
Comprehensive Proteome and Acetylome Analysis of Needle Senescence in Larix gmelinii
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
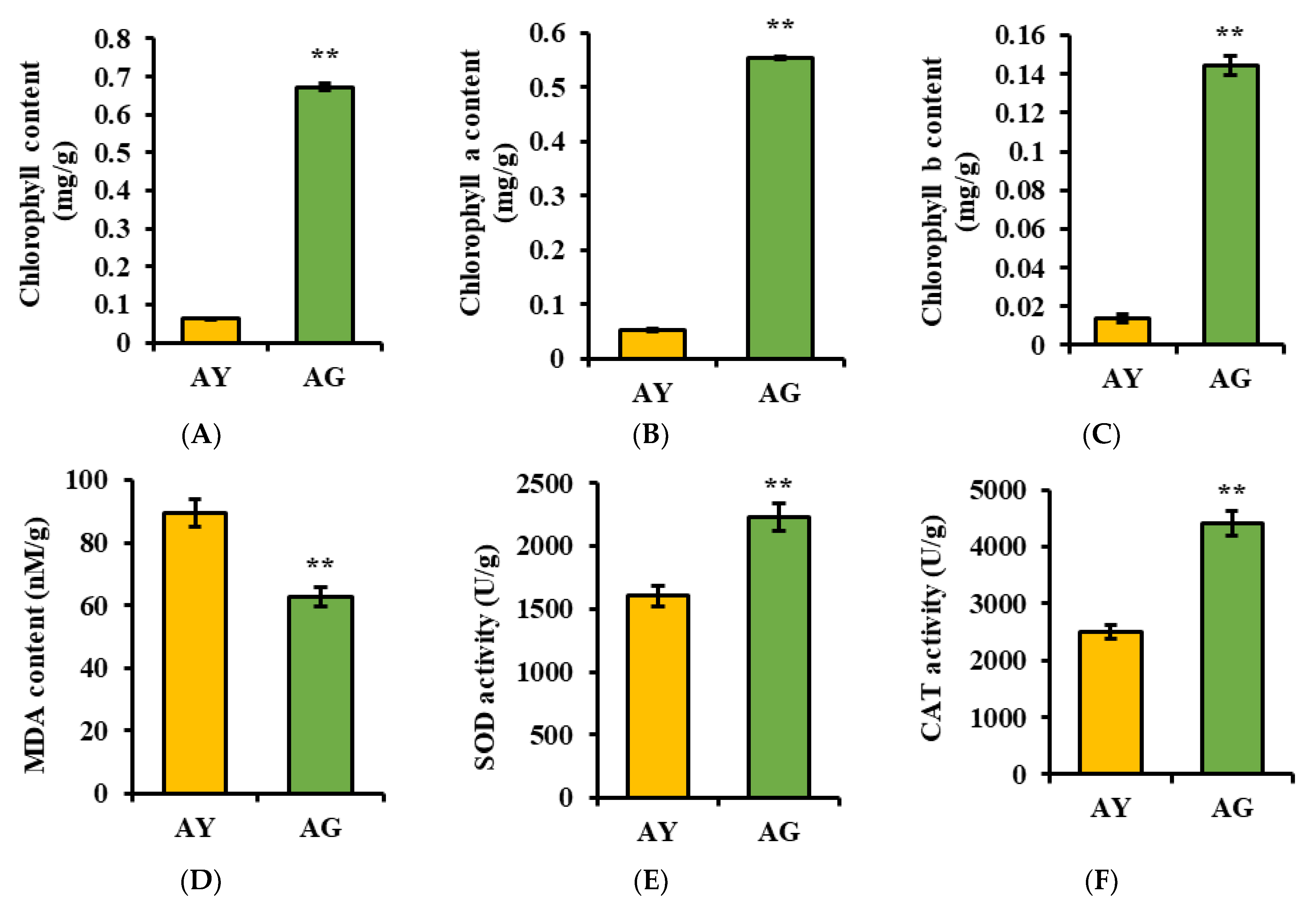
2.1. Changes in Physiological Indicators of L. gmelinii Needle Senescence
2.2. Identification of Proteins in Needle Senescence of L. gmelinii via Proteomics and DEPs Annotation
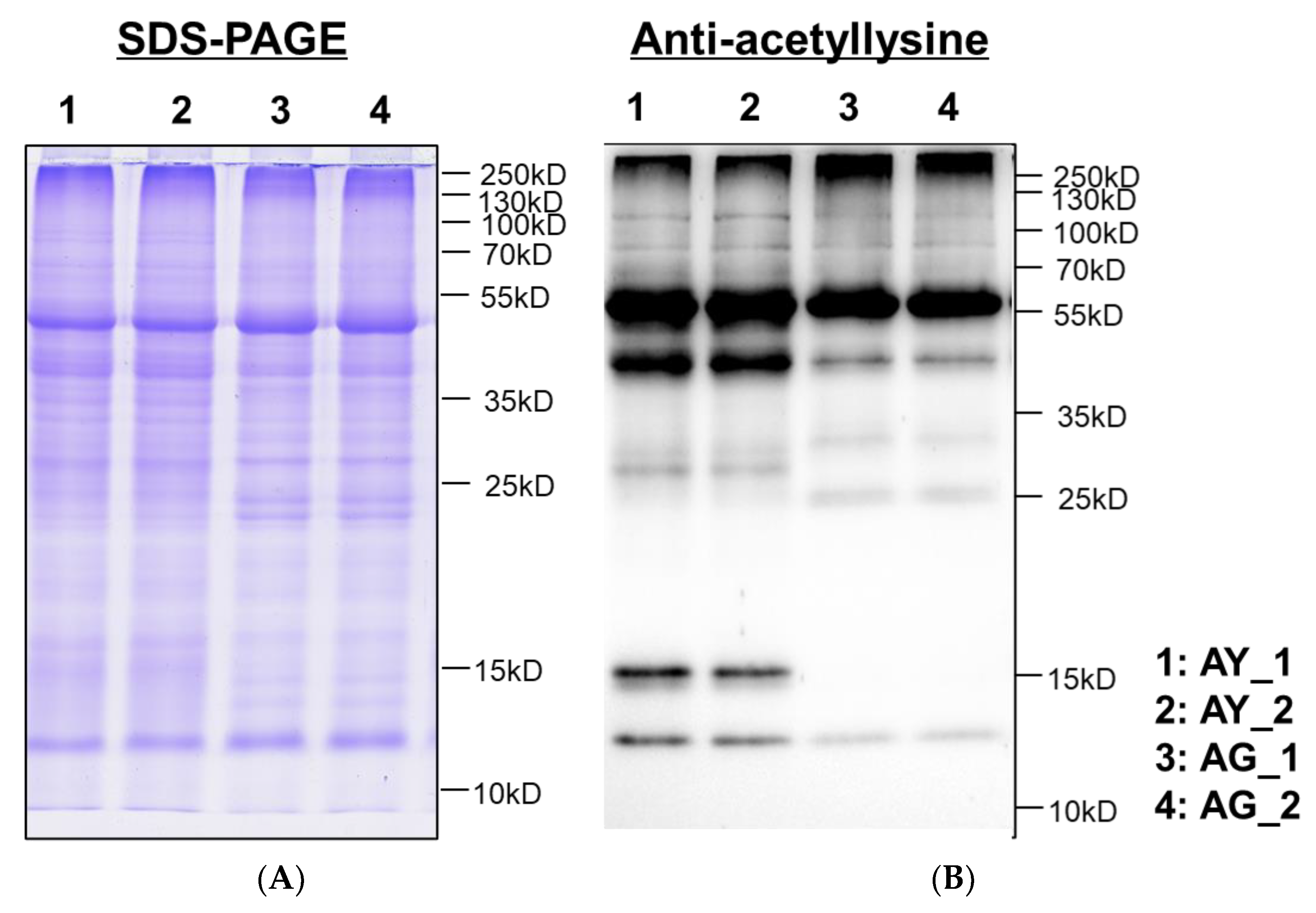
2.3. Lysine-Acetylation of Needle Senescence in L. gmelinii
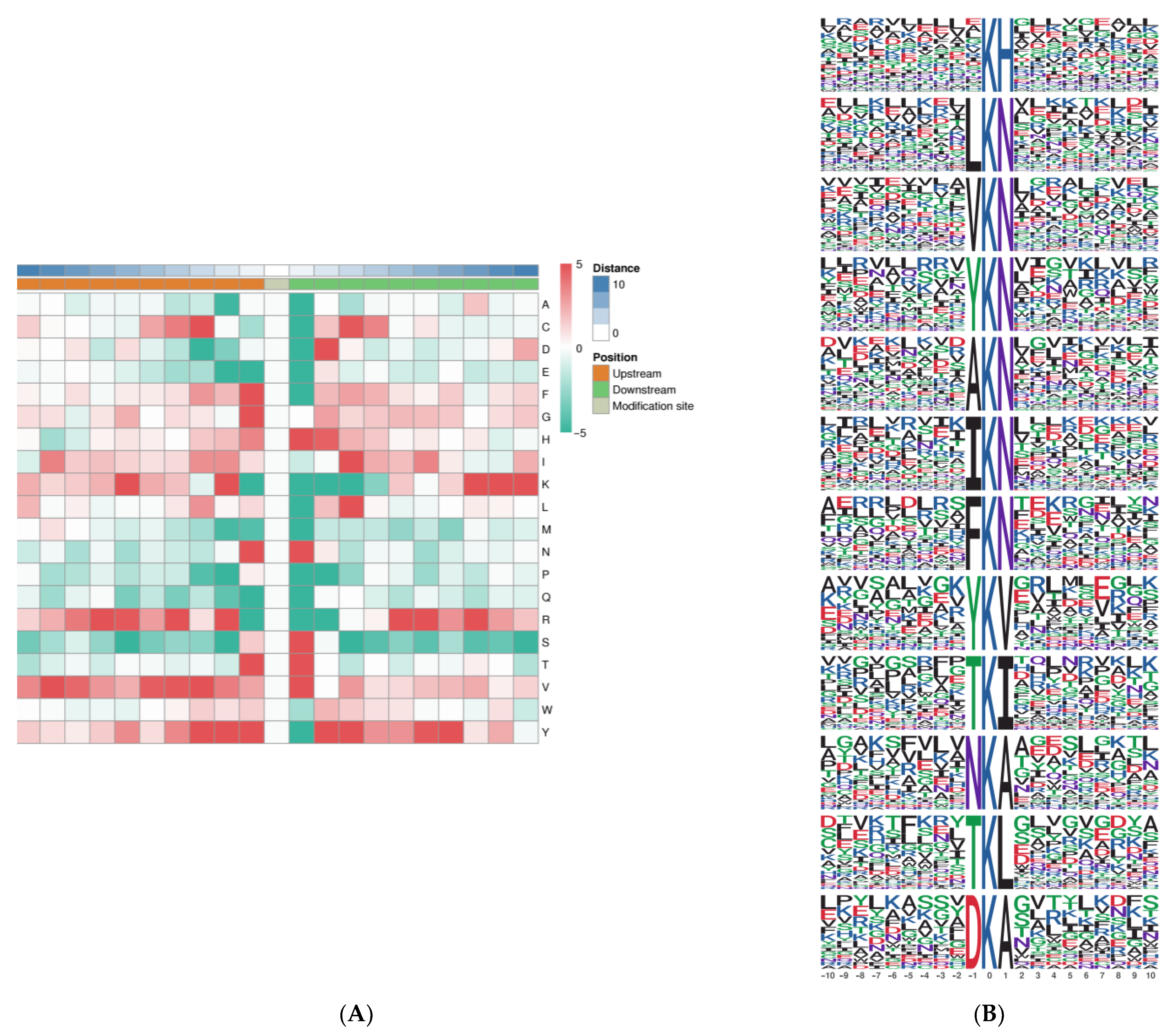
2.4. Analysis of LysAc Motifs in Needle Senescence of L. gmelinii
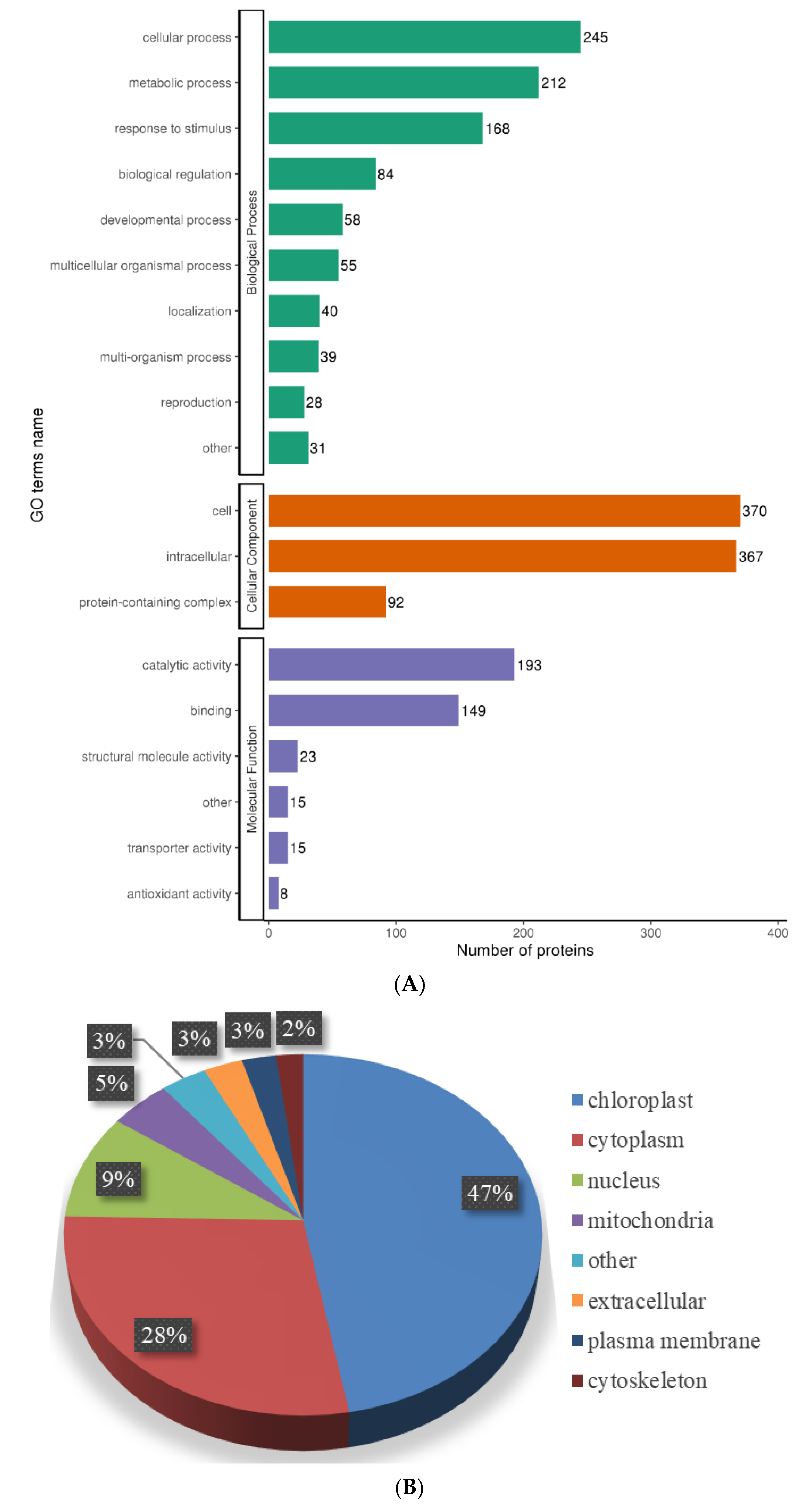
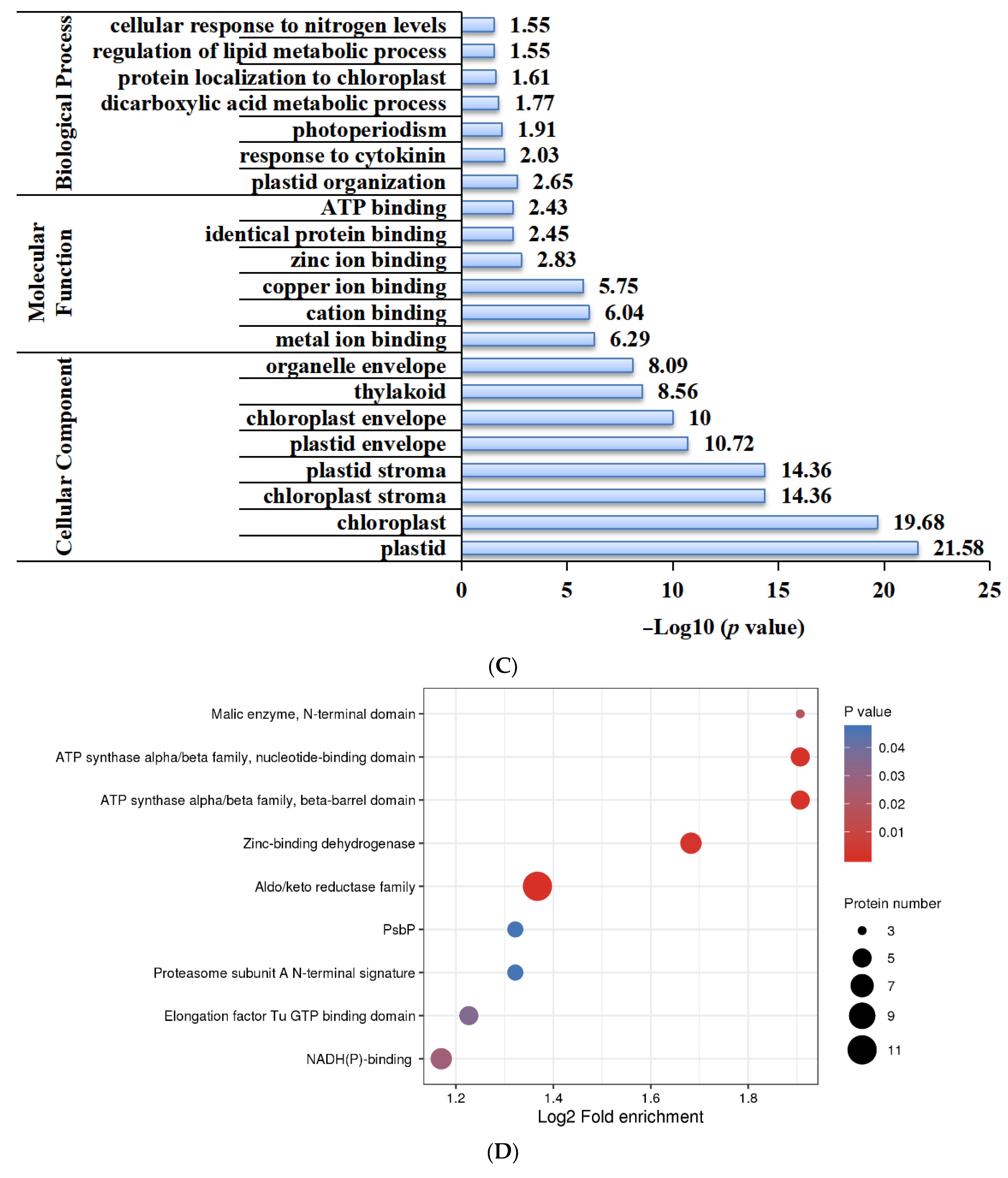
2.5. Functional Characterization of Lysine-Acetylated Proteins in Needle Senescence of L. gmelinii
2.6. Correlation between Proteome and Acetylome in Needle Senescence of L. gmelinii
3. Discussion
4. Materials and Methods
4.1. Plant Materials
4.2. Determination of Physiological Indexes Indicators
4.3. Protein Extraction
4.4. Western Blotting
4.5. Protein Trypsin Digestion
4.6. Lysine-Acetylated Peptide Enrichment
4.7. Proteomic Analysis by LC–MS/MS
4.8. Database Search and Date Analysis
4.9. Motif Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Li, W.; Lee, J.; Yu, S.; Wang, F.; Lv, W.; Zhang, X.; Li, C.; Yang, J. Characterization and analysis of the transcriptome response to drought in Larix kaempferi using PacBio full-length cDNA sequencing integrated with de novo RNA-seq reads. Planta 2021, 253, 28. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, H.G.; Pang, Q.Y. Physiological evaluation of the responses of Larix olgensis families to drought stress and proteomic analysis of the superior family. Genet. Mol. Res. 2015, 14, 15577–15586. