | CD (n = 5) | Control (n = 25) | UC (n = 24) | CD (n = 26) | BD (n = 25) | |||
| Age, median (range) | 47.2 (32.1–74.2) | 42.6 (19.5–68.6) | 22.7 (18.0–31.5) | 46.2 (25.8–76.5) | <0.001 | 22.0 (20.5–31.2) | 44.3 (18.4–56.3) | 18.0 (18.0–38.2) | 0.05 | 33.7 (26.0–43.6) | 37.9 (18.0–68.6) | 23.3 (18.0–42.6) | 44.3 (23.3–62.9) | <0.001 |
| Sex, male/female, n (%) | 6 (50.0)/6 (50.0) | 13 (54.2)/11 (45.8) | 11 (78.6)/3 (21.4) | 10 (43.5)/13 (56.5) | 0.21 | 3 (60.0)/2 (40.0) | 5 (55.6)/4 (44.4) | 5 (100)/0 | 0.21 | 8 (32.0)/17 (68.0) | 15 (62.5)/9 (37.5) | 22 (84.6)/4 (15.4) | 14 (56.0)/11 (44.0) | <0.01 |
| BMI, kg/m2, median (range) | 22.3 (15.6–26.8) | 21.8 (16.1–28.7) | 21.1 (13.9–22.9) | 23.0 (16.0–28.0) | 0.10 | 19.1 (18.9–22.8) | 20.7 (16.8–23.3) | 20.5 (16.3–29.6) | 0.87 | 21.8 (18.1–24.2) | 21.8 (16.1–28.7) | 20.6 (13.9–31.2) | 21.5 (16.5–24.7) | 0.53 |
| Bowel resection history, n (%) | 0 | 0 | 3 (21.4) | 1 (4.3) | 0.03 | 0 | 1 (11.1) | 1 (20.0) | 1.00 | 0 | 0 | 1 (3.8) | 3 (12.0) | 0.31 |
| Disease duration, months, median (range) | - | 39.6 (24.0–65.6) | 20.3 (7.5–28.3) | 27.5 (6.8–72.7) | 0.20 † | - | 2.6 (1.0–46.0) | 2.9 (2.0–12.0) | 0.95 † | - | 32.1 (1.0–155.0) | 7.2 (0–128.0) | 5.9 (0–110.0) | <0.01 † |
| Disease location, n (%) | - | |||||||||||||
| E1 (proctitis) | - | 6 (25) | - | - | - | 3 (33.3) | - | - | 7 (29.2) | - | - | |||
| E2 (left sided) | - | 7 (29.2) | - | - | - | 4 (44.4) | - | - | 5 (20.8) | - | - | |||
| E3 (pancolitis) | - | 11 (45.8) | - | - | - | 2 (22.2) | - | - | 12 (50.0) | - | - | |||
| L1 (ileal) | - | - | 0 | - | - | - | 1 (20) | - | - | 4 (15.4) | - | |||
| L2 (colonic) | - | - | 1 (7.1) | - | - | - | 0 | - | - | 2 (7.7) | - | |||
| L3 (ileocolic) | - | - | 13 (92.9) | - | - | - | 4 (80) | - | - | 20 (76.9) | - | |||
| With/without systemic BD, n (%) | - | - | - | 16 (69.6)/7 (30.4) | - | - | - | - | - | - | 16 (64.0)/9 (36.0) | |||
| Disease activity, remission/mild/moderate/severe, n (%) | - | 2 (8.3)/10 (41.7)/7 (29.2)/5 (20.8) | 7 (50.0)/2 (14.3)/5 (35.7)/0 | 4 (17.4)/6 (26.1)/7 (30.4)/6 (26.1) | 0.05 | - | 4 (44.4)/4 (44.4)/1 (33.3)/0 | 1 (20.0)/2 (40.0)/2 (40.0)/0 | 0.63 | - | 4 (16.7)/11 (45.8)/4 (16.7)/5 (20.8) | 8 (30.8)/10 (38.5)/8 (30.8)/0 | 6 (24.0)/6 (24.0)/7 (28.0)/6 (24.0) | 0.08 |
| Disease activity score, median (range) | ||||||||||||||
| Partial Mayo score | - | 4.5 (3.0–6.0) | - | - | - | 2.0 (1.0–6.0) | - | - | 3.5 (1.0–9.0) | - | - | |||
| CDAI | - | - | 148.5 (73.0–226.0) | - | - | - | 186.0 (128.0–358.0) | - | - | 184.5 (71.0–366.0) | - | |||
| DAIBD | - | - | - | 60.0 (22.5–70.0) | - | - | - | - | - | - | 40.0 (10.0–150.0) | |||
| Hb, g/dL, median (range) | 13.0 (11.7–16.0) | 13.5 (9.7–15.2) | 12.9 (10.7–15.8) | 13.1 (10.2–16.8) | 0.63 | 14.6 (13.2–15.2) | 14.1 (12.5–16.5) | 12.7 (10.6–16.0) | 0.17 | 12.4 (11.7–13.1) | 13.7 (9.7–15.2) | 13.9 (10.0–16.9) | 12.5 (6.5–15.3) | 0.52 |
| ESR, mm/h, median (range) | 14.0 (2.0–62.0) | 23.5 (2.0–52.0) | 37.0 (3.0–99.0) | 33.0 (8.0–106.0) | 0.07 | 7.5 (3.0–24.0) | 9.0 (2.0–45.0) | 47.0 (17.0–99.0) | 0.02 | - | 14.0 (2.0–84.0) | 21.0 (7.0–119.0) | 37.0 (2.0–120.0) | 0.02 † |
