
New Insight into Neuropathic Pain: The Relationship between α7nAChR, Ferroptosis, and Neuroinflammation

Abstract
:1. Introduction
2. Neuroinflammation
2.1. Role of Microglia in Neuroinflammation
2.2. Neuroinflammation and Neuropathic Pain
3. α7nAChR
3.1. The Biochemical Features of α7nAChR
3.2. Target α7nAChR in the Treatment of Neuropathic Pain
4. Ferroptosis
4.1. Mechanisms of Ferroptosis
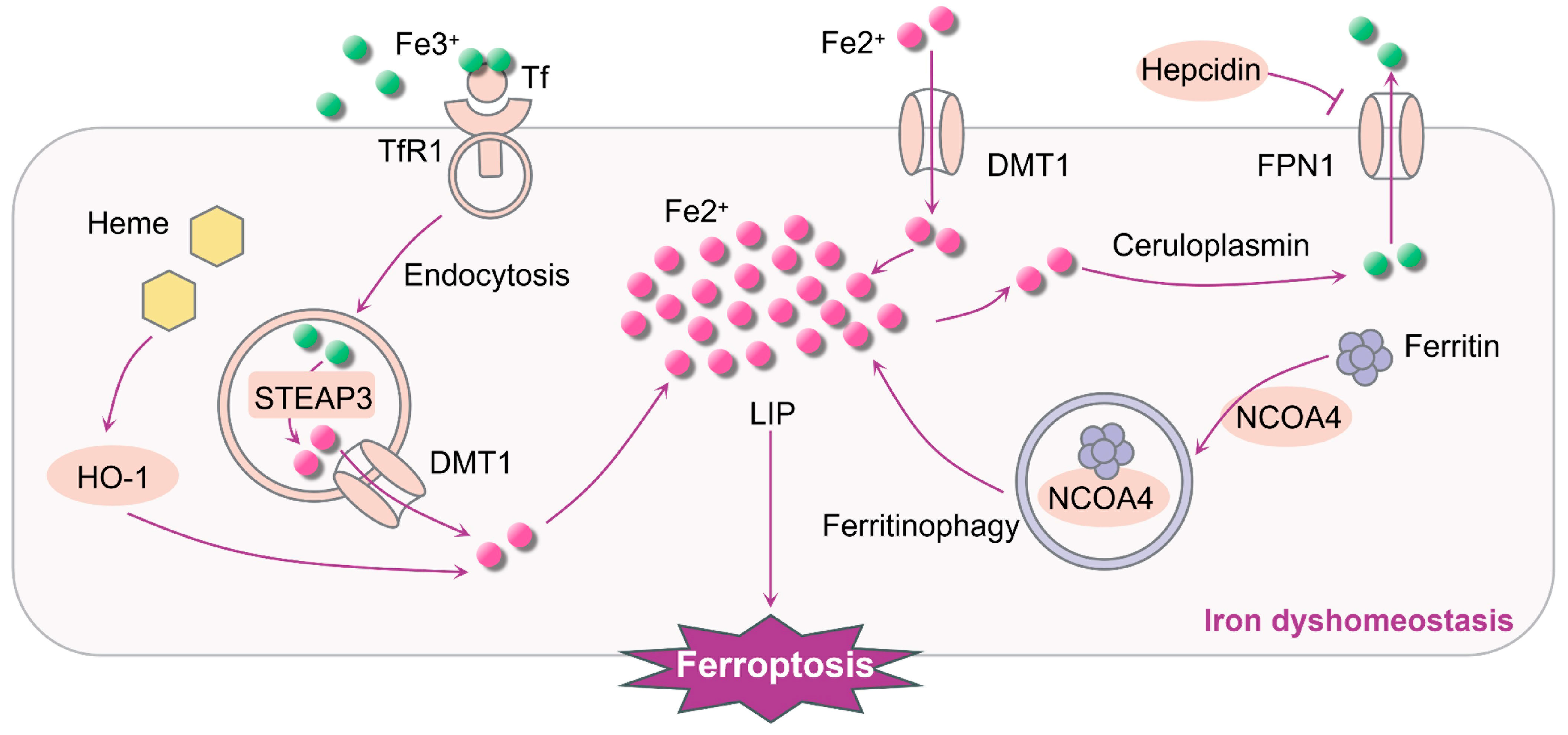
4.1.1. Iron Dyshomeostasis Contributes to Ferroptosis
4.1.2. System Xc-/GSH/GPX4 Pathway Regulates Ferroptosis
4.1.3. Lipid Peroxidation Induces Ferroptosis
4.2. Ferroptosis and Neuroinflammation

{kind=link}
{kind=link}
{kind=link}
| Targets | Effects on Ferroptosis | Effects on Neuroinflammation | References |
|---|---|---|---|
| LPS and Aβ | DMT1 and ferritin ↑ | Pro-inflammatory cytokines ↑ | [99] |
| IFN-γ | Microglia iron ↑ | TNF-α and iNOS mRNA expression ↑ | [100] |
| FAC and LPS | Microglia iron ↑ | IL-1β and TNF-α ↑ | [105] |
| NOX | Microglia iron ↑ | Superoxide and ROS ↑ | [106] |
| JNK pathway | DMT1 ↑, FPN1 ↓ | IL-6 ↑ | [109] |
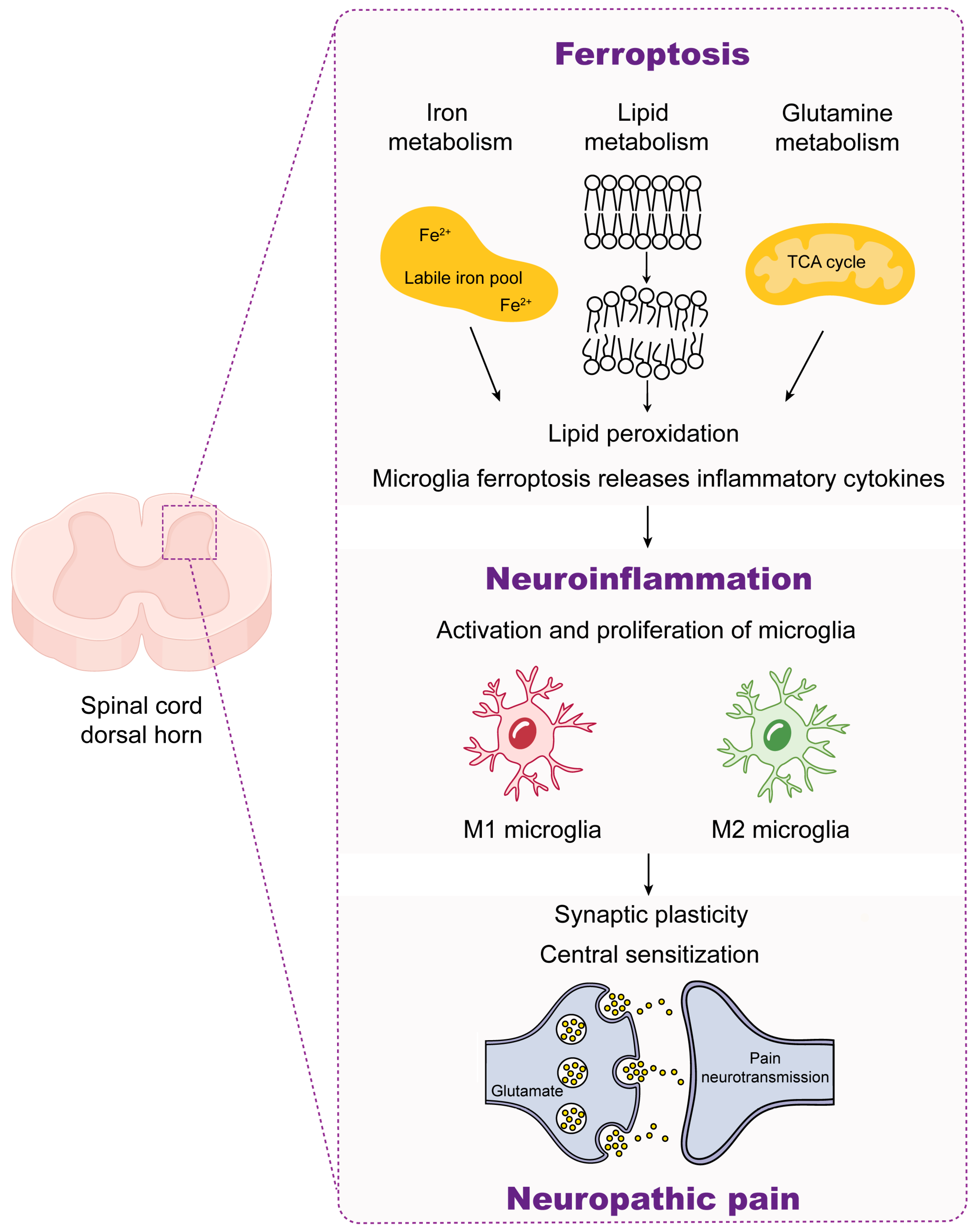
4.3. Ferroptosis and Neuropathic Pain
4.4. Ferroptosis and α7nAChR
5. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AA/AdA | Arachidonic acid |
| ACSL4 | Acyl-CoA synthetase long-chain family member 4 |
| ALOX | Arachidonate lipoxygenase |
| Aβ | β-amyloid |
| CAP | Cholinergic anti-inflammatory pathway |
| CCI | Chronic constriction injury |
| CFA | Complete Freund’s adjuvant |
| CNS | Central nervous system |
| DMT1 | Divalent metal transporter 1 |
| DRG | Dorsal root ganglion |
| EA | Electroacupuncture |
| FAC | Ferric ammonium citrate |
| FPN1 | Ferroportin-1 |
| GABAAR | γ-aminobutyric acid type A receptor |
| GlyR | Glycine receptor |
| GPX4 | Glutathione peroxidase 4 |
| GSH | Glutathione |
| IASP | International Association for the Study of Pain |
| IFN-γ | Interferon-γ |
| IL-10 | Interleukin-10 |
| IL-1β | Interleukin-1β |
| IL-6 | Interleukin-6 |
| iNOS | Inducible nitric oxide synthase |
| LIP | Labile intracellular iron pool |
| Lip-1 | Liproxstatin-1 |
| LPCAT3 | Lysophosphatidylcholine acyltransferase 3 |
| LPS | Lipopolysaccharide |
| MDA | Malondialdehyde |
| MFA | Methyl ferulic acid |
| nAChR | Nicotinic acetylcholine receptor |
| NF-κB | Nuclear factor-κB |
| NGF | Nerve growth factor |
| NOX | Non-phagocytic cell oxidase |
| Nrf2 | Nuclear factor E2-related factor 2 |
| PE | Phophatidyl ethanolamine |
| PG | Prostaglandin |
| PUFAs | Polyunsaturated fatty acids |
| RCD | Regulated cell death |
| ROS | Reactive oxygen species |
| RSL3 | Ras-selective-lethal compound 3 |
| SCP2-1 | Polysaccharides of Schisandra Chinensis Fructus |
| SNI | Spared nerve injury |
| SP | Sanfilippo syndrome |
| STEAP3 | Six-transmembrane epithelial antigen of the prostate 3 |
| TBI | Transferrin-binding iron |
| TfR1 | Transferrin receptor 1 |
| TGF-β | Transforming growth factor-β |
| TNF-α | Tumor necrosis factor-α |
| 5-HT3R | 5-hydroxytryptamine 3 receptor |
References
- Vlaeyen, J.W.S.; Crombez, G. Behavioral Conceptualization and Treatment of Chronic Pain. Annu. Rev. Clin. Psychol. 2020, 16, 187–212. [Google Scholar] [CrossRef] [PubMed]
- Theofilou, P.; Giannouli, V.; Kolias, S.; Tsolaki, M. Perception of Pain Self-Efficacy and Fatigue in Greek Patients with Multiple Sclerosis: A Study Protocol. Health Psychol. Res. 2015, 3, 1556. [Google Scholar] [CrossRef] [PubMed]
- Jensen, T.S.; Baron, R.; Haanpää, M.; Kalso, E.; Loeser, J.D.; Rice, A.S.C.; Treede, R.-D. A New Definition of Neuropathic Pain. Pain 2011, 152, 2204–2205. [Google Scholar] [CrossRef] [PubMed]
- Yam, M.F.; Loh, Y.C.; Tan, C.S.; Khadijah Adam, S.; Abdul Manan, N.; Basir, R. General Pathways of Pain Sensation and the Major Neurotransmitters Involved in Pain Regulation. Int. J. Mol. Sci. 2018, 19, 2164. [Google Scholar] [CrossRef] [PubMed]
- Nugraha, B.; Gutenbrunner, C.; Barke, A.; Karst, M.; Schiller, J.; Schäfer, P.; Falter, S.; Korwisi, B.; Rief, W.; Treede, R.-D.; et al. The IASP Classification of Chronic Pain for ICD-11: Functioning Properties of Chronic Pain. Pain 2019, 160, 88–94. [Google Scholar] [CrossRef]
- Colloca, L.; Ludman, T.; Bouhassira, D.; Baron, R.; Dickenson, A.H.; Yarnitsky, D.; Freeman, R.; Truini, A.; Attal, N.; Finnerup, N.B.; et al. Neuropathic Pain. Nat. Rev. Dis. Primers 2017, 3, 17002. [Google Scholar] [CrossRef]