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Sun, X.; Xie, Y.; Feng, J.; Zhang, S. Transcriptome and proteome profiling of adventitious root development in hybrid larch (Larix kaempferi × Larix olgensis). BMC Plant Biol. 2014, 14, 305. [Google Scholar] [CrossRef]
- Hou, J.; Wang, X.; Liu, W.; Jiang, X.; Gai, Y. Large-Scale Quantitative Proteomic Analysis during Different Stages of Somatic Embryogenesis in Larix olgensis. Curr. Issues Mol. Biol. 2023, 45, 2021–2034. [Google Scholar] [CrossRef]
- Teyssier, C.; Grondin, C.; Bonhomme, L.; Lomenech, A.M.; Vallance, M.; Morabito, D.; Label, P.; Lelu-Walter, M.A. Increased gelling agent concentration promotes somatic embryo maturation in hybrid larch (Larix × eurolepsis): A 2-DE proteomic analysis. Physiol. Plant 2011, 141, 152–165. [Google Scholar] [CrossRef] [PubMed]
- Teyssier, C.; Maury, S.; Beaufour, M.; Grondin, C.; Delaunay, A.; Le Metté, C.; Ader, K.; Cadene, M.; Label, P.; Lelu-Walter, M.A. In search of markers for somatic embryo maturation in hybrid larch (Larix × eurolepis): Global DNA methylation and proteomic analyses. Physiol. Plant 2014, 150, 271–291. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Li, H.; Fu, S.; Chen, B.; Sun, W.; Zhang, J.; Zhang, J. An iTRAQ-based proteomics approach to clarify the molecular physiology of somatic embryo development in Prince Rupprecht’s larch (Larix principis-rupprechtii Mayr). PLoS ONE 2015, 10, e0119987. [Google Scholar] [CrossRef]
- Hart, G.W.; Ball, L.E. Post-translational modifications: A major focus for the future of proteomics. Mol. Cell. Proteom. 2013, 12, 3443. [Google Scholar] [CrossRef]
