
Chloroplast Genomes Evolution and Phylogenetic Relationships of Caragana species


Abstract
:1. Introduction
2. Results
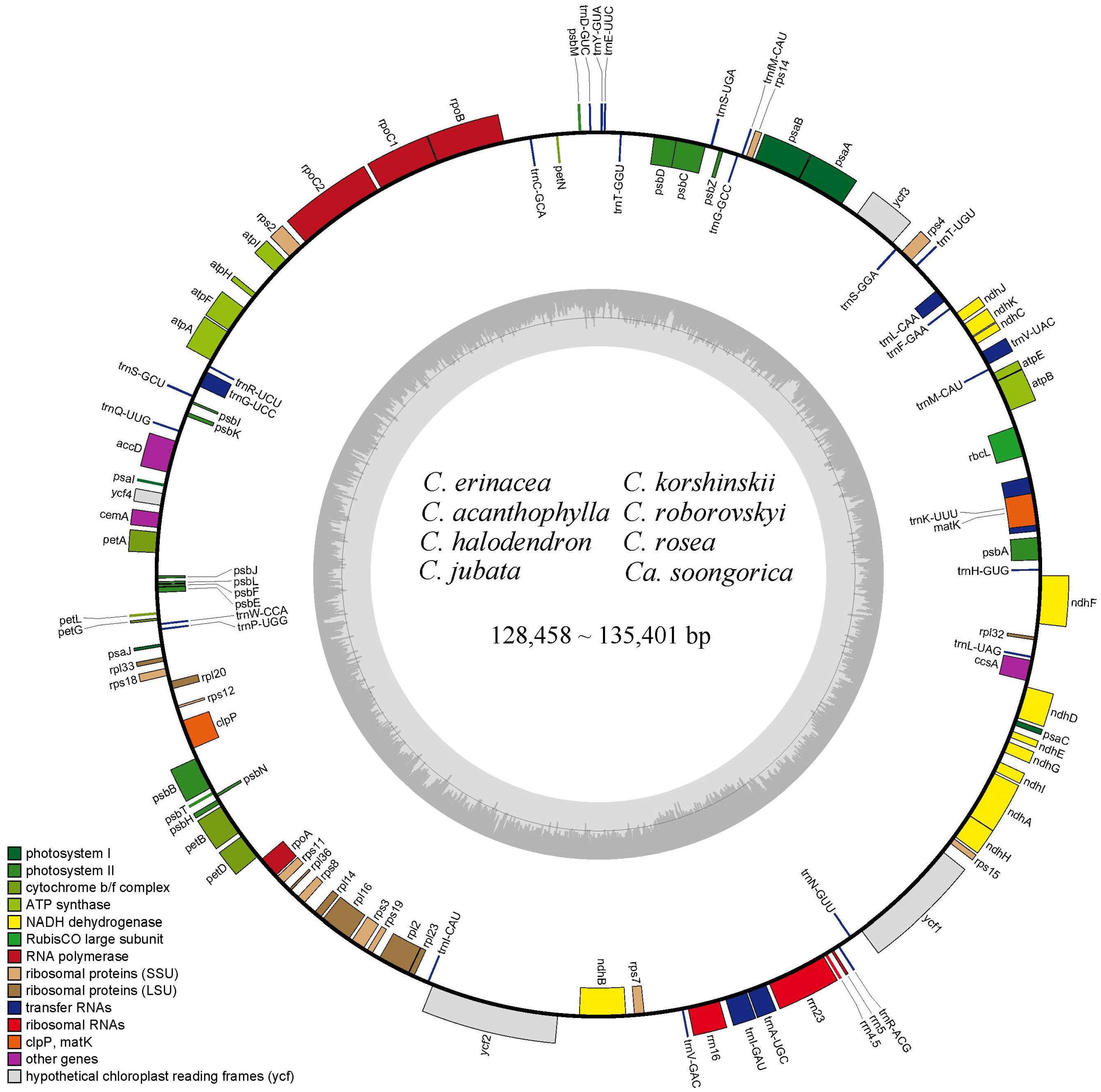
2.1. Characteristics of Caragana Chloroplast Genomes
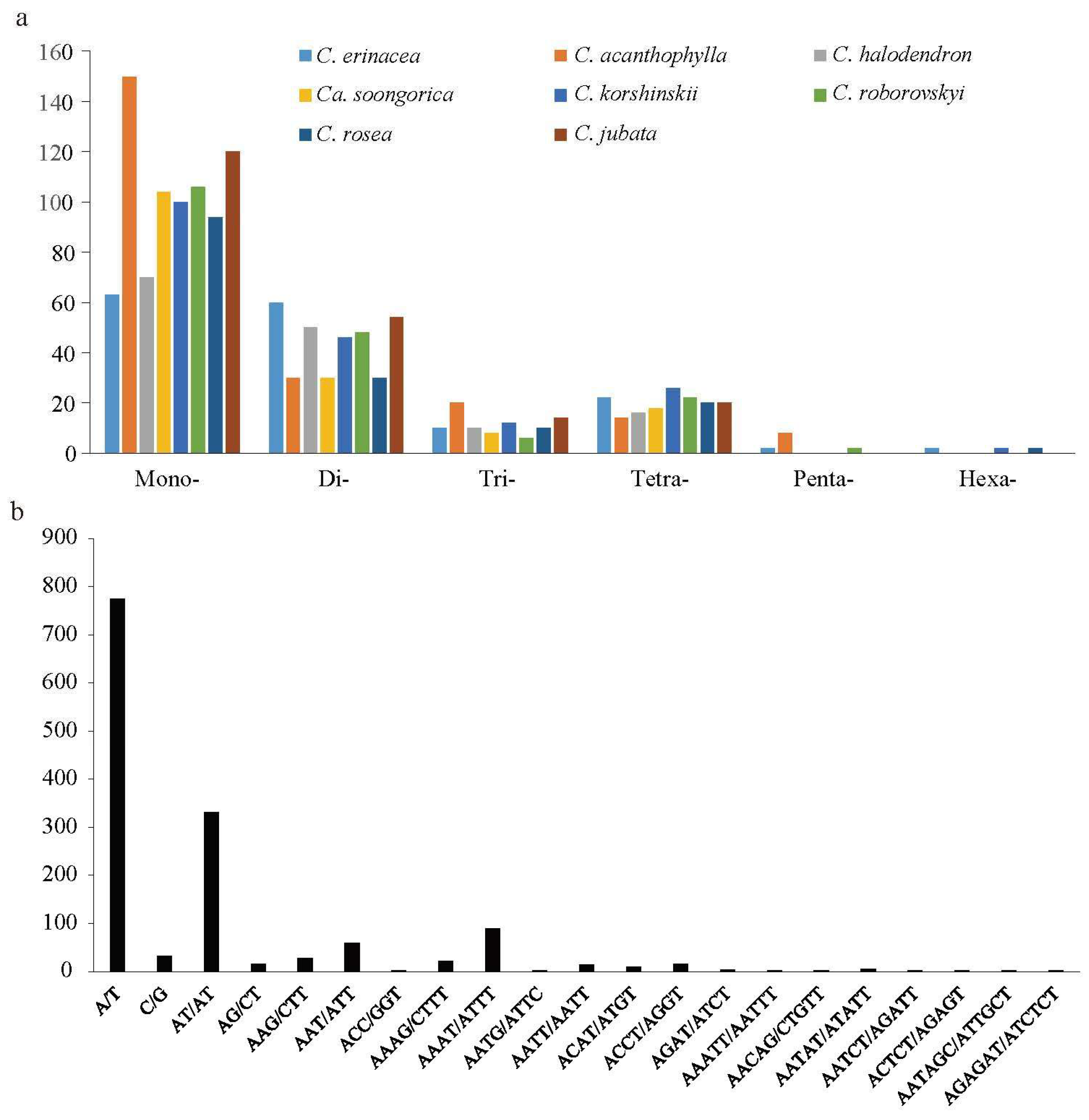
2.2. Analyses of Repeat Sequences
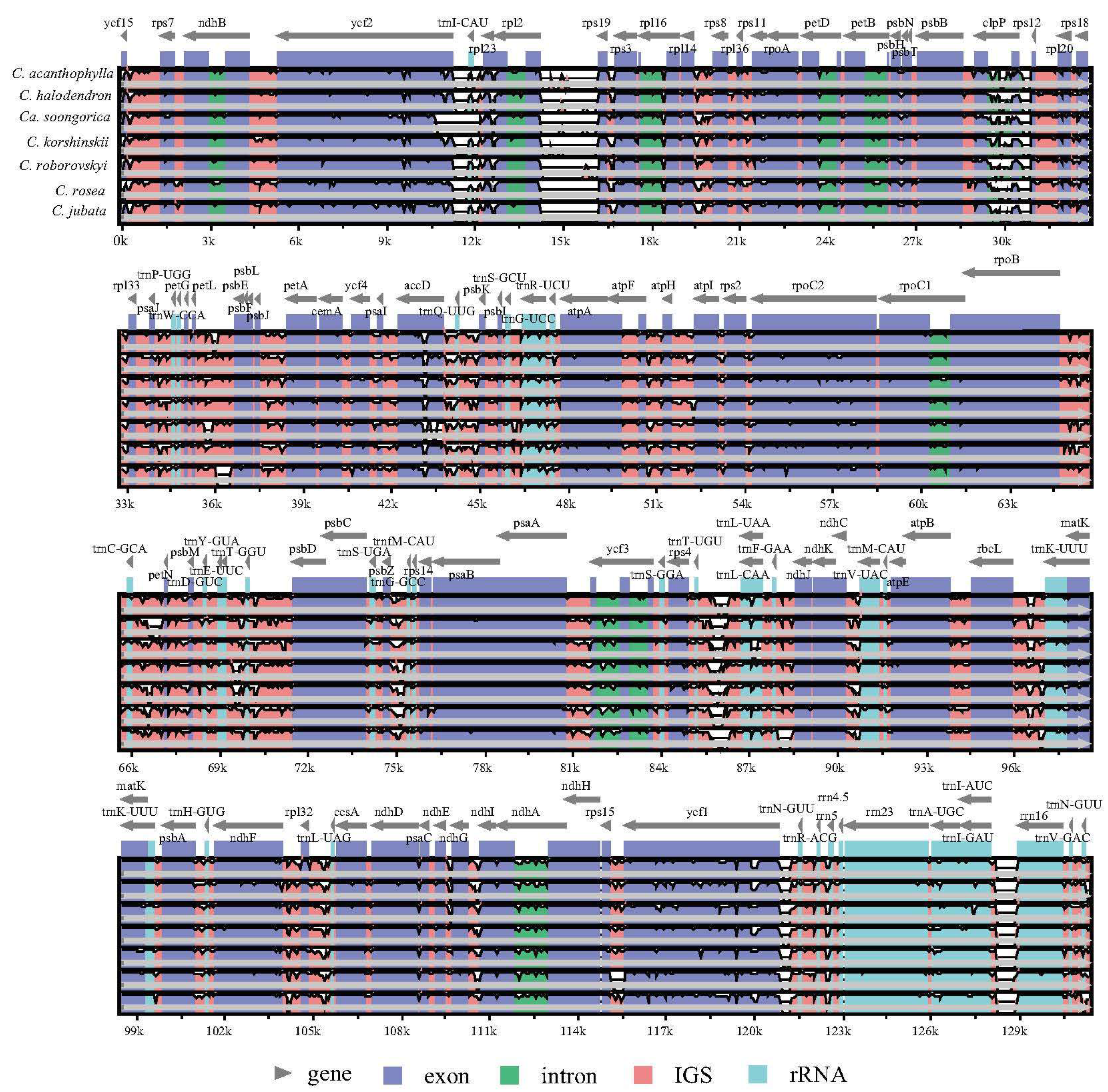
2.3. Genomic Sequence Divergence
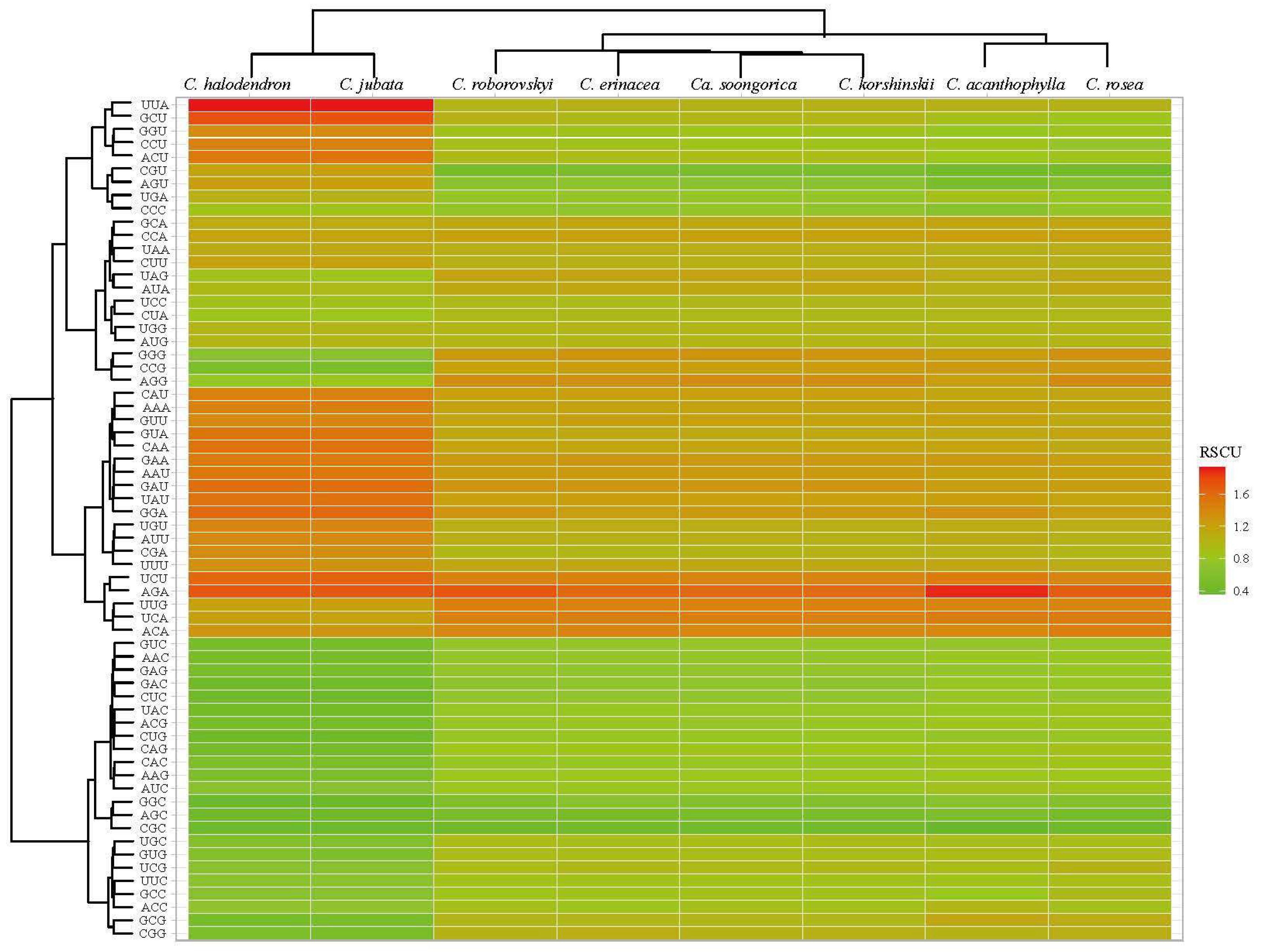
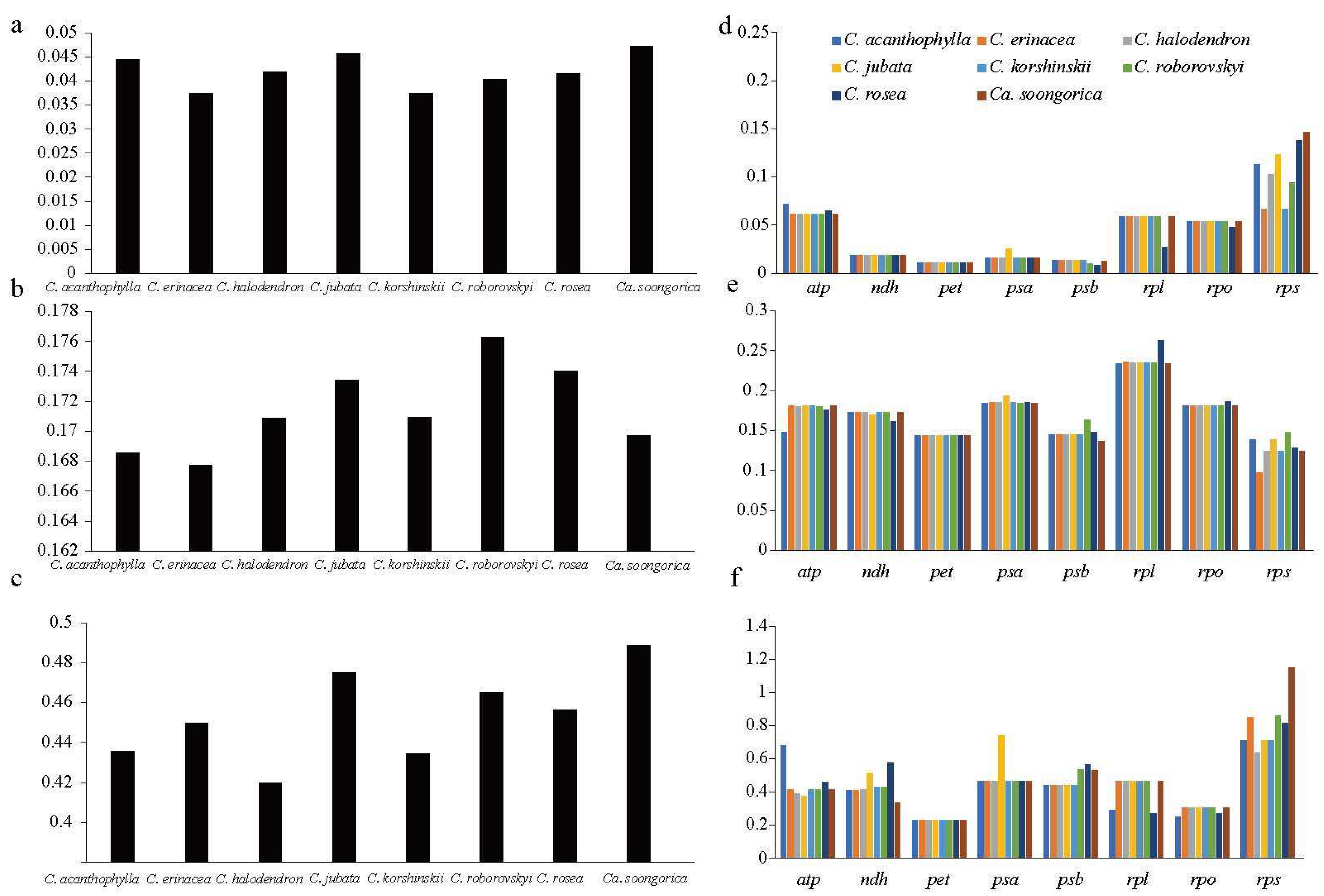
2.4. Codon Usage Pattern and Molecular Evolution
2.5. Molecular Evolution of the Caragana Chloroplast Genome
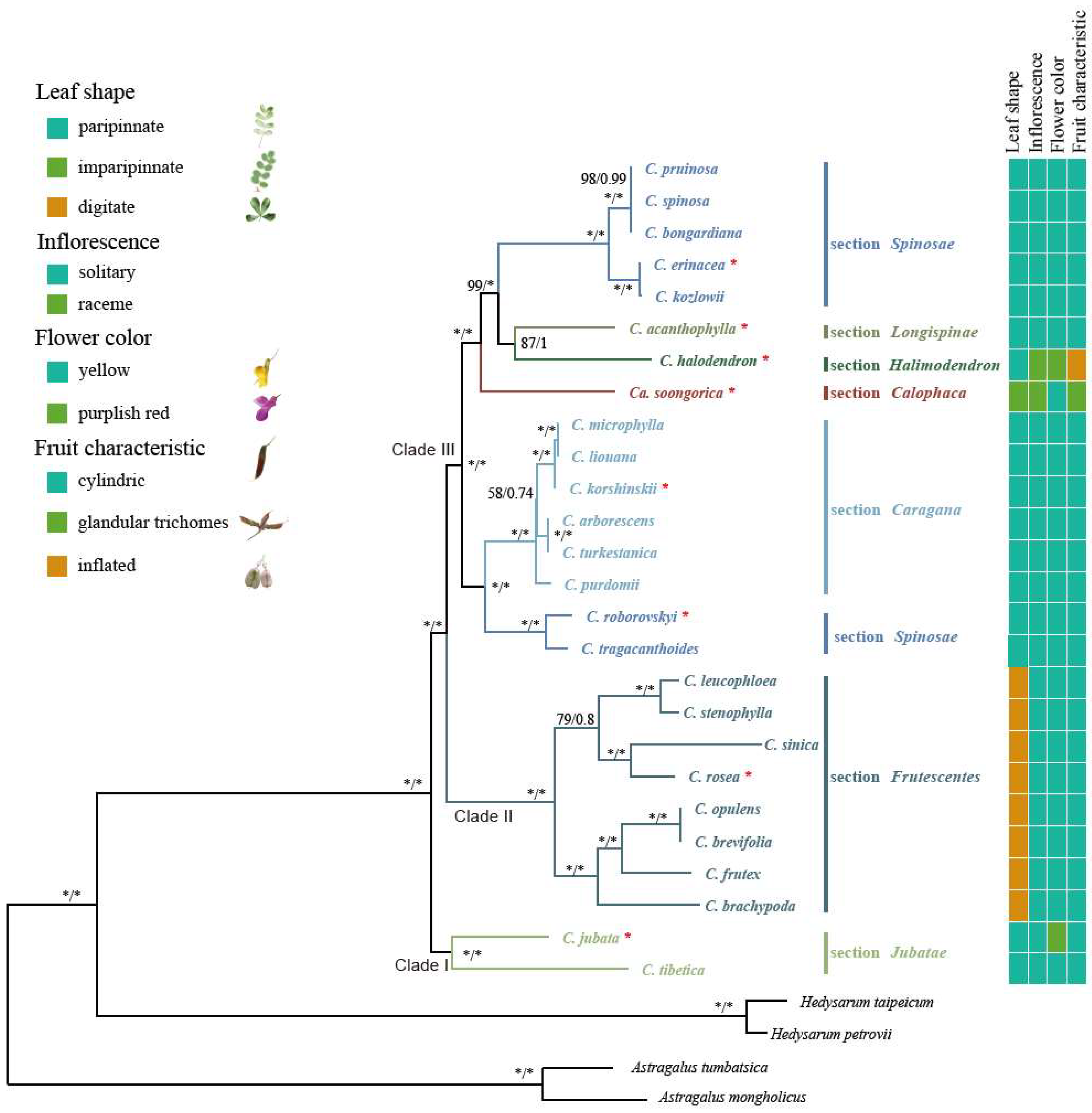
2.6. Phylogenetic Analysis
3. Discussion
3.1. Structure and Comparative Analysis of Caragana Chloroplast Genomes
3.2. Phylogenetic Relationships among the Sections of Caragana
4. Materials and Methods
4.1. Plant Material, DNA Extraction, and Sequencing
4.2. Chloroplast Genome Assembly and Annotation
4.3. Analysis of Repeat Sequences and SSRs
4.4. Comparative Genomic Analyses and Molecular Evolution Analysis
4.5. Phylogenetic Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Roskov, Y.; Bisby, F.; Zarucchi, J.; Schrire, B.; White, R.J.R. ILDIS World Database of Legumes: Draft Checklist, 10th ed.; Mini CD-ROM: Reading, UK, 2005; ISBN 070491-2481. [Google Scholar]
- Lewis, G.P.; Schrire, B.; Mackinder, B.; Lock, M. Legumes of the World; Royal Botanic Gardens Kew: London, UK, 2005. [Google Scholar]
- Du, L.-T.; Ma, L.-L.; Pan, H.-Z.; Qiao, C.-L.; Meng, C.; Wu, H.-Y.; Tian, J.; Yuan, H.-Y. Carbon–water coupling and its relationship with environmental and biological factors in a planted Caragana liouana shrub community in desert steppe, northwest China. J. Plant Ecol. 2022, 15, 947–960. [Google Scholar] [CrossRef]