- Fiore, N.T.; Debs, S.R.; Hayes, J.P.; Duffy, S.S.; Moalem-Taylor, G. Pain-Resolving Immune Mechanisms in Neuropathic Pain. Nat. Rev. Neurol. 2023, 19, 199–220. [Google Scholar] [CrossRef]
- Xu, Z.; Xie, W.; Feng, Y.; Wang, Y.; Li, X.; Liu, J.; Xiong, Y.; He, Y.; Chen, L.; Liu, G.; et al. Positive Interaction between GPER and β-Alanine in the Dorsal Root Ganglion Uncovers Potential Mechanisms: Mediating Continuous Neuronal Sensitization and Neuroinflammation Responses in Neuropathic Pain. J. Neuroinflammation 2022, 19, 164. [Google Scholar] [CrossRef]
- Salvemini, D.; Doyle, T.M. Targeting Neuroinflammation in Neuropathic Pain and Opioid Use. J. Exp. Med. 2023, 220, e20221244. [Google Scholar] [CrossRef]
- Di Cesare Mannelli, L.; Pacini, A.; Matera, C.; Zanardelli, M.; Mello, T.; De Amici, M.; Dallanoce, C.; Ghelardini, C. Involvement of A7 nAChR Subtype in Rat Oxaliplatin-Induced Neuropathy: Effects of Selective Activation. Neuropharmacology 2014, 79, 37–48. [Google Scholar] [CrossRef]
- Papke, R.L.; Lindstrom, J.M. Nicotinic Acetylcholine Receptors: Conventional and Unconventional Ligands and Signaling. Neuropharmacology 2020, 168, 108021. [Google Scholar] [CrossRef]
- Dineley, K.T.; Pandya, A.A.; Yakel, J.L. Nicotinic ACh Receptors as Therapeutic Targets in CNS Disorders. Trends Pharmacol. Sci. 2015, 36, 96–108. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Wang, X.; Li, S.; Yu, J.; Wang, W.; Du, Z.; Gao, S.; Ma, Y.; Tang, R.; Liu, T.; Ma, S.; et al. Saikosaponin B2 Ameliorates Depression-Induced Microglia Activation by Inhibiting Ferroptosis-Mediated Neuroinflammation and ER Stress. J. Ethnopharmacol. 2023, 316, 116729. [Google Scholar] [CrossRef]
- Xu, Y.; Jia, B.; Li, J.; Li, Q.; Luo, C. The Interplay between Ferroptosis and Neuroinflammation in Central Neurological Disorders. Antioxidants 2024, 13, 395. [Google Scholar] [CrossRef]
- Ou, M.; Jiang, Y.; Ji, Y.; Zhou, Q.; Du, Z.; Zhu, H.; Zhou, Z. Role and Mechanism of Ferroptosis in Neurological Diseases. Mol. Metab. 2022, 61, 101502. [Google Scholar] [CrossRef]
- Healy, S.; McMahon, J.; Owens, P.; FitzGerald, U. Significant Glial Alterations in Response to Iron Loading in a Novel Organotypic Hippocampal Slice Culture Model. Sci. Rep. 2016, 6, 36410. [Google Scholar] [CrossRef] [PubMed]
- Urrutia, P.; Aguirre, P.; Esparza, A.; Tapia, V.; Mena, N.P.; Arredondo, M.; González-Billault, C.; Núñez, M.T. Inflammation Alters the Expression of DMT1, FPN1 and Hepcidin, and It Causes Iron Accumulation in Central Nervous System Cells. J. Neurochem. 2013, 126, 541–549. [Google Scholar] [CrossRef]
- Sun, Y.; Chen, P.; Zhai, B.; Zhang, M.; Xiang, Y.; Fang, J.; Xu, S.; Gao, Y.; Chen, X.; Sui, X.; et al. The Emerging Role of Ferroptosis in Inflammation. Biomed. Pharmacother. 2020, 127, 110108. [Google Scholar] [CrossRef]
- Echeverria-Villalobos, M.; Tortorici, V.; Brito, B.E.; Ryskamp, D.; Uribe, A.; Weaver, T. The Role of Neuroinflammation in the Transition of Acute to Chronic Pain and the Opioid-Induced Hyperalgesia and Tolerance. Front. Pharmacol. 2023, 14, 1297931. [Google Scholar] [CrossRef]
- Li, L.; Li, T.; Qu, X.; Sun, G.; Fu, Q.; Han, G. Stress/Cell Death Pathways, Neuroinflammation, and Neuropathic Pain. Immunol. Rev. 2024, 321, 33–51. [Google Scholar] [CrossRef]
- Yang, Q.-Q.; Zhou, J.-W. Neuroinflammation in the Central Nervous System: Symphony of Glial Cells. Glia 2019, 67, 1017–1035. [Google Scholar] [CrossRef]
- Pajares, M.; I Rojo, A.; Manda, G.; Boscá, L.; Cuadrado, A. Inflammation in Parkinson’s Disease: Mechanisms and Therapeutic Implications. Cells 2020, 9, 1687. [Google Scholar] [CrossRef]
- Garaschuk, O.; Verkhratsky, A. Physiology of Microglia. Methods Mol. Biol. 2019, 2034, 27–40. [Google Scholar]
- Atta, A.A.; Ibrahim, W.W.; Mohamed, A.F.; Abdelkader, N.F. Microglia Polarization in Nociplastic Pain: Mechanisms and Perspectives. Inflammopharmacology 2023, 31, 1053–1067. [Google Scholar] [CrossRef]
- Borst, K.; Dumas, A.A.; Prinz, M. Microglia: Immune and Non-Immune Functions. Immunity 2021, 54, 2194–2208. [Google Scholar] [CrossRef]
- Thurgur, H.; Pinteaux, E. Microglia in the Neurovascular Unit: Blood-Brain Barrier-Microglia Interactions After Central Nervous System Disorders. Neuroscience 2019, 405, 55–67. [Google Scholar] [CrossRef]
