
Bcl-2 Up-Regulation Mediates Taxane Resistance Downstream of APC Loss

 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
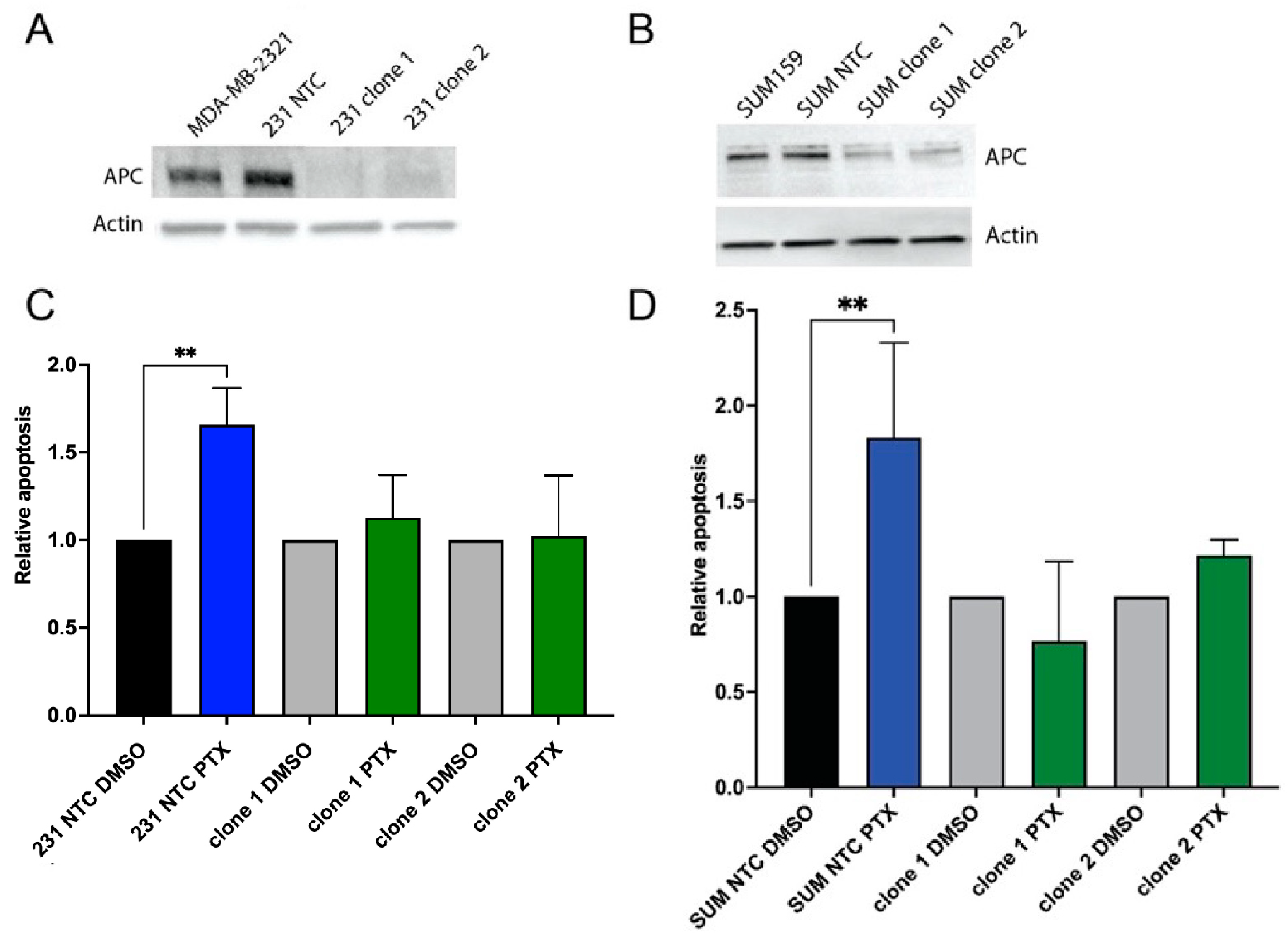
2.1. Loss of APC Leads to PTX Resistance
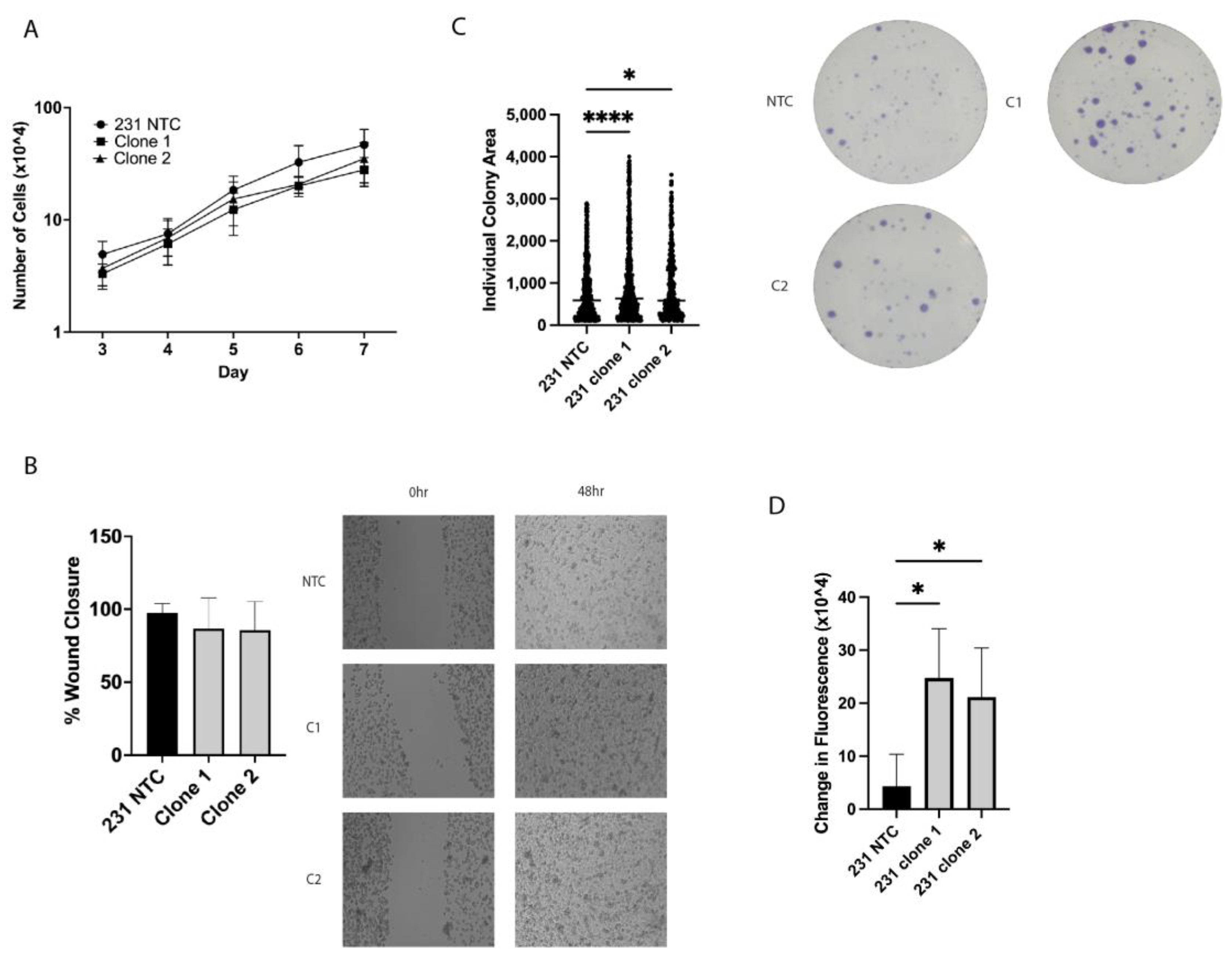
2.2. In Vitro Tumorigenic Assays Show Alterations in Clonogenic Growth and ALDH+ Cell Population
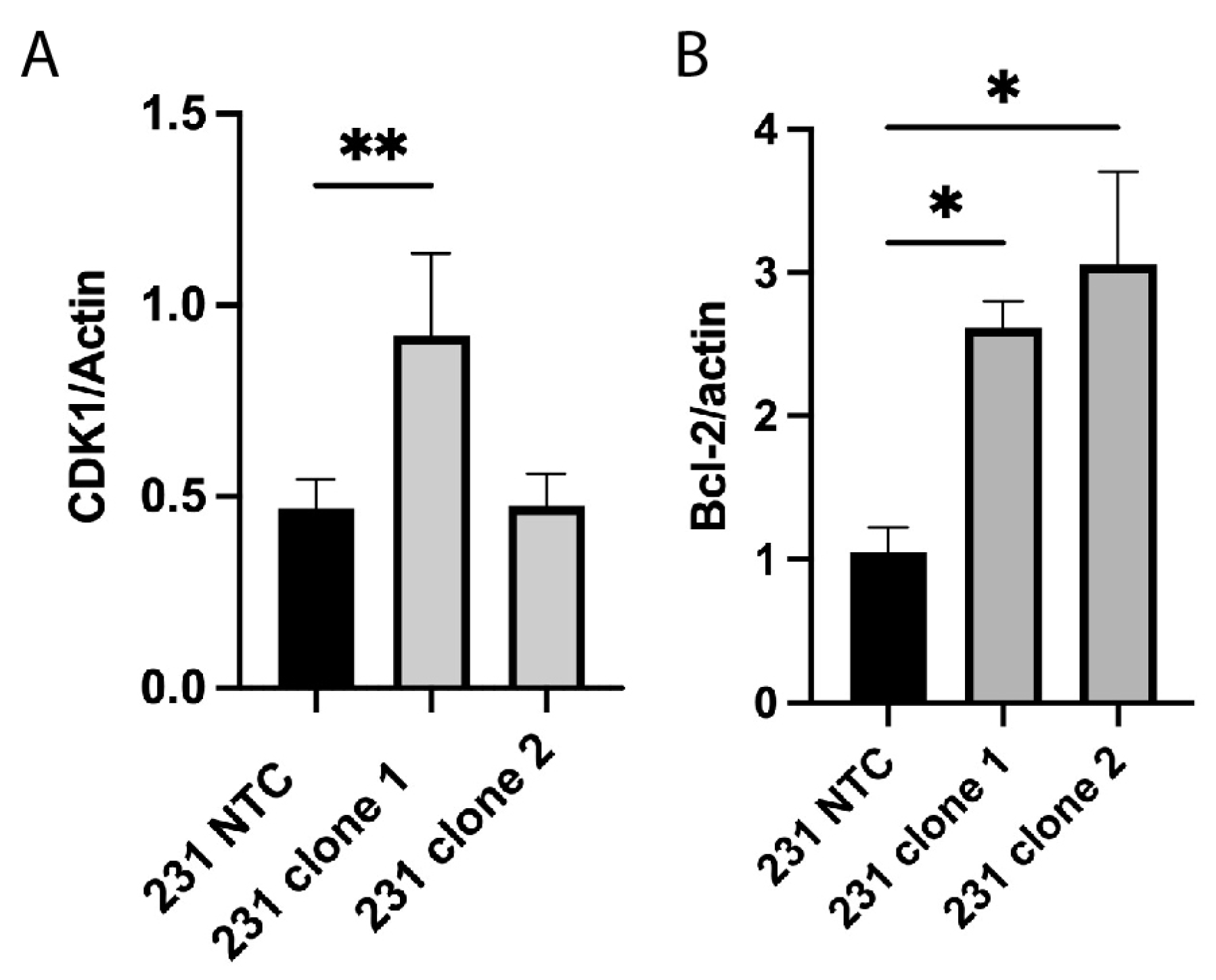
2.3. Alterations in Cell Cycle and Apoptosis Proteins
2.4. Blocking Bcl-2 Function Enhances Response to PTX in Cells Lacking APC Expression
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. CRISPR-Mediated Knockout
4.3. Protein Isolation and Western Blot Analysis
4.4. Clonogenic Assay
4.5. Migration Assay
4.6. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cheang, M.C.; Voduc, D.; Bajdik, C.; Leung, S.; McKinney, S.; Chia, S.K.; Perou, C.M.; Nielsen, T.O. Basal-like breast cancer defined by five biomarkers has superior prognostic value than triple-negative phenotype. Clin. Cancer Res. 2008, 14, 1368–1376. [Google Scholar] [CrossRef]
- Leidy, J.; Khan, A.; Kandil, D. Basal-like breast cancer: Update on clinicopathologic, immunohistochemical, and molecular features. Arch. Pathol. Lab. Med. 2014, 138, 37–43. [Google Scholar] [CrossRef]
- Brabender, J.; Usadel, H.; Danenberg, K.D.; Metzger, R.; Schneider, P.M.; Lord, R.V.; Wickramasinghe, K.; Lum, C.E.; Park, J.; Salonga, D.; et al. Adenomatous polyposis coli gene promoter hypermethylation in non-small cell lung cancer is associated with survival. Oncogene 2001, 20, 3528–3532. [Google Scholar] [CrossRef]