- Zhang, M.L.; Xiang, X.G.; Xue, J.J.; Sanderson, S.C.; Fritsch, P.W. Himalayan uplift shaped biomes in Miocene temperate Asia: Evidence from leguminous Caragana. Sci. Rep. 2016, 6, 36528. [Google Scholar] [CrossRef]
- Zhao, Y. Classification and Its Floristic Geography of Caragana Fabr. in the World; Inner Mongolia University Press: Hohhot, China, 2009. [Google Scholar]
- Duan, L.; Yang, X.; Liu, P.L.; Johnson, G.; Wen, J.; Chang, Z.Y. A molecular phylogeny of Caraganeae (Leguminosae, Papilionoideae) reveals insights into new generic and infrageneric delimitations. PhytoKeys 2016, 70, 111–137. [Google Scholar] [CrossRef]
- Zhang, M.L.; Wen, Z.B.; Fritsch, P.W.; Sanderson, S.C. Spatiotemporal evolution of Calophaca (Fabaceae) reveals multiple dispersals in central Asian mountains. PLoS ONE 2015, 10, e0123228. [Google Scholar] [CrossRef]
- Wu, Z.; Raven, P.; Hong, D. Flora of China, Volume 10: Fabaceae; Missouri Botanical Garden Press: St. Louis, MO, USA, 2010. [Google Scholar]
- Zhang, M.; Xue, J.; Zhang, Q.; Sanderson, S.C. Inferring ancestral distribution area and survival vegetation of Caragana (Fabaceae) in Tertiary. Plant Syst. Evol. 2015, 301, 1831–1842. [Google Scholar] [CrossRef]
- Hou, X.; Liu, J.E.; Zhao, Y.Z. Molecular phylogeny of Caragana (Fabaceae) in China. J. Syst. Evol. 2008, 46, 600–607. [Google Scholar]
- Zhang, M.; Fritsch, P.W.; Cruz, B.C. Phylogeny of Caragana (Fabaceae) based on DNA sequence data from rbcL, trnS-trnG, and ITS. Mol. Phylogenet. Evol. 2009, 50, 547–559. [Google Scholar] [CrossRef]