- Zhang, L.-Q.; Gao, S.-J.; Sun, J.; Li, D.-Y.; Wu, J.-Y.; Song, F.-H.; Liu, D.-Q.; Zhou, Y.-Q.; Mei, W. DKK3 Ameliorates Neuropathic Pain via Inhibiting ASK-1/JNK/p-38-Mediated Microglia Polarization and Neuroinflammation. J. Neuroinflammation 2022, 19, 129. [Google Scholar] [CrossRef]
- Shao, J.; Liu, T.; Xie, Q.R.; Zhang, T.; Yu, H.; Wang, B.; Ying, W.; Mruk, D.D.; Silvestrini, B.; Cheng, C.Y.; et al. Adjudin Attenuates Lipopolysaccharide (LPS)- and Ischemia-Induced Microglial Activation. J. Neuroimmunol. 2013, 254, 83–90. [Google Scholar] [CrossRef]
- Long, Y.; Li, X.-Q.; Deng, J.; Ye, Q.-B.; Li, D.; Ma, Y.; Wu, Y.-Y.; Hu, Y.; He, X.-F.; Wen, J.; et al. Modulating the Polarization Phenotype of Microglia—A Valuable Strategy for Central Nervous System Diseases. Ageing Res. Rev. 2024, 93, 102160. [Google Scholar] [CrossRef]
- Yu, H.; Chang, Q.; Sun, T.; He, X.; Wen, L.; An, J.; Feng, J.; Zhao, Y. Metabolic Reprogramming and Polarization of Microglia in Parkinson’s Disease: Role of Inflammasome and Iron. Ageing Res. Rev. 2023, 90, 102032. [Google Scholar] [CrossRef] [PubMed]
- Meacham, K.; Shepherd, A.; Mohapatra, D.P.; Haroutounian, S. Neuropathic Pain: Central vs. Peripheral Mechanisms. Curr. Pain. Headache Rep. 2017, 21, 28. [Google Scholar] [CrossRef] [PubMed]
- Ho, I.H.T.; Chan, M.T.V.; Wu, W.K.K.; Liu, X. Spinal Microglia-Neuron Interactions in Chronic Pain. J. Leukoc. Biol. 2020, 108, 1575–1592. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Xu, S.; Qian, Y.; Xiao, Q. Resveratrol Regulates Microglia M1/M2 Polarization via PGC-1α in Conditions of Neuroinflammatory Injury. Brain Behav. Immun. 2017, 64, 162–172. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Wang, J.; Zhang, X.; Yan, T.; Wu, B.; Bi, K.; Jia, Y. Polysaccharide from Schisandra Chinensis Acts via LRP-1 to Reverse Microglia Activation through Suppression of the NF-κB and MAPK Signaling. J. Ethnopharmacol. 2020, 256, 112798. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Shi, H.; Zhang, D.; Jing, B.; Chen, Z.; Zheng, Y.; Chang, S.; Gao, L.; Zhao, G. Paeonol Alleviates Neuropathic Pain by Modulating Microglial M1 and M2 Polarization via the RhoA/p38MAPK Signaling Pathway. CNS Neurosci. Ther. 2023, 29, 2666–2679. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Zheng, J.; Xu, S.; Fang, Y.; Wu, Y.; Zeng, J.; Shao, A.; Shi, L.; Lu, J.; Mei, S.; et al. Mer Regulates Microglial/Macrophage M1/M2 Polarization and Alleviates Neuroinflammation Following Traumatic Brain Injury. J. Neuroinflammation 2021, 18, 2. [Google Scholar] [CrossRef] [PubMed]
- Chen, O.; Luo, X.; Ji, R.-R. Macrophages and Microglia in Inflammation and Neuroinflammation Underlying Different Pain States. Med. Rev. 2023, 3, 381–407. [Google Scholar] [CrossRef] [PubMed]
- Zipp, F.; Bittner, S.; Schafer, D.P. Cytokines as Emerging Regulators of Central Nervous System Synapses. Immunity 2023, 56, 914–925. [Google Scholar] [CrossRef]
- Ji, R.-R.; Chamessian, A.; Zhang, Y.-Q. Pain Regulation by Non-Neuronal Cells and Inflammation. Science 2016, 354, 572–577. [Google Scholar] [CrossRef]
- Liu, Z.; Miao, G.; Wang, J.; Yang, C.; Fu, Z.; Sun, T. Resolvin D1 Inhibits Mechanical Hypersensitivity in Sciatica by Modulating the Expression of Nuclear Factor-κB, Phospho-Extracellular Signal-Regulated Kinase, and Pro- and Antiinflammatory Cytokines in the Spinal Cord and Dorsal Root Ganglion. Anesthesiology 2016, 124, 934–944. [Google Scholar] [CrossRef] [PubMed]
- Holló, K.; Ducza, L.; Hegyi, Z.; Dócs, K.; Hegedűs, K.; Bakk, E.; Papp, I.; Kis, G.; Mészár, Z.; Bardóczi, Z.; et al. Interleukin-1 Receptor Type 1 Is Overexpressed in Neurons but Not in Glial Cells within the Rat Superficial Spinal Dorsal Horn in Complete Freund Adjuvant-Induced Inflammatory Pain. J. Neuroinflammation 2017, 14, 125. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.M.; Lee, S.H.; An, S.M.; Park, D.Y.; Lee, G.W.; Noh, G.-J. The Time-Course and RNA Interference of TNF-α, IL-6, and IL-1β Expression on Neuropathic Pain Induced by L5 Spinal Nerve Transection in Rats. Korean J. Anesthesiol. 2015, 68, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Castelli, V.; Palumbo, P.; d’Angelo, M.; Moorthy, N.K.; Antonosante, A.; Catanesi, M.; Lombardi, F.; Iannotta, D.; Cinque, B.; Benedetti, E.; et al. Probiotic DSF Counteracts Chemotherapy Induced Neuropathic Pain. Oncotarget 2018, 9, 27998–28008. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, K.; Martin, D.P.; Schmelzer, J.D.; Mitsui, Y.; Low, P.A. Pro- and Anti-Inflammatory Cytokine Gene Expression in Rat Sciatic Nerve Chronic Constriction Injury Model of Neuropathic Pain. Exp. Neurol. 2001, 169, 386–391. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, C.R.; Andriessen, A.S.; Chen, G.; Wang, K.; Jiang, C.; Maixner, W.; Ji, R.-R. Central Nervous System Targets: Glial Cell Mechanisms in Chronic Pain. Neurotherapeutics 2020, 17, 846–860. [Google Scholar] [CrossRef]