- Millar, A.H.; Heazlewood, J.L.; Giglione, C.; Holdsworth, M.J.; Bachmair, A.; Schulze, W.X. The Scope, Functions, and Dynamics of Posttranslational Protein Modifications. Annu. Rev. Plant Biol. 2019, 70, 119–151. [Google Scholar] [CrossRef]
- Narita, T.; Weinert, B.T.; Choudhary, C. Functions and mechanisms of non-histone protein acetylation. Nat. Rev. Mol. Cell Biol. 2019, 20, 156–174. [Google Scholar] [CrossRef]
- Xia, L.; Kong, X.; Song, H.; Han, Q.; Zhang, S. Advances in proteome-wide analysis of plant lysine acetylation. Plant Commun. 2022, 3, 100266. [Google Scholar] [CrossRef] [PubMed]
- Xing, S.; Poirier, Y. The protein acetylome and the regulation of metabolism. Trends Plant Sci. 2012, 17, 423–430. [Google Scholar] [CrossRef] [PubMed]
- David Law, R.; Suttle, J.C. Changes in histone H3 and H4 multi-acetylation during natural and forced dormancy break in potato tubers. Physiol. Plant 2004, 120, 642–649. [Google Scholar] [CrossRef] [PubMed]
- Shahbazian, M.D.; Grunstein, M. Functions of site-specific histone acetylation and deacetylation. Annu. Rev. Biochem. 2007, 76, 75–100. [Google Scholar] [CrossRef] [PubMed]
- Finkemeier, I.; Laxa, M.; Miguet, L.; Howden, A.J.; Sweetlove, L.J. Proteins of diverse function and subcellular location are lysine acetylated in Arabidopsis. Plant Physiol. 2011, 155, 1779–1790. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Oh, M.H.; Schwarz, E.M.; Larue, C.T.; Sivaguru, M.; Imai, B.S.; Yau, P.M.; Ort, D.R.; Huber, S.C. Lysine acetylation is a widespread protein modification for diverse proteins in Arabidopsis. Plant Physiol. 2011, 155, 1769–1778. [Google Scholar] [CrossRef] [PubMed]