- Guo, C.; Liu, K.; Li, E.; Chen, Y.; He, J.; Li, W.; Dong, W.; Suo, Z. Maternal donor and genetic variation of Lagerstroemia indica cultivars. Int. J. Mol. Sci. 2023, 24, 3606. [Google Scholar] [CrossRef]
- Li, E.; Liu, K.; Deng, R.; Gao, Y.; Liu, X.; Dong, W.; Zhang, Z. Insights into the phylogeny and chloroplast genome evolution of Eriocaulon (Eriocaulaceae). BMC Plant Biol. 2023, 23, 32. [Google Scholar] [CrossRef]
- Liu, X.; Bai, Y.; Wang, Y.; Chen, Y.; Dong, W.; Zhang, Z. Complete Chloroplast Genome of Hypericum perforatum and Dynamic Evolution in Hypericum (Hypericaceae). Int. J. Mol. Sci. 2023, 24, 16130. [Google Scholar] [CrossRef]
- Dong, W.; Li, E.; Liu, Y.; Xu, C.; Wang, Y.; Liu, K.; Cui, X.; Sun, J.; Suo, Z.; Zhang, Z.; et al. Phylogenomic approaches untangle early divergences and complex diversifications of the olive plant family. BMC Biol. 2022, 20, 92. [Google Scholar] [CrossRef]
- Liu, L.; Li, H.; Li, J.; Li, X.; Hu, N.; Sun, J.; Zhou, W. Chloroplast genomes of Caragana tibetica and Caragana turkestanica: Structures and comparative analysis. BMC Plant Biol. 2024, 24, 254. [Google Scholar] [CrossRef]
- Jiang, M.; Chen, H.; He, S.; Wang, L.; Chen, A.J.; Liu, C. Sequencing, Characterization, and Comparative Analyses of the Plastome of Caragana rosea var. rosea. Int. J. Mol. Sci. 2018, 19, 1419. [Google Scholar] [CrossRef]
- Yuan, M.; Yin, X.; Gao, B.; Gu, R.; Jiang, G. The chloroplasts genomic analyses of four specific Caragana species. PLoS ONE 2022, 17, e0272990. [Google Scholar] [CrossRef]
- Olejniczak, S.A.; Lojewska, E.; Kowalczyk, T.; Sakowicz, T. Chloroplasts: State of research and practical applications of plastome sequencing. Planta 2016, 244, 517–527. [Google Scholar] [CrossRef]