- Ji, R.-R.; Berta, T.; Nedergaard, M. Glia and Pain: Is Chronic Pain a Gliopathy? Pain 2013, 154 (Suppl. 1), S10–S28. [Google Scholar] [CrossRef] [PubMed]
- Rugnath, R.; Orzechowicz, C.; Newell, C.; Carullo, V.; Rugnath, A. A Literature Review: The Mechanisms and Treatment of Neuropathic Pain-A Brief Discussion. Biomedicines 2024, 12, 204. [Google Scholar] [CrossRef] [PubMed]
- Smith, P.A. Neuropathic Pain: What We Know and What We Should Do about It. Front. Pain Res. 2023, 4, 1220034. [Google Scholar] [CrossRef]
- Matta, J.A.; Gu, S.; Davini, W.B.; Bredt, D.S. Nicotinic Acetylcholine Receptor Redux: Discovery of Accessories Opens Therapeutic Vistas. Science 2021, 373, eabg6539. [Google Scholar] [CrossRef]
- Hone, A.J.; McIntosh, J.M. Nicotinic Acetylcholine Receptors: Therapeutic Targets for Novel Ligands to Treat Pain and Inflammation. Pharmacol. Res. 2023, 190, 106715. [Google Scholar] [CrossRef] [PubMed]
- Hone, A.J.; McIntosh, J.M. Nicotinic Acetylcholine Receptors in Neuropathic and Inflammatory Pain. FEBS Lett. 2018, 592, 1045–1062. [Google Scholar] [CrossRef]
- Stokes, C.; Treinin, M.; Papke, R.L. Looking below the Surface of Nicotinic Acetylcholine Receptors. Trends Pharmacol. Sci. 2015, 36, 514–523. [Google Scholar] [CrossRef] [PubMed]
- Bagdas, D.; Gurun, M.S.; Flood, P.; Papke, R.L.; Damaj, M.I. New Insights on Neuronal Nicotinic Acetylcholine Receptors as Targets for Pain and Inflammation: A Focus on A7 nAChRs. Curr. Neuropharmacol. 2018, 16, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Lindstrom, J. Orthosteric and Allosteric Potentiation of Heteromeric Neuronal Nicotinic Acetylcholine Receptors. Br. J. Pharmacol. 2018, 175, 1805–1821. [Google Scholar] [CrossRef] [PubMed]
- Hoover, D.B. Cholinergic Modulation of the Immune System Presents New Approaches for Treating Inflammation. Pharmacol. Ther. 2017, 179, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Xiao, T.; Sun, Q.; Wang, K. The Current Agonists and Positive Allosteric Modulators of A7 nAChR for CNS Indications in Clinical Trials. Acta Pharm. Sin. B 2017, 7, 611–622. [Google Scholar] [CrossRef] [PubMed]
- Baranowska, U.; Wiśniewska, R.J. The A7-nACh Nicotinic Receptor and Its Role in Memory and Selected Diseases of the Central Nervous System. Postepy Hig. Med. Dosw. 2017, 71, 633–648. [Google Scholar] [CrossRef] [PubMed]
- Camacho-Hernandez, G.A.; Stokes, C.; Duggan, B.M.; Kaczanowska, K.; Brandao-Araiza, S.; Doan, L.; Papke, R.L.; Taylor, P. Synthesis, Pharmacological Characterization, and Structure-Activity Relationships of Noncanonical Selective Agonists for A7 nAChRs. J. Med. Chem. 2019, 62, 10376–10390. [Google Scholar] [CrossRef]
- Albuquerque, E.X.; Pereira, E.F.R.; Alkondon, M.; Rogers, S.W. Mammalian Nicotinic Acetylcholine Receptors: From Structure to Function. Physiol. Rev. 2009, 89, 73–120. [Google Scholar] [CrossRef]
- Cheng, Q.; Yakel, J.L. The Effect of A7 Nicotinic Receptor Activation on Glutamatergic Transmission in the Hippocampus. Biochem. Pharmacol. 2015, 97, 439–444. [Google Scholar] [CrossRef] [PubMed]
- Morioka, N.; Hisaoka-Nakashima, K.; Nakata, Y. Regulation by Nicotinic Acetylcholine Receptors of Microglial Glutamate Transporters: Role of Microglia in Neuroprotection. In Nicotinic Acetylcholine Receptor Signaling in Neuroprotection; Akaike, A., Shimohama, S., Misu, Y., Eds.; Springer: Singapore, 2018; ISBN 978-981-10-8487-4. [Google Scholar]