- Arnold, C.N.; Goel, A.; Niedzwiecki, D.; Dowell, J.M.; Wasserman, L.; Compton, C.; Mayer, R.J.; Bertagnolli, M.M.; Boland, C.R. APC promoter hypermethylation contributes to the loss of APC expression in colorectal cancers with allelic loss on 5q. Cancer Biol. Ther. 2004, 3, 960–964. [Google Scholar] [CrossRef]
- Esteller, M.; Sparks, A.; Toyota, M.; Sanchez-Cespedes, M.; Capella, G.; Peinado, M.A.; Gonzalez, S.; Tarafa, G.; Sidransky, D.; Meltzer, S.J.; et al. Analysis of adenomatous polyposis coli promoter hypermethylation in human cancer. Cancer Res. 2000, 60, 4366–4371. [Google Scholar]
- Jin, Z.; Tamura, G.; Tsuchiya, T.; Sakata, K.; Kashiwaba, M.; Osakabe, M.; Motoyama, T. Adenomatous polyposis coli (APC) gene promoter hypermethylation in primary breast cancers. Br. J. Cancer 2001, 85, 69–73. [Google Scholar] [CrossRef]
- Virmani, A.K.; Rathi, A.; Sathyanarayana, U.G.; Padar, A.; Huang, C.X.; Cunnigham, H.T.; Farinas, A.J.; Milchgrub, S.; Euhus, D.M.; Gilcrease, M.; et al. Aberrant methylation of the adenomatous polyposis coli (APC) gene promoter 1A in breast and lung carcinomas. Clin. Cancer Res. 2001, 7, 1998–2004. [Google Scholar]
- Li, B.Q.; Liu, P.P.; Zhang, C.H. Correlation between the methylation of APC gene promoter and colon cancer. Oncol. Lett. 2017, 14, 2315–2319. [Google Scholar] [CrossRef]
- Sarrio, D.; Moreno-Bueno, G.; Hardisson, D.; Sanchez-Estevez, C.; Guo, M.; Herman, J.G.; Gamallo, C.; Esteller, M.; Palacios, J. Epigenetic and genetic alterations of APC and CDH1 genes in lobular breast cancer: Relationships with abnormal E-cadherin and catenin expression and microsatellite instability. Int. J. Cancer J. Int. Du Cancer 2003, 106, 208–215. [Google Scholar] [CrossRef]