- Koskela, M.M.; Brünje, A.; Ivanauskaite, A.; Grabsztunowicz, M.; Lassowskat, I.; Neumann, U.; Dinh, T.V.; Sindlinger, J.; Schwarzer, D.; Wirtz, M.; et al. Chloroplast Acetyltransferase NSI Is Required for State Transitions in Arabidopsis thaliana. Plant Cell 2018, 30, 1695–1709. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.T.; Jiang, J.F.; Liu, X.N.; Jiang, J.Z.; Sun, L.; Duan, W.; Li, R.M.; Wang, Y.; Lecourieux, D.; Liu, C.H.; et al. New insights into the heat responses of grape leaves via combined phosphoproteomic and acetylproteomic analyses. Hortic. Res. 2019, 6, 100. [Google Scholar] [CrossRef]
- Melo-Braga, M.N.; Verano-Braga, T.; León, I.R.; Antonacci, D.; Nogueira, F.C.; Thelen, J.J.; Larsen, M.R.; Palmisano, G. Modulation of protein phosphorylation, N-glycosylation and Lys-acetylation in grape (Vitis vinifera) mesocarp and exocarp owing to Lobesia botrana infection. Mol. Cell. Proteom. 2012, 11, 945–956. [Google Scholar] [CrossRef]
- Smith-Hammond, C.L.; Hoyos, E.; Miernyk, J.A. The pea seedling mitochondrial Nε-lysine acetylome. Mitochondrion 2014, 19 Pt B, 154–165. [Google Scholar] [CrossRef]
- Li, G.; Zheng, B.; Zhao, W.; Ren, T.; Zhang, X.; Ning, T.; Liu, P. Global analysis of lysine acetylation in soybean leaves. Sci. Rep. 2021, 11, 17858. [Google Scholar] [CrossRef] [PubMed]
- Marx, H.; Minogue, C.E.; Jayaraman, D.; Richards, A.L.; Kwiecien, N.W.; Siahpirani, A.F.; Rajasekar, S.; Maeda, J.; Garcia, K.; Del Valle-Echevarria, A.R.; et al. A proteomic atlas of the legume Medicago truncatula and its nitrogen-fixing endosymbiont Sinorhizobium meliloti. Nat. Biotechnol. 2016, 34, 1198–1205. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Song, J.; Miao, W.; Yang, B.; Zhang, Z.; Chen, W.; Tan, F.; Suo, H.; Dai, X.; Zou, X.; et al. Comprehensive Proteome and Lysine Acetylome Analysis Reveals the Widespread Involvement of Acetylation in Cold Resistance of Pepper (Capsicum annuum L.). Front. Plant Sci. 2021, 12, 730489. [Google Scholar] [CrossRef] [PubMed]