| CRP, mg/L, median (range) | 0.4 (0.3–0.5) | 2.2 (0.8–3.9) | 7.9 (2.3–23.6) | 5.6 (1.5–13.9) | 0.02 | 2.3 (0.6–5.5) | 1.0 (0.3–25.5) | 18.6 (0.7–91.3) | 0.10 | - | 1.0 (0.1–9.2) | 6.5 (0.9–66.2) | 2.4 (0.3–28.4) | <0.01 † |
| Albumin, g/dL, median (range) | 4.3 (4.2–4.5) | 4.2 (4.0–4.5) | 4.0 (3.7–4.5) | 4.4 (4.2–4.5) | 0.14 | 4.4 (4.1–4.4) | 4.4 (4.1–4.8) | 3.7 (3.6–4.4) | 0.10 | 4.5 (4.4–4.6) | 4.4 (3.5–5.1) | 4.1 (3.4–4.8) | 4.2 (3.4–5.1) | 0.07 |
| Creatinine, mg/dL, median (range) | 0.8 (0.5–1.0) | 0.7 (0.5–1.1) | 0.8 (0.5–1.0) | 0.7 (0.5–1.4) | 0.62 | 0.76 (0.62–0.99) | 0.78 (0.56–0.94) | 0.67 (0.61–0.95) | 0.92 | 0.7 (0.6–0.8) | 0.7 (0.5–1.2) | 0.7 (0.5–1.1) | 0.8 (0.5–1.2) | 0.34 |
| ALT, U/L, median (range) | 18 (8.0–30.0) | 9 (5.0–34.0) | 9.5 (6.0–40.0) | 20.0 (7.0–40.0) | <0.01 | 14.0 (12.0–14.0) | 11.0 (9.0–20.0) | 10.0 (10.0–12.0) | 0.71 | 19 (17.0–21.0) | 11.0 (5.0–34.0) | 13.0 (5.0–47.0) | 14.0 (6.0–58.0) | 0.10 |
| Medication, n (%) | ||||||||||||||
| 5-ASA | - | 24 (100) | 12 (85.7) | 23 (100) | 0.05 † | - | 9 (100) | 4 (80.0) | 0.36 † | - | 24 (100) | 24 (92.3) | 25 (100) | 0.33 † |
| Steroid | - | 3 (12.5) | 1 (7.1) | 10 (43.5) | 0.01 † | - | 2 (22.2) | 1 (20.0) | 1.00 † | - | 6 (25.0) | 6 (23.1) | 13 (52.0) | 0.05 † |
| IMM | - | 5 (20.8) | 8 (57.1) | 5 (21.7) | 0.04 † | - | 0 | 4 (80) | 0.01 † | - | 7 (29.2) | 15 (57.7) | 7 (28.0) | 0.05 † |
| Anti-TNF | - | 4 (16.7) | 1 (7.1) | 0 | 0.10 † | - | 0 | 0 | - | - | 1 (4.2) | 1 (3.8) | 2 (8.0) | 0.84 † |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, Y.; Ahn, J.B.; Kim, D.H.; Park, I.S.; Son, M.; Kim, J.H.; Ma, H.W.; Kim, S.W.; Cheon, J.H. Integrated Analysis of Microbiome and Metabolome Reveals Disease-Specific Profiles in Inflammatory Bowel Diseases and Intestinal Behçet’s Disease. Int. J. Mol. Sci. 2024, 25, 6697. https://doi.org/10.3390/ijms25126697
Park Y, Ahn JB, Kim DH, Park IS, Son M, Kim JH, Ma HW, Kim SW, Cheon JH. Integrated Analysis of Microbiome and Metabolome Reveals Disease-Specific Profiles in Inflammatory Bowel Diseases and Intestinal Behçet’s Disease. International Journal of Molecular Sciences. 2024; 25(12):6697. https://doi.org/10.3390/ijms25126697
Chicago/Turabian StylePark, Yehyun, Jae Bum Ahn, Da Hye Kim, I Seul Park, Mijeong Son, Ji Hyung Kim, Hyun Woo Ma, Seung Won Kim, and Jae Hee Cheon. 2024. "Integrated Analysis of Microbiome and Metabolome Reveals Disease-Specific Profiles in Inflammatory Bowel Diseases and Intestinal Behçet’s Disease" International Journal of Molecular Sciences 25, no. 12: 6697. https://doi.org/10.3390/ijms25126697
APA StylePark, Y., Ahn, J. B., Kim, D. H., Park, I. S., Son, M., Kim, J. H., Ma, H. W., Kim, S. W., & Cheon, J. H. (2024). Integrated Analysis of Microbiome and Metabolome Reveals Disease-Specific Profiles in Inflammatory Bowel Diseases and Intestinal Behçet’s Disease. International Journal of Molecular Sciences, 25(12), 6697. https://doi.org/10.3390/ijms25126697