
The Role of the JAK–STAT Pathway in Childhood B-Cell Acute Lymphoblastic Leukemia
 , , ,
, , ,
Abstract
:1. Introduction
2. Background
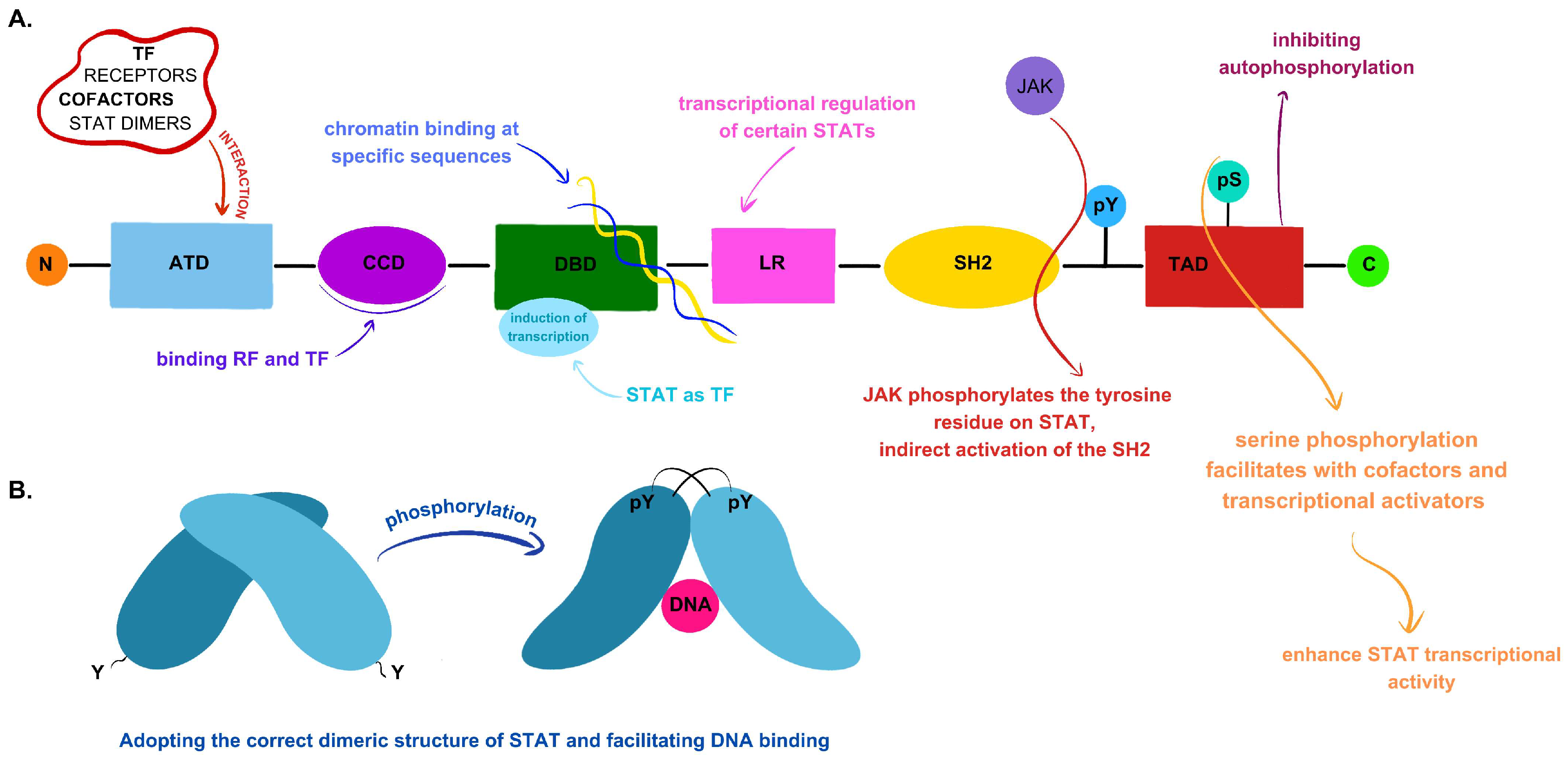
2.1. JAK–STAT Signaling Pathway Components and Their Primary Functions
2.2. The Regulation of the JAK–STAT Signaling Pathway
2.2.1. Signaling Activation via Cytokines
2.2.2. Negative Regulation of the JAK–STAT Pathway
3. Selected Genes Associated with Disorders of the JAK/STAT Pathway
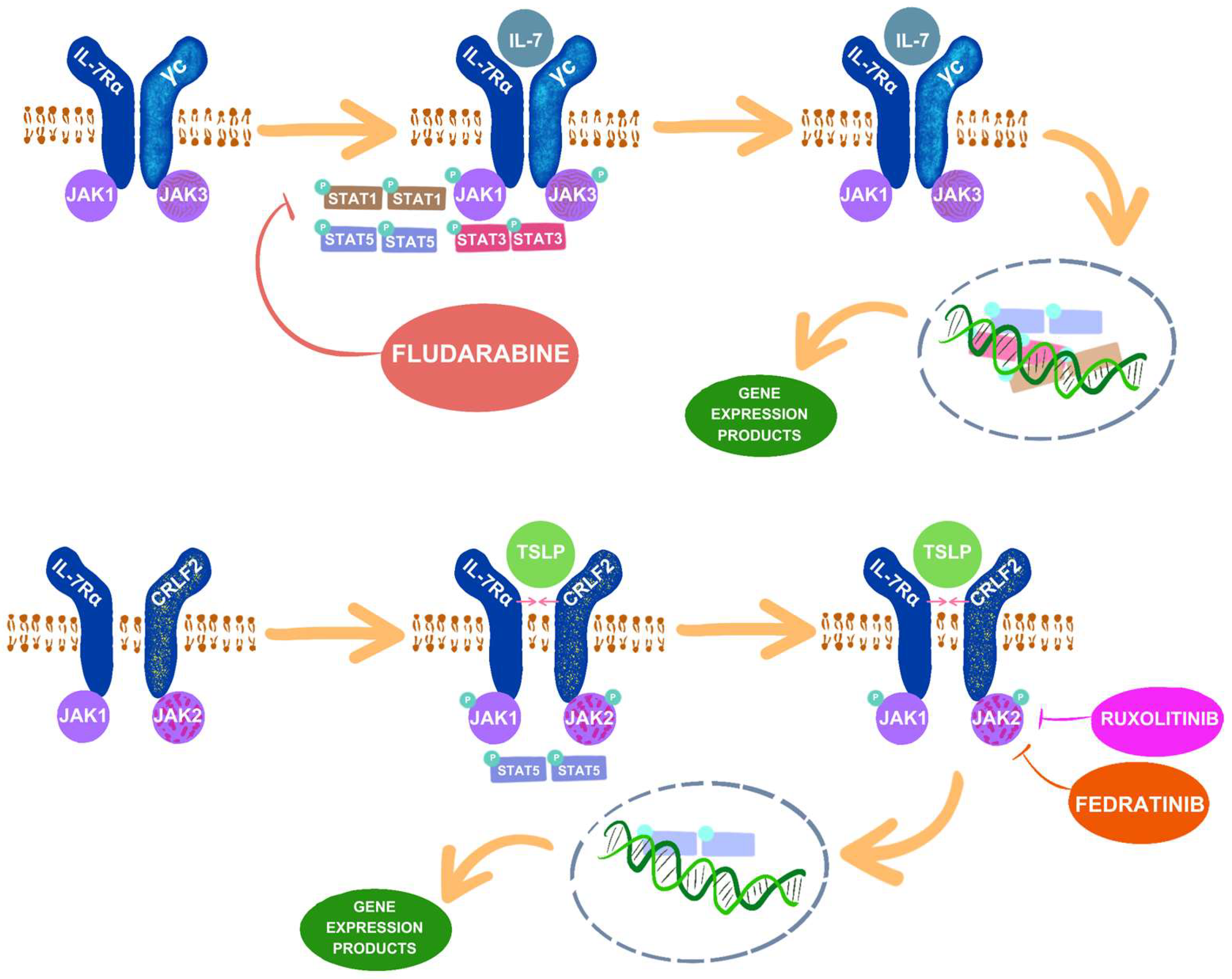
3.1. IL-7 and IL-7R
3.2. TLSP and CRLF2
3.2.1. Enhancement of CRLF2 Function
3.2.2. Clinical Significance of CRLF2 Mutations
3.3. JAK1 and JAK2
Clinical Significance of JAK2 Mutations
3.4. BCR::ABL1 Fusion
3.5. ETV6::RUNX Fusion
3.6. KMT2A
3.7. PAX5::JAK2
4. JAK/STAT Inhibitors as Therapeutic Targets
4.1. Ruxolitinib
4.2. Fedratinib
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Huang, J.; Chan, S.C.; Ngai, C.H.; Lok, V.; Zhang, L.; Lucero-Prisno, D.E., III; Xu, W.; Zheng, Z.-J.; Elcarte, E.; Withers, M.; et al. Global Incidence, Mortality and Temporal Trends of Cancer in Children: A Joinpoint Regression Analysis. Cancer Med. 2023, 12, 1903–1911. [Google Scholar] [CrossRef] [PubMed]
- Estimated Number of Prevalent Cases (5-Year), Both Sexes, Age [0–19], in 2022. Available online: https://gco.iarc.fr/today/en/dataviz/pie-prevalence?populations=900&mode=cancer&prev_time=5&types=2&group_populations=1&multiple_populations=0&age_end=3 (accessed on 4 June 2024).
- Onyije, F.M.; Olsson, A.; Baaken, D.; Erdmann, F.; Stanulla, M.; Wollschläger, D.; Schüz, J. Environmental Risk Factors for Childhood Acute Lymphoblastic Leukemia: An Umbrella Review. Cancers 2022, 14, 382. [Google Scholar] [CrossRef] [PubMed]
- Al Shamahy, H.A. Prevalence of Different Types of Leukemia and Associated Factors among Children with Leukemia in Children’s Cancer Units at Al-Kuwait Hospital, Sana’a City: A Cross-Sectional Study. Glob. J. Pediatr. Neonatal. Care 2021, 3, 000569. [Google Scholar] [CrossRef]