- Dulaimi, E.; Hillinck, J.; Ibanez de Caceres, I.; Al-Saleem, T.; Cairns, P. Tumor suppressor gene promoter hypermethylation in serum of breast cancer patients. Clin. Cancer Res. 2004, 10, 6189–6193. [Google Scholar] [CrossRef]
- Holleman, A.; Chung, I.; Olsen, R.R.; Kwak, B.; Mizokami, A.; Saijo, N.; Parissenti, A.; Duan, Z.; Voest, E.E.; Zetter, B.R. miR-135a contributes to paclitaxel resistance in tumor cells both in vitro and in vivo. Oncogene 2011, 30, 4386–4398. [Google Scholar] [CrossRef]
- Valeri, N.; Braconi, C.; Gasparini, P.; Murgia, C.; Lampis, A.; Paulus-Hock, V.; Hart, J.R.; Ueno, L.; Grivennikov, S.I.; Lovat, F.; et al. MicroRNA-135b promotes cancer progression by acting as a downstream effector of oncogenic pathways in colon cancer. Cancer Cell 2014, 25, 469–483. [Google Scholar] [CrossRef]
- Nagel, R.; le Sage, C.; Diosdado, B.; van der Waal, M.; Oude Vrielink, J.A.; Bolijn, A.; Meijer, G.A.; Agami, R. Regulation of the adenomatous polyposis coli gene by the miR-135 family in colorectal cancer. Cancer Res. 2008, 68, 5795–5802. [Google Scholar] [CrossRef]
- Jiang, D.; Zhou, B.; Xiong, Y.; Cai, H. miR-135 regulated breast cancer proliferation and epithelial-mesenchymal transition acts by the Wnt/beta-catenin signaling pathway. Int. J. Mol. Med. 2019, 43, 1623–1634. [Google Scholar] [CrossRef]
- Wang, L.P.; Ma, X.Q.; Cai, J.C. Clinicopathological significance and function of miR-135b in the occurrence and development of gastric cancer. Zhonghua Yi Xue Za Zhi 2012, 92, 3269–3273. [Google Scholar]
- Mukherjee, N.; Bhattacharya, N.; Alam, N.; Roy, A.; Roychoudhury, S.; Panda, C.K. Subtype-specific alterations of the Wnt signaling pathway in breast cancer: Clinical and prognostic significance. Cancer Sci. 2012, 103, 210–220. [Google Scholar] [CrossRef]