- Choi, I.S.; Jansen, R.; Ruhlman, T. Lost and Found: Return of the Inverted Repeat in the Legume Clade Defined by Its Absence. Genome Biol. Evol. 2019, 11, 1321–1333. [Google Scholar] [CrossRef]
- Jiao, Y.X.; He, X.F.; Song, R.; Wang, X.M.; Zhang, H.; Aili, R.; Chao, Y.H.; Shen, Y.H.; Yu, L.X.; Zhang, T.J.; et al. Recent structural variations in the Medicago chloroplast genomes and their horizontal transfer into nuclear chromosomes. J. Syst. Evol. 2023, 61, 627–642. [Google Scholar] [CrossRef]
- Lee, C.; Choi, I.S.; Cardoso, D.; de Lima, H.C.; de Queiroz, L.P.; Wojciechowski, M.F.; Jansen, R.K.; Ruhlman, T.A. The chicken or the egg? Plastome evolution and an independent loss of the inverted repeat in papilionoid legumes. Plant J. 2021, 107, 861–875. [Google Scholar]
- Ping, J.; Hao, J.; Li, J.; Yang, Y.; Su, Y.; Wang, T. Loss of the IR region in conifer plastomes: Changes in the selection pressure and substitution rate of protein-coding genes. Ecol. Evol. 2022, 12, e8499. [Google Scholar] [CrossRef]
- Jansen, R.K.; Wojciechowski, M.F.; Sanniyasi, E.; Lee, S.B.; Daniell, H. Complete plastid genome sequence of the chickpea (Cicer arietinum) and the phylogenetic distribution of rps12 and clpP intron losses among legumes (Leguminosae). Mol. Phylogenet. Evol. 2008, 48, 1204–1217. [Google Scholar] [CrossRef]
- Cai, Z.Q.; Guisinger, M.; Kim, H.G.; Ruck, E.; Blazier, J.C.; McMurtry, V.; Kuehl, J.V.; Boore, J.; Jansen, R.K. Extensive Reorganization of the Plastid Genome of Trifolium subterraneum (Fabaceae) Is Associated with Numerous Repeated Sequences and Novel DNA Insertions. J. Mol. Evol. 2008, 67, 696–704. [Google Scholar] [CrossRef]