- Lejman, M.; Chałupnik, A.; Chilimoniuk, Z.; Dobosz, M. Genetic Biomarkers and Their Clinical Implications in B-Cell Acute Lymphoblastic Leukemia in Children. Int. J. Mol. Sci. 2022, 23, 2755. [Google Scholar] [CrossRef] [PubMed]
- Tebbi, C.K. Etiology of Acute Leukemia: A Review. Cancers 2021, 13, 2256. [Google Scholar] [CrossRef] [PubMed]
- Malard, F.; Mohty, M. Acute Lymphoblastic Leukaemia. Lancet 2020, 395, 1146–1162. [Google Scholar] [CrossRef] [PubMed]
- Agashe, R.P.; Lippman, S.M.; Kurzrock, R. JAK: Not Just Another Kinase. Mol. Cancer Ther. 2022, 21, 1757–1764. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Li, J.; Fu, M.; Zhao, X.; Wang, W. The JAK/STAT Signaling Pathway: From Bench to Clinic. Signal Transduct. Target. Ther. 2021, 6, 402. [Google Scholar] [CrossRef] [PubMed]
- Valle-Mendiola, A.; Gutiérrez-Hoya, A.; Soto-Cruz, I. JAK/STAT Signaling and Cervical Cancer: From the Cell Surface to the Nucleus. Genes 2023, 14, 1141. [Google Scholar] [CrossRef]
- Imada, K.; Leonard, W.J. The Jak-STAT Pathway. Mol. Immunol. 2000, 37, 1–11. [Google Scholar] [CrossRef]
- Xin, P.; Xu, X.; Deng, C.; Liu, S.; Wang, Y.; Zhou, X.; Ma, H.; Wei, D.; Sun, S. The Role of JAK/STAT Signaling Pathway and Its Inhibitors in Diseases. Int. Immunopharmacol. 2020, 80, 106210. [Google Scholar] [CrossRef] [PubMed]
- Liongue, C.; O’Sullivan, L.A.; Trengove, M.C.; Ward, A.C. Evolution of JAK-STAT Pathway Components: Mechanisms and Role in Immune System Development. PLoS ONE 2012, 7, e0032777. [Google Scholar] [CrossRef] [PubMed]
- Yamaoka, K.; Saharinen, P.; Pesu, M.; Holt, V.E.T.; Silvennoinen, O.; O’Shea, J.J. The Janus Kinases (Jaks). Genome Biol. 2004, 5, 253. [Google Scholar] [CrossRef] [PubMed]
- Rane, S.G.; Reddy, E.P. Janus Kinases: Components of Multiple Signaling Pathways. Oncogene 2000, 19, 5662–5679. [Google Scholar] [CrossRef] [PubMed]
- JAK1 Janus Kinase 1 [Homo Sapiens (Human)]. Available online: https://www.ncbi.nlm.nih.gov/gene/3716 (accessed on 4 June 2024).