- Stefanski, C.D.; Arnason, A.; Maloney, S.; Kotsen, J.; Powers, E.; Zhang, J.T.; Prosperi, J.R. APC Loss Prevents Doxorubicin-Induced Cell Death by Increasing Drug Efflux and a Chemoresistant Cell Population in Breast Cancer. Int. J. Mol. Sci. 2023, 24, 7621. [Google Scholar] [CrossRef]
- Chandrashekar, D.S.; Bashel, B.; Balasubramanya, S.A.H.; Creighton, C.J.; Ponce-Rodriguez, I.; Chakravarthi, B.; Varambally, S. UALCAN: A Portal for Facilitating Tumor Subgroup Gene Expression and Survival Analyses. Neoplasia 2017, 19, 649–658. [Google Scholar] [CrossRef]
- Chandrashekar, D.S.; Karthikeyan, S.K.; Korla, P.K.; Patel, H.; Shovon, A.R.; Athar, M.; Netto, G.J.; Qin, Z.S.; Kumar, S.; Manne, U.; et al. UALCAN: An update to the integrated cancer data analysis platform. Neoplasia 2022, 25, 18–27. [Google Scholar] [CrossRef]
- VanKlompenberg, M.K.; Bedalov, C.O.; Soto, K.F.; Prosperi, J.R. APC selectively mediates response to chemotherapeutic agents in breast cancer. BMC Cancer 2015, 15, 457. [Google Scholar] [CrossRef]
- Creighton, C.J.; Li, X.; Landis, M.; Dixon, J.M.; Neumeister, V.M.; Sjolund, A.; Rimm, D.L.; Wong, H.; Rodriguez, A.; Herschkowitz, J.I.; et al. Residual breast cancers after conventional therapy display mesenchymal as well as tumor-initiating features. Proc. Natl. Acad. Sci. USA 2009, 106, 13820–13825. [Google Scholar] [CrossRef]
- Astarita, E.M.; Maloney, S.M.; Hoover, C.A.; Berkeley, B.J.; VanKlompenberg, M.K.; Nair, T.M.; Prosperi, J.R. Adenomatous Polyposis Coli loss controls cell cycle regulators and response to paclitaxel in MDA-MB-157 metaplastic breast cancer cells. PLoS ONE 2021, 16, e0255738. [Google Scholar] [CrossRef]