- He, D.; Wang, Q.; Li, M.; Damaris, R.N.; Yi, X.; Cheng, Z.; Yang, P. Global Proteome Analyses of Lysine Acetylation and Succinylation Reveal the Widespread Involvement of both Modification in Metabolism in the Embryo of Germinating Rice Seed. J. Proteome Res. 2016, 15, 879–890. [Google Scholar] [CrossRef] [PubMed]
- Xue, C.; Liu, S.; Chen, C.; Zhu, J.; Yang, X.; Zhou, Y.; Guo, R.; Liu, X.; Gong, Z. Global Proteome Analysis Links Lysine Acetylation to Diverse Functions in Oryza sativa. Proteomics 2018, 18, 1700036. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Finkemeier, I.; Guan, W.; Tossounian, M.A.; Wei, B.; Young, D.; Huang, J.; Messens, J.; Yang, X.; Zhu, J.; et al. Oxidative stress-triggered interactions between the succinyl- and acetyl-proteomes of rice leaves. Plant Cell Environ. 2018, 41, 1139–1153. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.R.; Yan, X.; Zhu, D.; Deng, X.; Wu, J.S.; Xia, J.; Yan, Y.M. Lysine acetylproteome profiling under water deficit reveals key acetylated proteins involved in wheat grain development and starch biosynthesis. J. Proteom. 2018, 185, 8–24. [Google Scholar] [CrossRef] [PubMed]
- Walley, J.W.; Shen, Z.; McReynolds, M.R.; Schmelz, E.A.; Briggs, S.P. Fungal-induced protein hyperacetylation in maize identified by acetylome profiling. Proc. Natl. Acad. Sci. USA 2018, 115, 210–215. [Google Scholar] [CrossRef]
- Jiang, J.; Gai, Z.; Wang, Y.; Fan, K.; Sun, L.; Wang, H.; Ding, Z. Comprehensive proteome analyses of lysine acetylation in tea leaves by sensing nitrogen nutrition. BMC Genom. 2018, 19, 840. [Google Scholar] [CrossRef]