- JAK2 Janus Kinase 2 [Homo Sapiens (Human)]. Available online: https://www.ncbi.nlm.nih.gov/gene/3717 (accessed on 4 June 2024).
- JAK3 Janus Kinase 3 [Homo Sapiens (Human)]. Available online: https://www.ncbi.nlm.nih.gov/gene/3718 (accessed on 4 June 2024).
- TYK2 Tyrosine Kinase 2 [Homo Sapiens (Human)]. Available online: https://www.ncbi.nlm.nih.gov/gene/7297 (accessed on 4 June 2024).
- Morris, R.; Kershaw, N.J.; Babon, J.J. The Molecular Details of Cytokine Signaling via the JAK/STAT Pathway. Protein Sci. 2018, 27, 1984–2009. [Google Scholar] [CrossRef]
- Ferrao, R.; Lupardus, P.J. The Janus Kinase (JAK) FERM and SH2 Domains: Bringing Specificity to JAK–Receptor Interactions. Front. Endocrinol. 2017, 8, 71. [Google Scholar] [CrossRef] [PubMed]
- Hammarén, H.M.; Virtanen, A.T.; Raivola, J.; Silvennoinen, O. The Regulation of JAKs in Cytokine Signaling and Its Breakdown in Disease. Cytokine 2019, 118, 48–63. [Google Scholar] [CrossRef]
- Raivola, J.; Haikarainen, T.; Abraham, B.G.; Silvennoinen, O. Janus Kinases in Leukemia. Cancers 2021, 13, 800. [Google Scholar] [CrossRef]
- Rah, B.; Rather, R.A.; Bhat, G.R.; Baba, A.B.; Mushtaq, I.; Farooq, M.; Yousuf, T.; Dar, S.B.; Parveen, S.; Hassan, R.; et al. JAK/STAT Signaling: Molecular Targets, Therapeutic Opportunities, and Limitations of Targeted Inhibitions in Solid Malignancies. Front. Pharmacol. 2022, 13, 821344. [Google Scholar] [CrossRef]
- Standing, D.; Feess, E.; Kodiyalam, S.; Kuehn, M.; Hamel, Z.; Johnson, J.; Thomas, S.M.; Anant, S. The Role of STATs in Ovarian Cancer: Exploring Their Potential for Therapy. Cancers 2023, 15, 2485. [Google Scholar] [CrossRef]
- Liu, J.; Wang, F.; Luo, F. The Role of JAK/STAT Pathway in Fibrotic Diseases: Molecular and Cellular Mechanisms. Biomolecules 2023, 13, 119. [Google Scholar] [CrossRef] [PubMed]
- Verhoeven, Y.; Tilborghs, S.; Jacobs, J.; De Waele, J.; Quatannens, D.; Deben, C.; Prenen, H.; Pauwels, P.; Trinh, X.B.; Wouters, A.; et al. The Potential and Controversy of Targeting STAT Family Members in Cancer. Semin. Cancer Biol. 2020, 60, 41–56. [Google Scholar] [CrossRef] [PubMed]
- Leonard, W.J. Role of Jak Kinases and STATs in Cytokine Signal Transduction. Int. J. Hematol. 2001, 73, 271–277. [Google Scholar] [CrossRef] [PubMed]
- Chou, W.-C.; Levy, D.E.; Lee, C.-K. STAT3 Positively Regulates an Early Step in B-Cell Development. Blood 2006, 108, 3005–3011. [Google Scholar] [CrossRef] [PubMed]
- Corfe, S.A.; Paige, C.J. The Many Roles of IL-7 in B Cell Development; Mediator of Survival, Proliferation and Differentiation. Semin. Immunol. 2012, 24, 198–208. [Google Scholar] [CrossRef]
- Chen, D.; Tang, T.-X.; Deng, H.; Yang, X.-P.; Tang, Z.-H. Interleukin-7 Biology and Its Effects on Immune Cells: Mediator of Generation, Differentiation, Survival, and Homeostasis. Front. Immunol. 2021, 12, 747324. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Wang, X. Role of IL-9 and STATs in Hematological Malignancies (Review). Oncol. Lett. 2014, 7, 602–610. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; van Besouw, N.M.; Shi, Y.; Hoogduijn, M.J.; Wang, L.; Baan, C.C. The Biological Effects of IL-21 Signaling on B-Cell-Mediated Responses in Organ Transplantation. Front. Immunol. 2016, 7, 319. [Google Scholar] [CrossRef]
- Yu, X.; Li, H.; Ren, X. Signaling Cascades Initiated by TSLP-Mediated Signals in Different Cell Types. Cell Immunol. 2012, 279, 174–179. [Google Scholar] [CrossRef]
- Lu, H.; Wu, X.; Peng, Y.; Sun, R.; Nie, Y.; Li, J.; Wang, M.; Luo, Y.; Peng, L.; Fei, Y.; et al. TSLP Promoting B Cell Proliferation and Polarizing Follicular Helper T Cell as a Therapeutic Target in IgG4-Related Disease. J. Transl. Med. 2022, 20, 414. [Google Scholar] [CrossRef]
- Horikawa, K.; Takatsu, K. Interleukin-5 Regulates Genes Involved in B-cell Terminal Maturation. Immunology 2006, 118, 497–508. [Google Scholar] [CrossRef] [PubMed]
- Kopalli, S.R.; Annamneedi, V.P.; Koppula, S. Potential Natural Biomolecules Targeting JAK/STAT/SOCS Signaling in the Management of Atopic Dermatitis. Molecules 2022, 27, 4660. [Google Scholar] [CrossRef]
- Liongue, C.; Ward, A.C. Evolution of the JAK-STAT Pathway. Jak-Stat 2013, 2, e22756. [Google Scholar] [CrossRef]
- Croker, B.A.; Kiu, H.; Nicholson, S.E. SOCS Regulation of the JAK/STAT Signalling Pathway. Semin. Cell Dev. Biol. 2008, 19, 414–422. [Google Scholar] [CrossRef] [PubMed]
- Cooney, R.N. Suppressors of Cytokine Signaling (SOCS): Inhibitors of the JAK/STAT Pathway. Shock 2002, 17, 83–90. [Google Scholar] [CrossRef]
- Seif, F.; Khoshmirsafa, M.; Aazami, H.; Mohsenzadegan, M.; Sedighi, G.; Bahar, M. The Role of JAK-STAT Signaling Pathway and Its Regulators in the Fate of T Helper Cells. Cell Commun. Signal. 2017, 15, 23. [Google Scholar] [CrossRef] [PubMed]