- Prosperi, J.R.; Khramtsov, A.I.; Khramtsova, G.F.; Goss, K.H. Apc mutation enhances PyMT-induced mammary tumorigenesis. PLoS ONE 2011, 6, e29339. [Google Scholar] [CrossRef]
- Gomez-Miragaya, J.; Palafox, M.; Pare, L.; Yoldi, G.; Ferrer, I.; Vila, S.; Galvan, P.; Pellegrini, P.; Perez-Montoyo, H.; Igea, A.; et al. Resistance to Taxanes in Triple-Negative Breast Cancer Associates with the Dynamics of a CD49f+ Tumor-Initiating Population. Stem Cell Rep. 2017, 8, 1392–1407. [Google Scholar] [CrossRef]
- Lacerda, L.; Pusztai, L.; Woodward, W.A. The role of tumor initiating cells in drug resistance of breast cancer: Implications for future therapeutic approaches. Drug Resist. Updat. 2010, 13, 99–108. [Google Scholar] [CrossRef]
- VanKlompenberg, M.K.; Leyden, E.; Arnason, A.H.; Zhang, J.T.; Stefanski, C.D.; Prosperi, J.R. APC loss in breast cancer leads to doxorubicin resistance via STAT3 activation. Oncotarget 2017, 8, 102868–102879. [Google Scholar] [CrossRef]
- Maloney, S.M.; Hoover, C.A.; Morejon-Lasso, L.V.; Prosperi, J.R. Mechanisms of Taxane Resistance. Cancers 2020, 12, 3323. [Google Scholar] [CrossRef]
- Ben-Hamo, R.; Zilberberg, A.; Cohen, H.; Bahar-Shany, K.; Wachtel, C.; Korach, J.; Aviel-Ronen, S.; Barshack, I.; Barash, D.; Levanon, K.; et al. Resistance to paclitaxel is associated with a variant of the gene BCL2 in multiple tumor types. NPJ Precis. Oncol. 2019, 3, 12. [Google Scholar] [CrossRef]