- Wang, H.; Yu, M.; Ochani, M.; Amella, C.A.; Tanovic, M.; Susarla, S.; Li, J.H.; Wang, H.; Yang, H.; Ulloa, L.; et al. Nicotinic Acetylcholine Receptor Alpha7 Subunit Is an Essential Regulator of Inflammation. Nature 2003, 421, 384–388. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Zhang, X.; Shi, P.; Yuan, J.; Jia, Q.; Pi, C.; Chen, T.; Xiong, L.; Chen, J.; Tang, J.; et al. A7 Nicotinic Acetylcholine Receptor: A Key Receptor in the Cholinergic Anti-Inflammatory Pathway Exerting an Antidepressant Effect. J. Neuroinflamm. 2023, 20, 84. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Hao, H.; Gao, Y.; Wang, Z.; Lu, W.; Liu, J. Expression and Localization Analyses of the Cholinergic Anti-Inflammatory Pathway and α7nAchR in Different Tissues of Rats with Rheumatoid Arthritis. Acta Histochem. 2019, 121, 742–749. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhang, Y.; Wang, J.; Li, S.; Wang, Y.; Zhang, Z.; Zhang, J.; Xin, C.; Wang, Y.; Rong, P. Anti-Neuroinflammation Effects of Transcutaneous Auricular Vagus Nerve Stimulation against Depression-like Behaviors via Hypothalamic α7nAchR/JAK2/STAT3/NF-κB Pathway in Rats Exposed to Chronic Unpredictable Mild Stress. CNS Neurosci. Ther. 2023, 29, 2634–2644. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Lu, Y.; Bian, H.; Guo, L.; Zhu, H. Activation of the A7 Nicotinic Receptor Promotes Lipopolysaccharide-Induced Conversion of M1 Microglia to M2. Am. J. Transl. Res. 2017, 9, 971–985. [Google Scholar] [PubMed]
- Jia, D.; Liu, G.; Sun, Y.; Hu, Z.; Huang, Z.; Huang, C. Trifluoro-Icaritin Ameliorates Spared Nerve Injury-Induced Neuropathic Pain by Inhibiting Microglial Activation through α7nAChR-Mediated Blockade of BDNF/TrkB/KCC2 Signaling in the Spinal Cord of Rats. Biomed. Pharmacother. 2023, 157, 114001. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Jiang, Q.; Xia, Y.; Huang, Z.; Huang, C. Involvement of α7nAChR in Electroacupuncture Relieving Neuropathic Pain in the Spinal Cord of Rat with Spared Nerve Injury. Brain Res. Bull. 2018, 137, 257–264. [Google Scholar] [CrossRef]
- Salvador, G.A. Iron in Neuronal Function and Dysfunction. Biofactors 2010, 36, 103–110. [Google Scholar] [CrossRef]
- Yan, H.-F.; Zou, T.; Tuo, Q.-Z.; Xu, S.; Li, H.; Belaidi, A.A.; Lei, P. Ferroptosis: Mechanisms and Links with Diseases. Signal Transduct. Target. Ther. 2021, 6, 49. [Google Scholar] [CrossRef]
- Sun, S.; Shen, J.; Jiang, J.; Wang, F.; Min, J. Targeting Ferroptosis Opens New Avenues for the Development of Novel Therapeutics. Signal Transduct. Target. Ther. 2023, 8, 372. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Lai, Y.; Hua, Z.-C. Apoptosis and Apoptotic Body: Disease Message and Therapeutic Target Potentials. Biosci. Rep. 2019, 39, BSR20180992. [Google Scholar] [CrossRef] [PubMed]
- Samson, A.L.; Zhang, Y.; Geoghegan, N.D.; Gavin, X.J.; Davies, K.A.; Mlodzianoski, M.J.; Whitehead, L.W.; Frank, D.; Garnish, S.E.; Fitzgibbon, C.; et al. MLKL Trafficking and Accumulation at the Plasma Membrane Control the Kinetics and Threshold for Necroptosis. Nat. Commun. 2020, 11, 3151. [Google Scholar] [CrossRef]
- Yang, Y.; Klionsky, D.J. Autophagy and Disease: Unanswered Questions. Cell Death Differ. 2020, 27, 858–871. [Google Scholar] [CrossRef]
- Demarco, B.; Chen, K.W.; Broz, P. Cross Talk between Intracellular Pathogens and Cell Death. Immunol. Rev. 2020, 297, 174–193. [Google Scholar] [CrossRef]
- Mizushima, N.; Levine, B. Autophagy in Human Diseases. N. Engl. J. Med. 2020, 383, 1564–1576. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.-L.; Ghosh, M.C.; Rouault, T.A. The Physiological Functions of Iron Regulatory Proteins in Iron Homeostasis—An Update. Front. Pharmacol. 2014, 5, 124. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Xu, Y.; Xu, H.; Jin, C.; Zhang, H.; Su, H.; Li, Y.; Zhou, K.; Ni, W. Progress in Understanding Ferroptosis and Its Targeting for Therapeutic Benefits in Traumatic Brain and Spinal Cord Injuries. Front. Cell Dev. Biol. 2021, 9, 705786. [Google Scholar] [CrossRef]
- Fang, X.; Ardehali, H.; Min, J.; Wang, F. The Molecular and Metabolic Landscape of Iron and Ferroptosis in Cardiovascular Disease. Nat. Rev. Cardiol. 2023, 20, 7–23. [Google Scholar] [CrossRef]
- Ji, C.; Kosman, D.J. Molecular Mechanisms of Non-Transferrin-Bound and Transferring-Bound Iron Uptake in Primary Hippocampal Neurons. J. Neurochem. 2015, 133, 668–683. [Google Scholar] [CrossRef]
- Gao, J.; Wang, Q.; Tang, Y.-D.; Zhai, J.; Hu, W.; Zheng, C. When Ferroptosis Meets Pathogenic Infections. Trends Microbiol. 2023, 31, 468–479. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Tao, Y.; Zhang, Z.; Guo, X.; An, P.; Shen, Y.; Wu, Q.; Yu, Y.; Wang, F. Metalloreductase Steap3 Coordinates the Regulation of Iron Homeostasis and Inflammatory Responses. Haematologica 2012, 97, 1826–1835. [Google Scholar] [CrossRef] [PubMed]