- Bang, I.S. JAK/STAT Signaling in Insect Innate Immunity. Entomol. Res. 2019, 49, 339–353. [Google Scholar] [CrossRef]
- Behl, T.; Gupta, A.; Sehgal, A.; Albarrati, A.; Albratty, M.; Meraya, A.M.; Najmi, A.; Bhatia, S.; Bungau, S. Exploring Protein Tyrosine Phosphatases (PTP) and PTP-1B Inhibitors in Management of Diabetes Mellitus. Biomed. Pharmacother. 2022, 153, 113405. [Google Scholar] [CrossRef]
- Welner, R.S.; Pelayo, R.; Kincade, P.W. Evolving views on the genealogy of B cells. Nat. Rev. Immunol. 2008, 8, 95–106. [Google Scholar] [CrossRef]
- Malin, S.; McManus, S.; Busslinger, M. STAT5 in B cell development and leukemia. Curr. Opin. Immunol. 2010, 22, 168–176. [Google Scholar] [CrossRef]
- Mackeh, R.; El Bsat, Y.; Elmi, A.; Bibawi, H.; Karim, M.Y.; Hassan, A.; Lo, B. Novel Synonymous Variant in IL7R Causes Preferential Expression of the Soluble Isoform. J. Clin. Immunol. 2024, 44, 96. [Google Scholar] [CrossRef] [PubMed]
- Campos, L.W.; Pissinato, L.G.; Yunes, J.A. Deleterious and Oncogenic Mutations in the IL7RA. Cancers 2019, 11, 1952. [Google Scholar] [CrossRef]
- Kovanen, P.E.; Leonard, W.J. Cytokines and immunodeficiency diseases: Critical roles of the gamma(c)-dependent cytokines interleukins 2, 4, 7, 9, 15, and 21, and their signaling pathways. Immunol. Rev. 2004, 202, 67–83. [Google Scholar] [CrossRef] [PubMed]
- Lodewijckx, I.; Cools, J. Deregulation of the Interleukin-7 Signaling Pathway in Lymphoid Malignancies. Pharmaceuticals 2021, 14, 443. [Google Scholar] [CrossRef] [PubMed]
- Hennighausen, L.; Robinson, G.W. Interpretation of cytokine signaling through the transcription factors STAT5A and STAT5B. Genes. Dev. 2008, 22, 711–721. [Google Scholar] [CrossRef]
- Wang, C.; Kong, L.; Kim, S.; Lee, S.; Oh, S.; Jo, S.; Jang, I.; Kim, T.D. The Role of IL-7 and IL-7R in Cancer Pathophysiology and Immunotherapy. Int. J. Mol. Sci. 2022, 23, 10412. [Google Scholar] [CrossRef]
- Nutt, S.L.; Kee, B.L. The transcriptional regulation of B cell lineage commitment. Immunity 2007, 26, 715–725. [Google Scholar] [CrossRef] [PubMed]
- Shochat, C.; Tal, N.; Bandapalli, O.R.; Palmi, C.; Ganmore, I.; te Kronnie, G.; Cario, G.; Cazzaniga, G.; Kulozik, A.E.; Stanulla, M.; et al. Gain-of-function mutations in interleukin-7 receptor-α (IL7R) in childhood acute lymphoblastic leukemias. J. Exp. Med. 2011, 208, 901–908. [Google Scholar] [CrossRef]
- Thomas, K.R.; Allenspach, E.J.; Camp, N.D.; Wray-Dutra, M.N.; Khim, S.; Zielinska-Kwiatkowska, A.; Timms, A.E.; Loftus, J.P.; Liggitt, H.D.; Georgopoulos, K.; et al. Activated interleukin-7 receptor signaling drives B-cell acute lymphoblastic leukemia in mice. Leukemia 2022, 36, 42–57. [Google Scholar] [CrossRef]
- Roberts, K.G.; Li, Y.; Payne-Turner, D.; Harvey, R.C.; Yang, Y.L.; Pei, D.; McCastlain, K.; Ding, L.; Lu, C.; Song, G.; et al. Targetable kinase-activating lesions in Ph-like acute lymphoblastic leukemia. N. Engl. J. Med. 2014, 371, 1005–1015. [Google Scholar] [CrossRef]
- Geron, I.; Savino, A.M.; Fishman, H.; Tal, N.; Brown, J.; Turati, V.A.; James, C.; Sarno, J.; Hameiri-Grossman, M.; Lee, Y.N.; et al. An instructive role for Interleukin-7 receptor α in the development of human B-cell precursor leukemia. Nat. Commun. 2022, 13, 659. [Google Scholar] [CrossRef] [PubMed]
- Zhong, J.; Sharma, J.; Raju, R.; Palapetta, S.M.; Prasad, T.S.; Huang, T.C.; Yoda, A.; Tyner, J.W.; van Bodegom, D.; Weinstock, D.M.; et al. TSLP signaling pathway map: A platform for analysis of TSLP-mediated signaling. Database 2014, 2014, bau007. [Google Scholar] [CrossRef] [PubMed]
- Harvey, R.C.; Mullighan, C.G.; Chen, I.M.; Wharton, W.; Mikhail, F.M.; Carroll, A.J.; Kang, H.; Liu, W.; Dobbin, K.K.; Smith, M.A.; et al. Rearrangement of CRLF2 is associated with mutation of JAK kinases, alteration of IKZF1, Hispanic/Latino ethnicity, and a poor outcome in pediatric B-progenitor acute lymphoblastic leukemia. Blood 2010, 115, 5312–5321. [Google Scholar] [CrossRef] [PubMed]
- Isaksen, D.E.; Baumann, H.; Trobridge, P.A.; Farr, A.G.; Levin, S.D.; Ziegler, S.F. Requirement for stat5 in thymic stromal lymphopoietin-mediated signal transduction. J. Immunol. 1999, 163, 5971–5977. [Google Scholar] [CrossRef] [PubMed]
- Wohlmann, A.; Sebastian, K.; Borowski, A.; Krause, S.; Friedrich, K. Signal transduction by the atopy-associated human thymic stromal lymphopoietin (TSLP) receptor depends on Janus kinase function. Biol. Chem. 2010, 391, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Dou, H.; Chen, X.; Huang, Y.; Su, Y.; Lu, L.; Yu, J.; Yin, Y.; Bao, L. Prognostic significance of P2RY8-CRLF2 and CRLF2 overexpression may vary across risk subgroups of childhood B-cell acute lymphoblastic leukemia. Genes. Chromosomes Cancer 2017, 56, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Levin, S.D.; Koelling, R.M.; Friend, S.L.; Isaksen, D.E.; Ziegler, S.F.; Perlmutter, R.M.; Farr, A.G. Thymic stromal lymphopoietin: A cytokine that promotes the development of IgM+ B cells in vitro and signals via a novel mechanism. J. Immunol. 1999, 162, 677–683. [Google Scholar] [CrossRef] [PubMed]