- Xu, Y.X.; Chen, W.; Ma, C.L.; Shen, S.Y.; Zhou, Y.Y.; Zhou, L.Q.; Chen, L. Proteome and Acetyl-Proteome Profiling of Camellia sinensis cv. ‘Anjin Baicha’ during Periodic Albinism Reveals Alterations in Photosynthetic and Secondary Metabolite Biosynthetic Pathways. Front. Plant Sci. 2017, 8, 2104. [Google Scholar] [CrossRef]
- Liao, X.; Li, Y.; Hu, Z.; Lin, Y.; Zheng, B.; Ding, J. Poplar acetylome profiling reveals lysine acetylation dynamics in seasonal bud dormancy release. Plant Cell Environ. 2021, 44, 1830–1845. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Jing, D.; Kong, L.; Zhang, J.; OuYang, F.; Zhang, H.; Wang, J.; Zhang, S. Global Lysine Acetylome Analysis of Desiccated Somatic Embryos of Picea asperata. Front. Plant Sci. 2016, 7, 1927. [Google Scholar] [CrossRef] [PubMed]
- Hörtensteiner, S. Chlorophyll degradation during senescence. Annu. Rev. Plant Biol. 2006, 57, 55–77. [Google Scholar] [CrossRef] [PubMed]
- Keskitalo, J.; Bergquist, G.; Gardeström, P.; Jansson, S. A cellular timetable of autumn senescence. Plant Physiol. 2005, 139, 1635–1648. [Google Scholar] [CrossRef] [PubMed]
- Andersson, A.; Keskitalo, J.; Sjödin, A.; Bhalerao, R.; Sterky, F.; Wissel, K.; Tandre, K.; Aspeborg, H.; Moyle, R.; Ohmiya, Y.; et al. A transcriptional timetable of autumn senescence. Genome Biol. 2004, 5, R24. [Google Scholar] [CrossRef] [PubMed]
- Smeekens, S.; Ma, J.; Hanson, J.; Rolland, F. Sugar signals and molecular networks controlling plant growth. Curr. Opin. Plant Biol. 2010, 13, 274–279. [Google Scholar] [CrossRef] [PubMed]
- Quirino, B.F.; Noh, Y.S.; Himelblau, E.; Amasino, R.M. Molecular aspects of leaf senescence. Trends Plant Sci. 2000, 5, 278–282. [Google Scholar] [CrossRef] [PubMed]