- Sveinsson, S.; Cronk, Q. Evolutionary origin of highly repetitive plastid genomes within the clover genus (Trifolium). BMC Evol. Biol. 2014, 14, 228. [Google Scholar] [CrossRef]
- Jo, I.-H.; Han, S.; Shim, D.; Ryu, H.; Hyun, T.K.; Lee, Y.; Kim, D.; So, Y.-S.; Chung, J.-W. Complete Chloroplast Genome of the Inverted Repeat-Lacking Species Vicia bungei and Development of Polymorphic Simple Sequence Repeat Markers. Front. Plant Sci. 2022, 13, 891783. [Google Scholar] [CrossRef]
- Millen, R.S.; Olmstead, R.G.; Adams, K.L.; Palmer, J.D.; Lao, N.T.; Heggie, L.; Kavanagh, T.A.; Hibberd, J.M.; Gray, J.C.; Morden, C.W.; et al. Many parallel losses of infA from chloroplast DNA during angiosperm evolution with multiple independent transfers to the nucleus. Plant Cell 2001, 13, 645–658. [Google Scholar] [CrossRef]
- Jansen, R.K.; Saski, C.; Lee, S.B.; Hansen, A.K.; Daniell, H. Complete plastid genome sequences of three Rosids (Castanea, Prunus, Theobroma): Evidence for at least two independent transfers of rpl22 to the nucleus. Mol. Biol. Evol. 2011, 28, 835–847. [Google Scholar] [CrossRef]
- Moghaddam, M.; Ohta, A.; Shimizu, M.; Terauchi, R.; Kazempour-Osaloo, S. The complete chloroplast genome of Onobrychis gaubae (Fabaceae-Papilionoideae): Comparative analysis with related IR-lacking clade species. BMC Plant Biol. 2022, 22, 75. [Google Scholar] [CrossRef] [PubMed]
- Weng, M.-L.; Blazier, J.C.; Govindu, M.; Jansen, R.K. Reconstruction of the Ancestral Plastid Genome in Geraniaceae Reveals a Correlation between Genome Rearrangements, Repeats, and Nucleotide Substitution Rates. Mol. Biol. Evol. 2014, 31, 645–659. [Google Scholar] [CrossRef]