- Astrakhan, A.; Omori, M.; Nguyen, T.; Becker-Herman, S.; Iseki, M.; Aye, T.; Hudkins, K.; Dooley, J.; Farr, A.; Alpers, C.E.; et al. Local increase in thymic stromal lymphopoietin induces systemic alterations in B cell development. Nat. Immunol. 2007, 8, 522–531. [Google Scholar] [CrossRef] [PubMed]
- Russell, L.J.; Capasso, M.; Vater, I.; Akasaka, T.; Bernard, O.A.; Calasanz, M.J.; Chandrasekaran, T.; Chapiro, E.; Gesk, S.; Griffiths, M.; et al. Deregulated expression of cytokine receptor gene, CRLF2, is involved in lymphoid transformation in B-cell precursor acute lymphoblastic leukemia. Blood 2009, 114, 2688–2698. [Google Scholar] [CrossRef]
- Scott, L.M. Lymphoid malignancies: Another face to the Janus kinases. Blood Rev. 2013, 27, 63–70. [Google Scholar] [CrossRef]
- Schmäh, J.; Fedders, B.; Panzer-Grümayer, R.; Fischer, S.; Zimmermann, M.; Dagdan, E.; Bens, S.; Schewe, D.; Moericke, A.; Alten, J.; et al. Molecular characterization of acute lymphoblastic leukemia with high CRLF2 gene expression in childhood. Pediatr. Blood Cancer 2017, 64, e26359. [Google Scholar] [CrossRef]
- Yano, M.; Imamura, T.; Asai, D.; Moriya-Saito, A.; Suenobu, S.; Hasegawa, D.; Deguchi, T.; Hashii, Y.; Kawasaki, H.; Hori, H.; et al. An overall characterization of pediatric acute lymphoblastic leukemia with CRLF2 overexpression. Genes. Chromosomes Cancer 2014, 53, 815–823. [Google Scholar] [CrossRef] [PubMed]
- Hassan, N.M.; Abdellateif, M.S.; Radwan, E.M.; Hameed, S.A.; Desouky, E.D.E.; Kamel, M.M.; Gameel, A.M. Prognostic significance of CRLF2 overexpression and JAK2 mutation in Egyptian pediatric patients with B-precursor acute lymphoblastic leukemia. Clin. Lymphoma Myeloma Leuk. 2022, 22, e376–e385. [Google Scholar] [CrossRef] [PubMed]
- Tasian, S.K.; Doral, M.Y.; Borowitz, M.J.; Wood, B.L.; Chen, I.M.; Harvey, R.C.; Gastier-Foster, J.M.; Willman, C.L.; Hunger, S.P.; Mullighan, C.G.; et al. Aberrant STAT5 and PI3K/mTOR pathway signaling occurs in human CRLF2-rearranged B-precursor acute lymphoblastic leukemia. Blood 2012, 120, 833–842. [Google Scholar] [CrossRef] [PubMed]
- Juárez-Velázquez, M.D.R.; Moreno-Lorenzana, D.L.; Martínez Anaya, D.A.; Hernández Monterde, E.A.; Aguilar-Hernández, M.M.; Reyes-León, A.; Chávez-González, M.A.; López Santiago, N.; Zapata Tarrés, M.; Juárez Villegas, L.; et al. High occurrence of CRLF2 abnormalities in Mexican children with B-cell acute lymphoblastic leukemia. Cytokine 2022, 155, 155896. [Google Scholar] [CrossRef]
- Ghazavi, F.; Lammens, T.; Van Roy, N.; Poppe, B.; Speleman, F.; Benoit, Y.; Van Vlierberghe, P.; De Moerloose, B. Molecular basis and clinical significance of genetic aberrations in B-cell precursor acute lymphoblastic leukemia. Exp. Hematol. 2015, 43, 640–653. [Google Scholar] [CrossRef]
- Hunger, S.P.; Mullighan, C.G. Acute Lymphoblastic Leukemia in Children. N. Engl. J. Med. 2015, 373, 1541–1552. [Google Scholar] [CrossRef]
- Cui, L.; Gao, C.; Wang, C.J.; Zhao, X.X.; Li, W.J.; Li, Z.G.; Zheng, H.Y.; Wang, T.Y.; Zhang, R.D. Combined analysis of IKZF1 deletions and CRLF2 expression on prognostic impact in pediatric B-cell precursor acute lymphoblastic leukemia. Leuk. Lymphoma 2021, 62, 410–418. [Google Scholar] [CrossRef]
- Chen, I.M.; Harvey, R.C.; Mullighan, C.G.; Gastier-Foster, J.; Wharton, W.; Kang, H.; Borowitz, M.J.; Camitta, B.M.; Carroll, A.J.; Devidas, M.; et al. Outcome modeling with CRLF2, IKZF1, JAK, and minimal residual disease in pediatric acute lymphoblastic leukemia: A Children’s Oncology Group study. Blood 2012, 119, 3512–3522. [Google Scholar] [CrossRef]
- Palmi, C.; Vendramini, E.; Silvestri, D.; Longinotti, G.; Frison, D.; Cario, G.; Shochat, C.; Stanulla, M.; Rossi, V.; Di Meglio, A.M.; et al. Poor prognosis for P2RY8-CRLF2 fusion but not for CRLF2 over-expression in children with intermediate risk B-cell precursor acute lymphoblastic leukemia. Leukemia 2012, 26, 2245–2253. [Google Scholar] [CrossRef]
- Pastorczak, A.; Sedek, L.; Braun, M.; Madzio, J.; Sonsala, A.; Twardoch, M.; Fendler, W.; Nebral, K.; Taha, J.; Bielska, M.; et al. Surface expression of Cytokine Receptor-Like Factor 2 increases risk of relapse in pediatric acute lymphoblastic leukemia patients harboring IKZF1 deletions. Oncotarget 2018, 9, 25971–25982. [Google Scholar] [CrossRef]