- Lok, S.W.; Whittle, J.R.; Vaillant, F.; Teh, C.E.; Lo, L.L.; Policheni, A.N.; Bergin, A.R.T.; Desai, J.; Ftouni, S.; Gandolfo, L.C.; et al. A Phase Ib Dose-Escalation and Expansion Study of the BCL2 Inhibitor Venetoclax Combined with Tamoxifen in ER and BCL2-Positive Metastatic Breast Cancer. Cancer Discov. 2019, 9, 354–369. [Google Scholar] [CrossRef] [PubMed]
- Lindeman, G.J.; Fernando, T.M.; Bowen, R.; Jerzak, K.J.; Song, X.; Decker, T.; Boyle, F.; McCune, S.; Armstrong, A.; Shannon, C.; et al. VERONICA: Randomized Phase II Study of Fulvestrant and Venetoclax in ER-Positive Metastatic Breast Cancer Post-CDK4/6 Inhibitors-Efficacy, Safety, and Biomarker Results. Clin Cancer Res. 2022, 28, 3256–3267. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Sanjana, N.E.; Shalem, O.; Zhang, F. Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 2014, 11, 783–784. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wise, A.R.; Maloney, S.; Hering, A.; Zabala, S.; Richmond, G.E.; VanKlompenberg, M.K.; Nair, M.T.; Prosperi, J.R. Bcl-2 Up-Regulation Mediates Taxane Resistance Downstream of APC Loss. Int. J. Mol. Sci. 2024, 25, 6745. https://doi.org/10.3390/ijms25126745
Wise AR, Maloney S, Hering A, Zabala S, Richmond GE, VanKlompenberg MK, Nair MT, Prosperi JR. Bcl-2 Up-Regulation Mediates Taxane Resistance Downstream of APC Loss. International Journal of Molecular Sciences. 2024; 25(12):6745. https://doi.org/10.3390/ijms25126745
Chicago/Turabian StyleWise, Angelique R., Sara Maloney, Adam Hering, Sarah Zabala, Grace E. Richmond, Monica K. VanKlompenberg, Murlidharan T. Nair, and Jenifer R. Prosperi. 2024. "Bcl-2 Up-Regulation Mediates Taxane Resistance Downstream of APC Loss" International Journal of Molecular Sciences 25, no. 12: 6745. https://doi.org/10.3390/ijms25126745