- Liu, H.; Zhao, W.; Hua, W.; Liu, J. A large-scale population based organelle pan-genomes construction and phylogeny analysis reveal the genetic diversity and the evolutionary origins of chloroplast and mitochondrion in Brassica napus L. BMC Genom. 2022, 23, 339. [Google Scholar] [CrossRef]
- Xiong, Y.; Xiong, Y.L.; Jia, X.J.; Zhao, J.M.; Yan, L.J.; Sha, L.N.; Liu, L.; Yu, Q.Q.; Lei, X.; Bai, S.Q. Divergence in Elymus sibiricus is related to geography and climate oscillation: A new look from pan-chloroplast genome data. J. Syst. Evol. 2023. [Google Scholar] [CrossRef]
- Zhang, J.; Huang, H.; Qu, C.; Meng, X.; Meng, F.; Yao, X.; Wu, J.; Guo, X.; Han, B.; Xing, S. Comprehensive analysis of chloroplast genome of Albizia julibrissin Durazz. (Leguminosae sp.). Planta 2021, 255, 26. [Google Scholar] [CrossRef]
- Yen, L.-T.; Kousar, M.; Park, J. Comparative Analysis of Chloroplast Genome of Desmodium stryacifolium with Closely Related Legume Genome from the Phaseoloid Clade. Int. J. Mol. Sci. 2023, 24, 6072. [Google Scholar] [CrossRef] [PubMed]
- Ge, S. A review of recent studies of plant systematics and evolution in China. Biodivers. Sci. 2022, 30, 22385. [Google Scholar] [CrossRef]
- Li, J.L.; Wang, S.; Yu, J.; Wang, L.; Zhou, S.L. A Modified CTAB Protocol for Plant DNA Extraction. Chin. Bull. Bot. 2013, 48, 72–78. [Google Scholar]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.-J.; Yu, W.-B.; Yang, J.-B.; Song, Y.; dePamphilis, C.W.; Yi, T.-S.; Li, D.-Z. GetOrganelle: A fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 2020, 21, 241. [Google Scholar] [CrossRef] [PubMed]