- Downes, C.E.; McClure, B.J.; McDougal, D.P.; Heatley, S.L.; Bruning, J.B.; Thomas, D.; Yeung, D.T.; White, D.L. JAK2 Alterations in Acute Lymphoblastic Leukemia: Molecular Insights for Superior Precision Medicine Strategies. Front. Cell Dev. Biol. 2022, 10, 942053. [Google Scholar] [CrossRef]
- Hubbard, S.R. Mechanistic Insights into Regulation of JAK2 Tyrosine Kinase. Front. Endocrinol. 2018, 8, 361. [Google Scholar] [CrossRef] [PubMed]
- Hammarén, H.M.; Virtanen, A.T.; Abraham, B.G.; Peussa, H.; Hubbard, S.R.; Silvennoinen, O. Janus kinase 2 activation mechanisms revealed by analysis of suppressing mutations. J. Allergy Clin. Immunol. 2019, 143, 1549–1559. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.K.; Knight, D.A.; Jones, L.R.; Vervoort, S.; Ng, A.P.; Seymour, J.F.; Bradner, J.E.; Waibel, M.; Kats, L.; Johnstone, R.W. JAK2 is dispensable for maintenance of JAK2 mutant B-cell acute lymphoblastic leukemias. Genes Dev. 2018, 32, 849–864. [Google Scholar] [CrossRef]
- Francis, O.L.; Milford, T.A.; Martinez, S.R.; Baez, I.; Coats, J.S.; Mayagoitia, K.; Concepcion, K.R.; Ginelli, E.; Beldiman, C.; Benitez, A.; et al. A novel xenograft model to study the role of TSLP-induced CRLF2 signals in normal and malignant human B lymphopoiesis. Haematologica 2016, 101, 417–426. [Google Scholar] [CrossRef]
- Akin-Bali, D.F.; Doganay Erdogan, B.; Aslar Oner, D.; Mahmud, A.; Tasdelen, S.; Kurekci, E.; Akar, N.; Ozdag Sevgili, H. Genetic Profiling of Pediatric Patients with B-Cell Precursor Acute Lymphoblastic Leukemia. J. Pediatr. Genet. 2022, 12, 288–300. [Google Scholar] [CrossRef] [PubMed]
- Peroni, E.; Gottardi, M.; D’Antona, L.; Randi, M.L.; Rosato, A.; Coltro, G. Hematologic Neoplasms Associated with Down Syndrome: Cellular and Molecular Heterogeneity of the Diseases. Int. J. Mol. Sci. 2023, 24, 15325. [Google Scholar] [CrossRef] [PubMed]
- Gil, J.V.; Miralles, A.; de Las Heras, S.; Such, E.; Avetisyan, G.; Díaz-González, Á.; Santiago, M.; Fuentes, C.; Fernández, J.M.; Lloret, P.; et al. Comprehensive detection of CRLF2 alterations in acute lymphoblastic leukemia: A rapid and accurate novel approach. Front. Mol. Biosci. 2024, 11, 1362081. [Google Scholar] [CrossRef]
- Böhm, J.W.; Sia, K.C.S.; Jones, C.; Evans, K.; Mariana, A.; Pang, I.; Failes, T.; Zhong, L.; Mayoh, C.; Landman, R.; et al. Combination efficacy of ruxolitinib with standard-of-care drugs in CRLF2-rearranged Ph-like acute lymphoblastic leukemia. Leukemia 2021, 35, 3101–3112. [Google Scholar] [CrossRef] [PubMed]
- Al Hamad, M. Contribution of BCR-ABL molecular variants and leukemic stem cells in response and resistance to tyrosine kinase inhibitors: A review. F1000Research 2021, 10, 1288. [Google Scholar] [CrossRef] [PubMed]
- Komorowski, L.; Fidyt, K.; Patkowska, E.; Firczuk, M. Philadelphia Chromosome-Positive Leukemia in the Lymphoid Lineage-Similarities and Differences with the Myeloid Lineage and Specific Vulnerabilities. Int. J. Mol. Sci. 2020, 21, 5776. [Google Scholar] [CrossRef] [PubMed]
- NCBI National Center for Biotechnology Information. ABL1 ABL Proto-Oncogene 1, Non-Receptor Tyrosine Kinase [Homo Sapiens (Human)]—Gene. Available online: https://www.ncbi.nlm.nih.gov/gene/25 (accessed on 4 June 2024).
- NCBI National Center for Biotechnology Information. BCR Aktywator BCR RhoGEF i GTPazy [Homo Sapiens (Człowiek)]. Available online: https://www.ncbi.nlm.nih.gov/gene/613 (accessed on 5 May 2024).
- Wang, W.; Wang, L.; Zha, B. The roles of STAT6 in regulating B cell fate, activation, and function. Immunol. Lett. 2021, 233, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Cuesta-Domínguez, Á.; Ortega, M.; Ormazábal, C.; Santos-Roncero, M.; Galán-Díez, M.; Steegmann, J.L.; Figuera, Á.; Arranz, E.; Vizmanos, J.L.; Bueren, J.A.; et al. Transforming and tumorigenic activity of JAK2 by fusion to BCR: Molecular mechanisms of action of a novel BCR-JAK2 tyrosine-kinase. PLoS ONE 2012, 7, e32451. [Google Scholar] [CrossRef]
- Qin, R.; Wang, T.; He, W.; Wei, W.; Liu, S.; Gao, M.; Huang, Z. Jak2/STAT6/c-Myc pathway is vital to the pathogenicity of Philadelphia-positive acute lymphoblastic leukemia caused by P190BCR-ABL. Cell Commun. Signal. 2023, 21, 27. [Google Scholar] [CrossRef]
- Sundaresh, A.; Williams, O. Mechanism of ETV6-runx1 leukemia. In Advances in Experimental Medicine and Biology; Springer: Berlin/Heidelberg, Germany, 2017; pp. 201–216. [Google Scholar] [CrossRef]
- NCBI National Center for Biotechnology Information. ETV6 ETS Variant Transcription Factor 6 [Homo Sapiens (Human)]—Gene. Available online: https://www.ncbi.nlm.nih.gov/gene/2120 (accessed on 5 June 2024).
- NCBI National Center for Biotechnology Information. Runx1 RUNX Family Transcription Factor 1 [Homo Sapiens (Human)]—Gene. Available online: https://www.ncbi.nlm.nih.gov/gene/861 (accessed on 5 June 2024).