- Lane, D.J.R.; Merlot, A.M.; Huang, M.L.-H.; Bae, D.-H.; Jansson, P.J.; Sahni, S.; Kalinowski, D.S.; Richardson, D.R. Cellular Iron Uptake, Trafficking and Metabolism: Key Molecules and Mechanisms and Their Roles in Disease. Biochim. Biophys. Acta 2015, 1853, 1130–1144. [Google Scholar] [CrossRef] [PubMed]
- Camaschella, C.; Nai, A.; Silvestri, L. Iron Metabolism and Iron Disorders Revisited in the Hepcidin Era. Haematologica 2020, 105, 260–272. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Li, X.; Ge, C.; Min, J.; Wang, F. The Multifaceted Role of Ferroptosis in Liver Disease. Cell Death Differ. 2022, 29, 467–480. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Stockwell, B.R.; Conrad, M. Ferroptosis: Mechanisms, Biology and Role in Disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 266–282. [Google Scholar] [CrossRef] [PubMed]
- Yan, R.; Xie, E.; Li, Y.; Li, J.; Zhang, Y.; Chi, X.; Hu, X.; Xu, L.; Hou, T.; Stockwell, B.R.; et al. The Structure of Erastin-Bound xCT-4F2hc Complex Reveals Molecular Mechanisms Underlying Erastin-Induced Ferroptosis. Cell Res. 2022, 32, 687–690. [Google Scholar] [CrossRef]
- Cheng, Y.; Song, Y.; Chen, H.; Li, Q.; Gao, Y.; Lu, G.; Luo, C. Ferroptosis Mediated by Lipid Reactive Oxygen Species: A Possible Causal Link of Neuroinflammation to Neurological Disorders. Oxid. Med. Cell Longev. 2021, 2021, 5005136. [Google Scholar] [CrossRef]
- Su, Y.; Zhao, B.; Zhou, L.; Zhang, Z.; Shen, Y.; Lv, H.; AlQudsy, L.H.H.; Shang, P. Ferroptosis, a Novel Pharmacological Mechanism of Anti-Cancer Drugs. Cancer Lett. 2020, 483, 127–136. [Google Scholar] [CrossRef]
- Yu, Y.; Yan, Y.; Niu, F.; Wang, Y.; Chen, X.; Su, G.; Liu, Y.; Zhao, X.; Qian, L.; Liu, P.; et al. Ferroptosis: A Cell Death Connecting Oxidative Stress, Inflammation and Cardiovascular Diseases. Cell Death Discov. 2021, 7, 193. [Google Scholar] [CrossRef]
- Hassannia, B.; Van Coillie, S.; Vanden Berghe, T. Ferroptosis: Biological Rust of Lipid Membranes. Antioxid. Redox Signal 2021, 35, 487–509. [Google Scholar] [CrossRef] [PubMed]
- Liang, D.; Minikes, A.M.; Jiang, X. Ferroptosis at the Intersection of Lipid Metabolism and Cellular Signaling. Mol. Cell 2022, 82, 2215–2227. [Google Scholar] [CrossRef] [PubMed]
- Doll, S.; Conrad, M. Iron and Ferroptosis: A Still Ill-Defined Liaison. IUBMB Life 2017, 69, 423–434. [Google Scholar] [CrossRef] [PubMed]
- Kagan, V.E.; Mao, G.; Qu, F.; Angeli, J.P.F.; Doll, S.; Croix, C.S.; Dar, H.H.; Liu, B.; Tyurin, V.A.; Ritov, V.B.; et al. Oxidized Arachidonic and Adrenic PEs Navigate Cells to Ferroptosis. Nat. Chem. Biol. 2017, 13, 81–90. [Google Scholar] [CrossRef]
- Doll, S.; Proneth, B.; Tyurina, Y.Y.; Panzilius, E.; Kobayashi, S.; Ingold, I.; Irmler, M.; Beckers, J.; Aichler, M.; Walch, A.; et al. ACSL4 Dictates Ferroptosis Sensitivity by Shaping Cellular Lipid Composition. Nat. Chem. Biol. 2017, 13, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Doll, S.; Freitas, F.P.; Shah, R.; Aldrovandi, M.; da Silva, M.C.; Ingold, I.; Goya Grocin, A.; Xavier da Silva, T.N.; Panzilius, E.; Scheel, C.H.; et al. FSP1 Is a Glutathione-Independent Ferroptosis Suppressor. Nature 2019, 575, 693–698. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Hyun, D.-H. The Interplay between Intracellular Iron Homeostasis and Neuroinflammation in Neurodegenerative Diseases. Antioxidants 2023, 12, 918. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, R.C.; Sosa, J.C.; Gardeck, A.M.; Baez, A.S.; Lee, C.-H.; Wessling-Resnick, M. Inflammation-Induced Iron Transport and Metabolism by Brain Microglia. J. Biol. Chem. 2018, 293, 7853–7863. [Google Scholar] [CrossRef] [PubMed]
- Holland, R.; McIntosh, A.L.; Finucane, O.M.; Mela, V.; Rubio-Araiz, A.; Timmons, G.; McCarthy, S.A.; Gun’ko, Y.K.; Lynch, M.A. Inflammatory Microglia Are Glycolytic and Iron Retentive and Typify the Microglia in APP/PS1 Mice. Brain Behav. Immun. 2018, 68, 183–196. [Google Scholar] [CrossRef]
- Rathore, K.I.; Redensek, A.; David, S. Iron Homeostasis in Astrocytes and Microglia Is Differentially Regulated by TNF-α and TGF-Β1. Glia 2012, 60, 738–750. [Google Scholar] [CrossRef]