- Kanojia, A.; Gupta, S.; Benina, M.; Fernie, A.R.; Mueller-Roeber, B.; Gechev, T.; Dijkwel, P.P. Developmentally controlled changes during Arabidopsis leaf development indicate causes for loss of stress tolerance with age. J. Exp. Bot. 2020, 71, 6340–6354. [Google Scholar] [CrossRef]
- Li, L.; Zhao, J.; Zhao, Y.; Lu, X.; Zhou, Z.; Zhao, C.; Xu, G. Comprehensive investigation of tobacco leaves during natural early senescence via multi-platform metabolomics analyses. Sci. Rep. 2016, 6, 37976. [Google Scholar] [CrossRef]
- Moschen, S.; Luoni, S.B.; Di Rienzo, J.A.; Caro, M.D.P.; Tohge, T.; Watanabe, M.; Hollmann, J.; González, S.; Rivarola, M.; García-García, F.; et al. Integrating transcriptomic and metabolomic analysis to understand natural leaf senescence in sunflower. Plant Biotechnol. J. 2016, 14, 719–734. [Google Scholar] [CrossRef]
- Breeze, E.; Harrison, E.; McHattie, S.; Hughes, L.; Hickman, R.; Hill, C.; Kiddle, S.; Kim, Y.S.; Penfold, C.A.; Jenkins, D.; et al. High-resolution temporal profiling of transcripts during Arabidopsis leaf senescence reveals a distinct chronology of processes and regulation. Plant Cell 2011, 23, 873–894. [Google Scholar] [CrossRef] [PubMed]
- Woo, H.R.; Koo, H.J.; Kim, J.; Jeong, H.; Yang, J.O.; Lee, I.H.; Jun, J.H.; Choi, S.H.; Park, S.J.; Kang, B.; et al. Programming of Plant Leaf Senescence with Temporal and Inter-Organellar Coordination of Transcriptome in Arabidopsis. Plant Physiol. 2016, 171, 452–467. [Google Scholar] [CrossRef] [PubMed]