- Hiebert, S.W.; Sun, W.; Nathan Davis, J.; Golub, T.; Shurtleff, S.; Buijs, A.; Downing, J.R.; Grosveld, G.; Roussel, M.F.; Gary Gilliland, D.; et al. The T(12;21) Translocation Converts AML-1B from an Activator to a Repressor of Transcription. Mol. Cell. Biol. 1996, 16, 1349–1355. [Google Scholar] [CrossRef] [PubMed]
- McLean, T.; Ringold, S.; Neuberg, D.; Stegmaier, K.; Tantravahi, R.; Ritz, J.; Koeffler, H.; Takeuchi, S.; Janssen, J.; Seriu, T.; et al. Tel/AML-1 Dimerizes and Is Associated with a Favorable Outcome in Childhood Acute Lymphoblastic Leukemia. Blood 1996, 88, 4252–4258. [Google Scholar] [CrossRef]
- Inthal, A.; Krapf, G.; Beck, D.; Joas, R.; Kauer, M.O.; Orel, L.; Fuka, G.; Mann, G.; Panzer-Grümayer, E.R. Role of the Erythropoietin Receptor in ETV6/RUNX1-Positive Acute Lymphoblastic Leukemia. Clin. Cancer Res. 2008, 14, 7196–7204. [Google Scholar] [CrossRef]
- Mangolini, M.; de Boer, J.; Walf-Vorderwülbecke, V.; Pieters, R.; den Boer, M.L.; Williams, O. STAT3 Mediates Oncogenic Addiction to Tel-AML1 in T(12;21) Acute Lymphoblastic Leukemia. Blood 2013, 122, 542–549. [Google Scholar] [CrossRef]
- Shi, Y.; Zhang, Z.; Qu, X.; Zhu, X.; Zhao, L.; Wei, R.; Guo, Q.; Sun, L.; Yin, X.; Zhang, Y.; et al. Roles of STAT3 in Leukemia (Review). Int. J. Oncol. 2018, 53, 7–20. [Google Scholar] [CrossRef]
- Ma, L.; Zhou, M.; Wen, S.; Ni, C.; Jiang, L.; Fan, J.; Xia, L. Effects of STAT3 Silencing on Fate of Chronic Myelogenous Leukemia K562 Cells. Leuk. Lymphoma 2010, 51, 1326–1336. [Google Scholar] [CrossRef]
- Torrano, V.; Procter, J.; Cardus, P.; Greaves, M.; Ford, A.M. ETV6-RUNX1 Promotes Survival of Early B Lineage Progenitor Cells via a Dysregulated Erythropoietin Receptor. Blood 2011, 118, 4910–4918. [Google Scholar] [CrossRef] [PubMed]
- Tasian, S.K.; Loh, M.L.; Hunger, S.P. Philadelphia Chromosome–like Acute Lymphoblastic Leukemia. Blood 2017, 130, 2064–2072. [Google Scholar] [CrossRef] [PubMed]
- NCBI National Center for Biotechnology Information. EPOR Erythropoietin Receptor [Homo Sapiens (Human)]—Gene. Available online: https://www.ncbi.nlm.nih.gov/gene/2057 (accessed on 5 June 2024).
- NCBI National Center for Biotechnology Information. KMT2A Lysine Methyltransferase 2A [Homo Sapiens (Human)]—Gene. Available online: https://www.ncbi.nlm.nih.gov/gene/4297 (accessed on 5 June 2024).
- Castiglioni, S.; Di Fede, E.; Bernardelli, C.; Lettieri, A.; Parodi, C.; Grazioli, P.; Colombo, E.A.; Ancona, S.; Milani, D.; Ottaviano, E.; et al. KMT2A: Umbrella Gene for Multiple Diseases. Genes 2022, 13, 514. [Google Scholar] [CrossRef] [PubMed]
- El Chaer, F.; Keng, M.; Ballen, K.K. MLL-Rearranged Acute Lymphoblastic Leukemia. Curr. Hematol. Malig. Rep. 2020, 15, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Qiu, K.; Zhou, D.; Liao, X.; Huang, K.; Li, Y.; Xu, H.; Weng, W.; Xu, L.; Fang, J. Prognostic Value and Outcome for Acute Lymphocytic Leukemia in Children with MLL Rearrangement: A Case-Control Study. BMC Cancer 2022, 22, 1257. [Google Scholar] [CrossRef] [PubMed]
- Liao, X.-Y.; Zhou, D.-H.; Fang, J.-P.; Qiu, K.-Y. Ruxolitinib Inhibits the Proliferation and Induces the Apoptosis of MLL-R All Cells through Inactivating JAK/Stat Signaling Pathway. Transl. Pediatr. 2023, 12, 1088–1097. [Google Scholar] [CrossRef]
- NCBI National Center for Biotechnology Information. Pax5 Paired Box 5 [Homo Sapiens (Human)]—Gene. Available online: https://www.ncbi.nlm.nih.gov/gene/5079 (accessed on 5 June 2024).
- Vainchenker, W.; Constantinescu, S.N. JAK/STAT Signaling in Hematological Malignancies. Oncogene 2012, 32, 2601–2613. [Google Scholar] [CrossRef] [PubMed]
- Nebral, K.; Denk, D.; Attarbaschi, A.; König, M.; Mann, G.; Haas, O.A.; Strehl, S. Incidence and Diversity of PAX5 Fusion Genes in Childhood Acute Lymphoblastic Leukemia. Leukemia 2008, 23, 134–143. [Google Scholar] [CrossRef]
- Schinnerl, D.; Fortschegger, K.; Kauer, M.; Marchante, J.R.; Kofler, R.; Den Boer, M.L.; Strehl, S. The Role of the Janus-Faced Transcription Factor Pax5-JAK2 in Acute Lymphoblastic Leukemia. Blood 2015, 125, 1282–1291. [Google Scholar] [CrossRef]
- Liang, D.; Wang, Q.; Zhang, W.; Tang, H.; Song, C.; Yan, Z.; Liang, Y.; Wang, H. Jak/STAT in Leukemia: A Clinical Update. Mol. Cancer 2024, 23, 25. [Google Scholar] [CrossRef] [PubMed]
- Faderl, S.; Ferrajoli, A.; Harris, D.; Van, Q.; Kantarjian, H.M.; Estrov, Z. ATIPRIMOD Blocks Phosphorylation of Jak-STAT and Inhibits Proliferation of Acute Myeloid Leukemia (AML) Cells. Leuk. Res. 2007, 31, 91–95. [Google Scholar] [CrossRef]
- Shawky, A.M.; Almalki, F.A.; Abdalla, A.N.; Abdelazeem, A.H.; Gouda, A.M. A Comprehensive Overview of Globally Approved JAK Inhibitors. Pharmaceutics 2022, 14, 1001. [Google Scholar] [CrossRef] [PubMed]
- Becker, H.; Engelhardt, M.; von Bubnoff, N.; Wäsch, R. Ruxolitinib. Recent. Results Cancer Res. 2014, 201, 249–257. [Google Scholar] [PubMed]