- Arfi, A.; Richard, M.; Gandolphe, C.; Bonnefont-Rousselot, D.; Thérond, P.; Scherman, D. Neuroinflammatory and Oxidative Stress Phenomena in MPS IIIA Mouse Model: The Positive Effect of Long-Term Aspirin Treatment. Mol. Genet. Metab. 2011, 103, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Ausseil, J.; Desmaris, N.; Bigou, S.; Attali, R.; Corbineau, S.; Vitry, S.; Parent, M.; Cheillan, D.; Fuller, M.; Maire, I.; et al. Early Neurodegeneration Progresses Independently of Microglial Activation by Heparan Sulfate in the Brain of Mucopolysaccharidosis IIIB Mice. PLoS ONE 2008, 3, e2296. [Google Scholar] [CrossRef] [PubMed]
- Puy, V.; Darwiche, W.; Trudel, S.; Gomila, C.; Lony, C.; Puy, L.; Lefebvre, T.; Vitry, S.; Boullier, A.; Karim, Z.; et al. Predominant Role of Microglia in Brain Iron Retention in Sanfilippo Syndrome, a Pediatric Neurodegenerative Disease. Glia 2018, 66, 1709–1723. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Song, N.; Jiang, H.; Wang, J.; Xie, J. Pro-Inflammatory Cytokines Modulate Iron Regulatory Protein 1 Expression and Iron Transportation through Reactive Oxygen/Nitrogen Species Production in Ventral Mesencephalic Neurons. Biochim. Biophys. Acta 2013, 1832, 618–625. [Google Scholar] [CrossRef]
- Yauger, Y.J.; Bermudez, S.; Moritz, K.E.; Glaser, E.; Stoica, B.; Byrnes, K.R. Iron Accentuated Reactive Oxygen Species Release by NADPH Oxidase in Activated Microglia Contributes to Oxidative Stress in Vitro. J. Neuroinflammation 2019, 16, 41. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Yan, Z.; Gao, J.; Sun, L.; Huang, X.; Liu, Z.; Yu, S.; Cao, C.-J.; Zuo, L.; Chen, Z.-J.; et al. Role and Mechanism of Microglial Activation in Iron-Induced Selective and Progressive Dopaminergic Neurodegeneration. Mol. Neurobiol. 2014, 49, 1153–1165. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Li, J.; Kang, R.; Klionsky, D.J.; Tang, D. Ferroptosis: Machinery and Regulation. Autophagy 2021, 17, 2054–2081. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Du, X.; Xie, J.; Wang, J. Interleukin-6 Regulates Iron-Related Proteins through c-Jun N-Terminal Kinase Activation in BV2 Microglial Cell Lines. PLoS ONE 2017, 12, e0180464. [Google Scholar] [CrossRef]
- Merighi, S.; Nigro, M.; Travagli, A.; Gessi, S. Microglia and Alzheimer’s Disease. Int. J. Mol. Sci. 2022, 23, 12990. [Google Scholar] [CrossRef]
- Yang, Y.; Wang, Y.; Guo, L.; Gao, W.; Tang, T.-L.; Yan, M. Interaction between Macrophages and Ferroptosis. Cell Death Dis. 2022, 13, 355. [Google Scholar] [CrossRef]
- Wu, T.; Wang, X.; Cheng, J.; Liang, X.; Li, Y.; Chen, M.; Kong, L.; Tang, M. Nitrogen-Doped Graphene Quantum Dots Induce Ferroptosis through Disrupting Calcium Homeostasis in Microglia. Part. Fibre Toxicol. 2022, 19, 22. [Google Scholar] [CrossRef] [PubMed]
- Kroner, A.; Greenhalgh, A.D.; Zarruk, J.G.; Passos Dos Santos, R.; Gaestel, M.; David, S. TNF and Increased Intracellular Iron Alter Macrophage Polarization to a Detrimental M1 Phenotype in the Injured Spinal Cord. Neuron 2014, 83, 1098–1116. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Mendívil, C.; Luengo, E.; Trigo-Alonso, P.; García-Magro, N.; Negredo, P.; López, M.G. Protective Role of Microglial HO-1 Blockade in Aging: Implication of Iron Metabolism. Redox Biol. 2021, 38, 101789. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Zhang, Y.; Zhao, X.; Shao, L.; Liu, G.; Sun, C.; Xu, R.; Zhang, Z. ACSL4 Exacerbates Ischemic Stroke by Promoting Ferroptosis-Induced Brain Injury and Neuroinflammation. Brain Behav. Immun. 2021, 93, 312–321. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Zhang, Z.; Zhou, X.; Zhao, Z.; Zhao, R.; Xu, X.; Kong, X.; Ren, J.; Yao, X.; Wen, Q.; et al. Microglia and Macrophage Exhibit Attenuated Inflammatory Response and Ferroptosis Resistance after RSL3 Stimulation via Increasing Nrf2 Expression. J. Neuroinflammation 2021, 18, 249. [Google Scholar] [CrossRef]
- Li, H.; Shen, Y.; Xiao, H.; Sun, W. Resveratrol Attenuates Rotenone-Induced Inflammation and Oxidative Stress via STAT1 and Nrf2/Keap1/SLC7A11 Pathway in a Microglia Cell Line. Pathol. Res. Pract. 2021, 225, 153576. [Google Scholar] [PubMed]
- Jia, S.; Chen, G.; Liang, Y.; Liang, X.; Meng, C. GCH1-Regulated miRNAs Are Potential Targets for Microglial Activation in Neuropathic Pain. Biosci. Rep. 2021, 41, BSR20210051. [Google Scholar] [CrossRef] [PubMed]