- Uhrig, R.G.; Schläpfer, P.; Roschitzki, B.; Hirsch-Hoffmann, M.; Gruissem, W. Diurnal changes in concerted plant protein phosphorylation and acetylation in Arabidopsis organs and seedlings. Plant J. 2019, 99, 176–194. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.; Lv, Y.; Mujahid, H.; Edelmann, M.J.; Zhao, H.; Peng, X.; Peng, Z. Proteome-wide lysine acetylation identification in developing rice (Oryza sativa) seeds and protein co-modification by acetylation, succinylation, ubiquitination, and phosphorylation. Biochim. Biophys. Acta Proteins Proteom. 2018, 1866, 451–463. [Google Scholar] [CrossRef]
- Rohde, A.; Prinsen, E.; De Rycke, R.; Engler, G.; Van Montagu, M.; Boerjan, W. PtABI3 impinges on the growth and differentiation of embryonic leaves during bud set in poplar. Plant Cell 2002, 14, 1885–1901. [Google Scholar] [CrossRef]
- Lichtenthaler, H.K. Chlorophylls and carotenoids: Pigments of photosynthetic biomembranes. Methods Enzymol. 1987, 148C, 350–382. [Google Scholar]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, X.; Shan, J.; Wang, J.; Zhang, Y.; Yang, F.; Liu, B.; Zhang, L.; Li, G.; Wang, R. Comprehensive Proteome and Acetylome Analysis of Needle Senescence in Larix gmelinii. Int. J. Mol. Sci. 2024, 25, 6824. https://doi.org/10.3390/ijms25136824
Zhang X, Shan J, Wang J, Zhang Y, Yang F, Liu B, Zhang L, Li G, Wang R. Comprehensive Proteome and Acetylome Analysis of Needle Senescence in Larix gmelinii. International Journal of Molecular Sciences. 2024; 25(13):6824. https://doi.org/10.3390/ijms25136824
Chicago/Turabian StyleZhang, Xuting, Jinyuan Shan, Jiaxiu Wang, Yanxia Zhang, Feiyun Yang, Bin Liu, Lifeng Zhang, Guojing Li, and Ruigang Wang. 2024. "Comprehensive Proteome and Acetylome Analysis of Needle Senescence in Larix gmelinii" International Journal of Molecular Sciences 25, no. 13: 6824. https://doi.org/10.3390/ijms25136824