- Dong, W.; Liu, Y.; Li, E.; Xu, C.; Sun, J.; Li, W.; Zhou, S.; Zhang, Z.; Suo, Z. Phylogenomics and biogeography of Catalpa (Bignoniaceae) reveal incomplete lineage sorting and three dispersal events. Mol. Phylogenet. Evol. 2022, 166, 107330. [Google Scholar] [CrossRef] [PubMed]
- Qu, X.-J.; Moore, M.J.; Li, D.-Z.; Yi, T.-S. PGA: A software package for rapid, accurate, and flexible batch annotation of plastomes. Plant Methods 2019, 15, 50. [Google Scholar] [CrossRef] [PubMed]
- Beier, S.; Thiel, T.; Munch, T.; Scholz, U.; Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef] [PubMed]
- Frazer, K.A.; Pachter, L.; Poliakov, A.; Rubin, E.M.; Dubchak, I. VISTA: Computational tools for comparative genomics. Nucleic Acids Res. 2004, 32, W273–W279. [Google Scholar] [CrossRef]
- Suzuki, R.; Shimodaira, H. Pvclust: An R package for assessing the uncertainty in hierarchical clustering. Bioinformatics 2006, 22, 1540–1542. [Google Scholar] [CrossRef]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Gao, F.; Jakovlic, I.; Zou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Yang, Z. PAMLX: A graphical user interface for PAML. Mol. Biol. Evol. 2013, 30, 2723–2724. [Google Scholar] [CrossRef] [PubMed]
- Dong, W.; Xu, C.; Cheng, T.; Zhou, S. Complete chloroplast genome of Sedum sarmentosum and chloroplast genome evolution in Saxifragales. PLoS ONE 2013, 8, e77965. [Google Scholar] [CrossRef] [PubMed]