- Xue, C.; Kui, W.; Huang, A.; Li, Y.; Li, L.; Gu, Z.; Xie, L.; Kong, S.; Yu, J.; Ruan, H.; et al. Electroacupuncture Suppresses Neuronal Ferroptosis to Relieve Chronic Neuropathic Pain. J. Cell Mol. Med. 2024, 28, e18240. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Huo, X.; Han, C.; Ning, J.; Chen, H.; Li, B.; Liu, J.; Ma, W.; Li, Q.; Yu, Y.; et al. Ferroptosis Is Involved in the Development of Neuropathic Pain and Allodynia. Mol. Cell Biochem. 2021, 476, 3149–3161. [Google Scholar] [CrossRef]
- Yang, R.; Shi, L.; Si, H.; Hu, Z.; Zou, L.; Li, L.; Xu, X.; Schmalzing, G.; Nie, H.; Li, G.; et al. Gallic Acid Improves Comorbid Chronic Pain and Depression Behaviors by Inhibiting P2X7 Receptor-Mediated Ferroptosis in the Spinal Cord of Rats. ACS Chem. Neurosci. 2023, 14, 667–676. [Google Scholar] [CrossRef]
- Liu, T.; Wang, R.; Qi, W.; Jia, L.; Ma, K.; Si, J.; Yin, J.; Zhao, Y.; Dai, Z.; Yin, J. Methyl Ferulic Acid Alleviates Neuropathic Pain by Inhibiting Nox4-Induced Ferroptosis in Dorsal Root Ganglia Neurons in Rats. Mol. Neurobiol. 2023, 60, 3175–3189. [Google Scholar] [CrossRef]
- Wan, K.; Jia, M.; Zhang, H.; Lan, Y.; Wang, S.; Zhang, K.; Wang, Z.; Zhu, H.; Zheng, X.; Luo, Y.; et al. Electroacupuncture Alleviates Neuropathic Pain by Suppressing Ferroptosis in Dorsal Root Ganglion via SAT1/ALOX15 Signaling. Mol. Neurobiol. 2023, 60, 6121–6132. [Google Scholar] [CrossRef]
- Chen, X.; Wang, J.; He, Z.; Liu, X.; Liu, H.; Wang, X. Analgesic and Anxiolytic Effects of Gastrodin and Its Influences on Ferroptosis and Jejunal Microbiota in Complete Freund’s Adjuvant-Injected Mice. Front. Microbiol. 2022, 13, 841662. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.-F.; Xiang, P.; Du, J.-Y.; Liang, J.-F.; Li, X. Intrathecal Liproxstatin-1 Delivery Inhibits Ferroptosis and Attenuates Mechanical and Thermal Hypersensitivities in Rats with Complete Freund’s Adjuvant-Induced Inflammatory Pain. Neural Regen. Res. 2023, 18, 456–462. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Song, T.; Zhao, M.; Tao, X.; Zhang, B.; Sun, C.; Wang, P.; Wang, K.; Zhao, L. Sirtuin 2 Alleviates Chronic Neuropathic Pain by Suppressing Ferroptosis in Rats. Front. Pharmacol. 2022, 13, 827016. [Google Scholar] [CrossRef]
- Zhang, Y.; Zheng, L.; Deng, H.; Feng, D.; Hu, S.; Zhu, L.; Xu, W.; Zhou, W.; Wang, Y.; Min, K.; et al. Electroacupuncture Alleviates LPS-Induced ARDS Through A7 Nicotinic Acetylcholine Receptor-Mediated Inhibition of Ferroptosis. Front. Immunol. 2022, 13, 832432. [Google Scholar] [CrossRef]
- Billesbølle, C.B.; Azumaya, C.M.; Kretsch, R.C.; Powers, A.S.; Gonen, S.; Schneider, S.; Arvedson, T.; Dror, R.O.; Cheng, Y.; Manglik, A. Structure of Hepcidin-Bound Ferroportin Reveals Iron Homeostatic Mechanisms. Nature 2020, 586, 807–811. [Google Scholar] [CrossRef] [PubMed]
- Cortes, M.; Cao, M.; Liu, H.L.; Moore, C.S.; Durosier, L.D.; Burns, P.; Fecteau, G.; Desrochers, A.; Barreiro, L.B.; Antel, J.P.; et al. A7 Nicotinic Acetylcholine Receptor Signaling Modulates the Inflammatory Phenotype of Fetal Brain Microglia: First Evidence of Interference by Iron Homeostasis. Sci. Rep. 2017, 7, 10645. [Google Scholar] [CrossRef]
- Zhao, C.; Chen, J.; Liu, Z.; Liang, H.; Chen, X.; Cheng, L.; Xie, S.; Lin, Z.; Wu, R.; Zhao, Q.; et al. Activation of Nicotinic Acetylcholine Receptor A7 Subunit Limits Zika Viral Infection via Promoting Autophagy and Ferroptosis. Mol. Ther. 2024, 32, S1525001624003393. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luo, F.; Huang, C. New Insight into Neuropathic Pain: The Relationship between α7nAChR, Ferroptosis, and Neuroinflammation. Int. J. Mol. Sci. 2024, 25, 6716. https://doi.org/10.3390/ijms25126716
Luo F, Huang C. New Insight into Neuropathic Pain: The Relationship between α7nAChR, Ferroptosis, and Neuroinflammation. International Journal of Molecular Sciences. 2024; 25(12):6716. https://doi.org/10.3390/ijms25126716
Chicago/Turabian StyleLuo, Fangting, and Cheng Huang. 2024. "New Insight into Neuropathic Pain: The Relationship between α7nAChR, Ferroptosis, and Neuroinflammation" International Journal of Molecular Sciences 25, no. 12: 6716. https://doi.org/10.3390/ijms25126716
APA StyleLuo, F., & Huang, C. (2024). New Insight into Neuropathic Pain: The Relationship between α7nAChR, Ferroptosis, and Neuroinflammation. International Journal of Molecular Sciences, 25(12), 6716. https://doi.org/10.3390/ijms25126716