- Appeldoorn, T.Y.; Munnink, T.H.; Morsink, L.M.; Hooge, M.N.; Touw, D.J. Pharmacokinetics and Pharmacodynamics of Ruxolitinib: A Review. Clin. Pharmacokinet. 2023, 62, 559–571. [Google Scholar] [CrossRef]
- Ajayi, S.; Becker, H.; Reinhardt, H.; Engelhardt, M.; Zeiser, R.; von Bubnoff, N.; Wäsch, R. Ruxolitinib. Small Mol. Hematol. 2018, 119–132. [Google Scholar] [CrossRef] [PubMed]
- Kołodrubiec, J.; Kozłowska, M.; Irga-Jaworska, N.; Sędek, Ł.; Pastorczak, A.; Trelińska, J.; Młynarski, W. Efficacy of Ruxolitinib in Acute Lymphoblastic Leukemia: A Systematic Review. Leuk. Res. 2022, 121, 106925. [Google Scholar] [CrossRef] [PubMed]
- Downes, C.E.; McClure, B.J.; Bruning, J.B.; Page, E.; Breen, J.; Rehn, J.; Yeung, D.T.; White, D.L. Acquired JAK2 Mutations Confer Resistance to JAK Inhibitors in Cell Models of Acute Lymphoblastic Leukemia. NPJ Precis. Oncol. 2021, 5, 75. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.A.; Odenike, O. The next Generation of JAK Inhibitors: An Update on Fedratinib, Momelotonib, and Pacritinib. Curr. Hematol. Malig. Rep. 2020, 15, 409–418. [Google Scholar] [CrossRef]
- Rinella, S.P.; Bell, H.C.; Turicek, D.P.; Shi, L.; Hoang, N.-M.; Rui, L.; Hess, N.J.; Capitini, C.M. Combinatorial fedratinib and Venetoclax treatment is effective on human B cell acute lymphoblastic leukemia with high FLT3 expression. bioRxiv 2023. [Google Scholar] [CrossRef]
- Poubel, C.P.; Mansur, M.B.; Boroni, M.; Emerenciano, M. FLT3 Overexpression in Acute Leukaemias: New Insights into the Search for Molecular Mechanisms. Biochim. Biophys. Acta (BBA)—Rev. Cancer 2019, 1872, 80–88. [Google Scholar] [CrossRef]
- Wallen, H.; Thompson, J.A.; Reilly, J.Z.; Rodmyre, R.M.; Cao, J.; Yee, C. Fludarabine Modulates Immune Response and Extends in Vivo Survival of Adoptively Transferred CD8 T Cells in Patients with Metastatic Melanoma. PLoS ONE 2009, 4, e4794. [Google Scholar] [CrossRef]
- Langenhorst, J.B.; Dorlo, T.P.; van Maarseveen, E.M.; Nierkens, S.; Kuball, J.; Boelens, J.J.; van Kesteren, C.; Huitema, A.D. Population Pharmacokinetics of Fludarabine in Children and Adults during Conditioning Prior to Allogeneic Hematopoietic Cell Transplantation. Clin. Pharmacokinet. 2018, 58, 627–637. [Google Scholar] [CrossRef]
- Hui, Z.; Tretiakova, M.; Zhang, Z.; Li, Y.; Wang, X.; Zhu, J.X.; Gao, Y.; Mai, W.; Furge, K.; Qian, C.-N.; et al. Radiosensitization by Inhibiting STAT1 in Renal Cell Carcinoma. Int. J. Radiat. Oncol. Biol. Phys. 2009, 73, 288–295. [Google Scholar] [CrossRef]
- Feng, Z.; Zheng, W.; Tang, Q.; Cheng, L.; Li, H.; Ni, W.; Pan, X. FLUDARABINE Inhibits STAT1-Mediated up-Regulation of Caspase-3 Expression in Dexamethasone-Induced Osteoblasts Apoptosis and Slows the Progression of Steroid-Induced Avascular Necrosis of the Femoral Head in Rats. Apoptosis 2017, 22, 1001–1012. [Google Scholar] [CrossRef]
- Dekker, L.; Calkoen, F.G.; Jiang, Y.; Blok, H.; Veldkamp, S.R.; De Koning, C.; Spoon, M.; Admiraal, R.; Hoogerbrugge, P.; Vormoor, B.; et al. Fludarabine Exposure Predicts Outcome after CD19 Car T-Cell Therapy in Children and Young Adults with Acute Leukemia. Blood Advances 2022, 6, 1969–1976. [Google Scholar] [CrossRef]
- Inaba, H.; Pui, C.-H. Immunotherapy in Pediatric Acute Lymphoblastic Leukemia. J. Cancer Immunol. 2020, 2. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Cytokine | JAKs | STATs | Functions | References |
|---|---|---|---|---|
| IL-7 | JAK1, JAK3 | STAT1, STAT3, STAT5a, STAT5b | B-cell progenitor growth, differentiation, survival, and proliferation | [9,15,20,28,29,30,31,32,33,34,35,36] |
| IL-9 | JAK1, JAK3 | STAT1, STAT3, STAT5a, STAT5b | Development and stimulation of B-cells | |
| IL-21 | JAK1, JAK3 | STAT3, STAT5a, STAT5b | Maturation and development of B-cells; production of memory B-cells from naive precursors | |
| IL-5 | JAK1, JAK2 | STAT1, STAT3, STAT5a, STAT5b | B-cell growth, differentiation, and survival | |
| TLSP | JAK1, JAK2 | STAT1, STAT3, STAT5a, STAT5b | B-cell proliferation and stimulation |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ziętara, K.J.; Wróblewska, K.; Zajączkowska, M.; Taczała, J.; Lejman, M. The Role of the JAK–STAT Pathway in Childhood B-Cell Acute Lymphoblastic Leukemia. Int. J. Mol. Sci. 2024, 25, 6844. https://doi.org/10.3390/ijms25136844
Ziętara KJ, Wróblewska K, Zajączkowska M, Taczała J, Lejman M. The Role of the JAK–STAT Pathway in Childhood B-Cell Acute Lymphoblastic Leukemia. International Journal of Molecular Sciences. 2024; 25(13):6844. https://doi.org/10.3390/ijms25136844
Chicago/Turabian StyleZiętara, Karolina Joanna, Kinga Wróblewska, Monika Zajączkowska, Joanna Taczała, and Monika Lejman. 2024. "The Role of the JAK–STAT Pathway in Childhood B-Cell Acute Lymphoblastic Leukemia" International Journal of Molecular Sciences 25, no. 13: 6844. https://doi.org/10.3390/ijms25136844