- Duan, L.; Li, S.J.; Su, C.; Sirichamorn, Y.; Han, L.N.; Ye, W.; Loc, P.K.; Wen, J.; Compton, J.A.; Schrire, B.; et al. Phylogenomic framework of the IRLC legumes (Leguminosae subfamily Papilionoideae) and intercontinental biogeography of tribe Wisterieae. Mol. Phylogenet. Evol. 2021, 163, 107235. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef] [PubMed]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [PubMed]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Hohna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Length (bp) | GC (%) | Number of Genes | |||
|---|---|---|---|---|---|---|
| Total | Coding Genes | tRNA | rRNA | |||
| Caragana erinacea | 131,345 | 34.6 | 110 | 76 | 30 | 4 |
| Caragana acanthophyllaa | 130,523 | 34.5 | 110 | 76 | 30 | 4 |
| Caragana halodendron | 129,144 | 34.4 | 110 | 76 | 30 | 4 |
| Calophaca soongorica | 128,813 | 34.7 | 110 | 76 | 30 | 4 |
| Caragana korshinskii | 130,052 | 34.3 | 110 | 76 | 30 | 4 |
| Caragana roborovskyi | 130,392 | 34.4 | 110 | 76 | 30 | 4 |
| Caragana rosea | 135,401 | 34.8 | 110 | 76 | 30 | 4 |
| Caragana jubata | 128,458 | 34.4 | 110 | 76 | 30 | 4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cui, X.; Liu, K.; Li, E.; Zhang, Z.; Dong, W. Chloroplast Genomes Evolution and Phylogenetic Relationships of Caragana species. Int. J. Mol. Sci. 2024, 25, 6786. https://doi.org/10.3390/ijms25126786
Cui X, Liu K, Li E, Zhang Z, Dong W. Chloroplast Genomes Evolution and Phylogenetic Relationships of Caragana species. International Journal of Molecular Sciences. 2024; 25(12):6786. https://doi.org/10.3390/ijms25126786
Chicago/Turabian StyleCui, Xingyong, Kangjia Liu, Enze Li, Zhixiang Zhang, and Wenpan Dong. 2024. "Chloroplast Genomes Evolution and Phylogenetic Relationships of Caragana species" International Journal of Molecular Sciences 25, no. 12: 6786. https://doi.org/10.3390/ijms25126786