
GCPII Inhibition Promotes Remyelination after Peripheral Nerve Injury in Aged Mice

 , , and
, , and
Abstract
:1. Introduction
2. Results
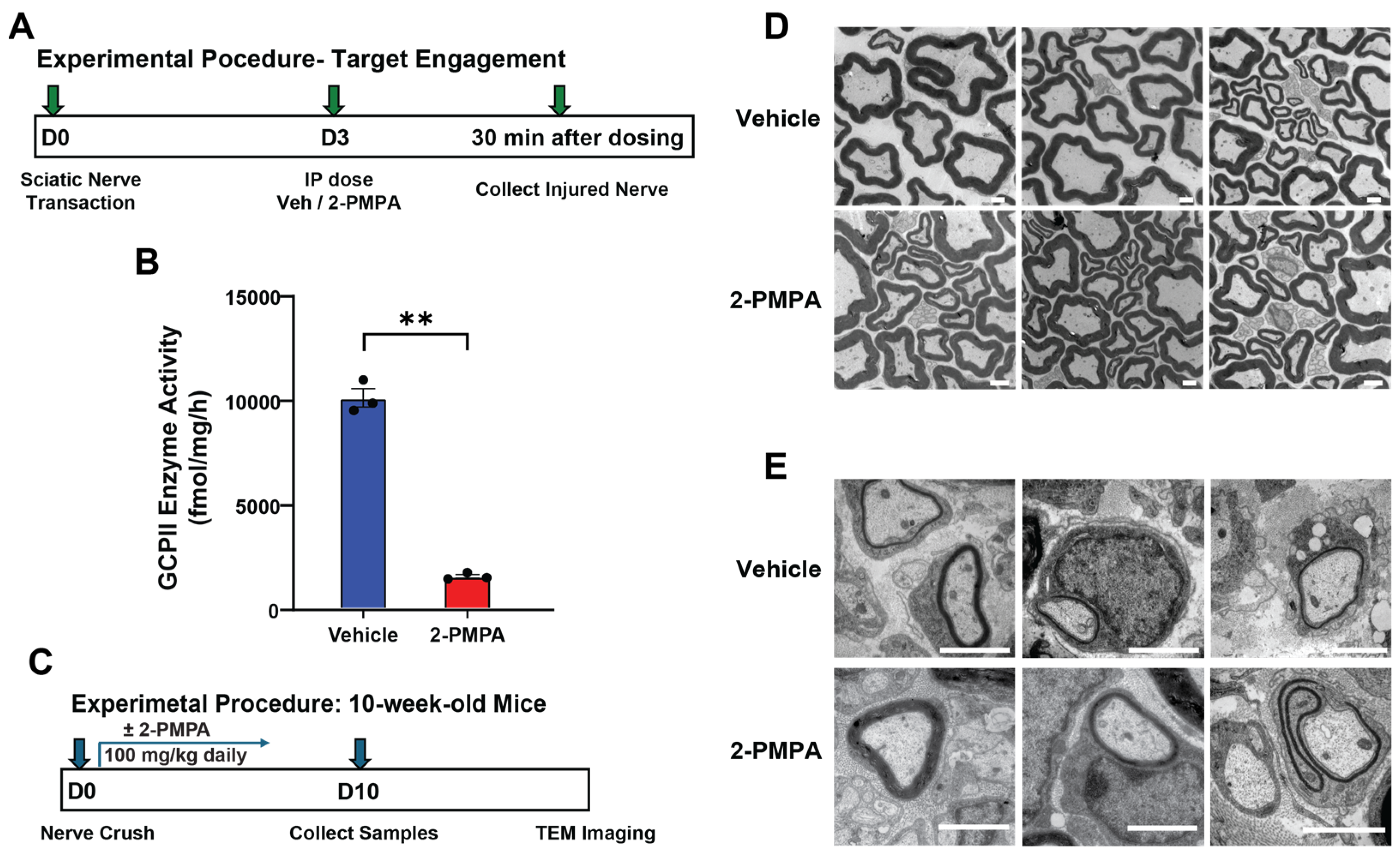
2.1. GCPII Activity and Expression Are Elevated in Peripheral Nerves after Injury
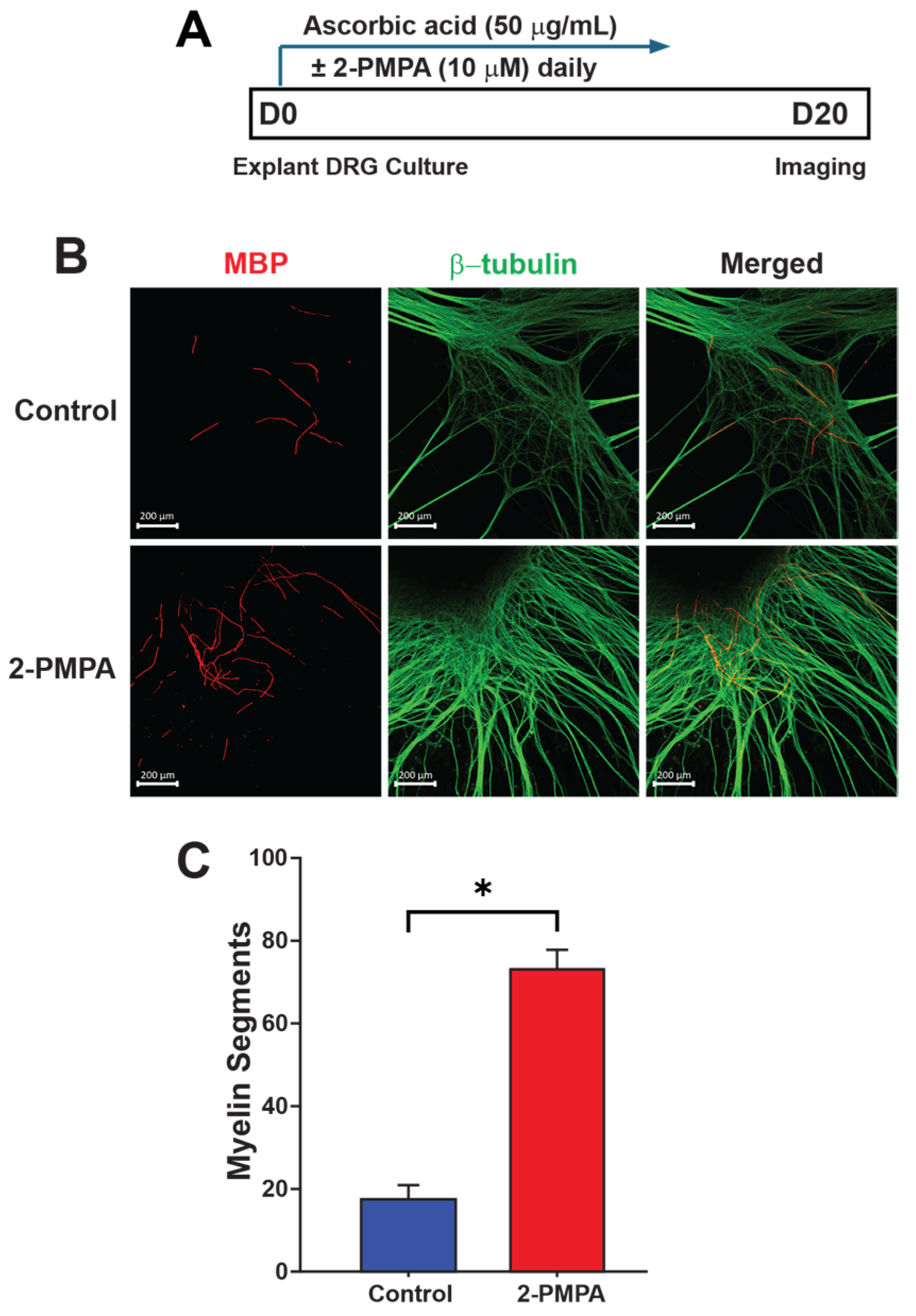
2.2. GCPII Inhibition with 2-PMPA Promotes Myelination In Vitro
2.3. GCPII Inhibition Accelerates Remyelination after PNI in 3-Month-Old Mice
2.4. GCPII Inhibition Enhances Remyelination after PNI in 20-Month-Old Mice
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Sciatic Nerve Injury
4.3. GCPII Enzyme Activity Assay
4.4. Immunofluorescent Staining
4.5. Dorsal Root Ganglia Explants
4.6. Morphological Analyses of Nerves
4.7. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Li, J.; Shi, H.; Liu, H.; Dong, F.; Liu, Z.; Lu, Y.; Chen, L.; Bao, L.; Zhang, X. Nerve Injury-Induced Neuronal PAP-I Maintains Neuropathic Pain by Activating Spinal Microglia. J. Neurosci. 2020, 40, 297–310. [Google Scholar] [CrossRef] [PubMed]
- Zainul, Z.; Ma, B.; Koka, M.; Wilkerson, J.L.; Ortiz, Y.T.; Kerosuo, L.; Chandran, V. Novel Roles of Phentolamine in Protecting Axon Myelination, Muscle Atrophy, and Functional Recovery Following Nerve Injury. Sci. Rep. 2022, 12, 3344. [Google Scholar] [CrossRef] [PubMed]
- Pestronk, A.; Drachman, D.B.; Griffin, J.W. Effects of Aging on Nerve Sprouting and Regeneration. Exp. Neurol. 1980, 70, 65–82. [Google Scholar] [CrossRef]
- Verdú, E.; Ceballos, D.; Vilches, J.J.; Navarro, X. Influence of Aging on Peripheral Nerve Function and Regeneration. J. Peripher. Nerv. Syst. 2000, 5, 191–208. [Google Scholar] [CrossRef] [PubMed]
- Welleford, A.S.; Quintero, J.E.; Seblani, N.E.; Blalock, E.; Gunewardena, S.; Shapiro, S.M.; Riordan, S.M.; Huettl, P.; Guduru, Z.; Stanford, J.A.; et al. RNA Sequencing of Human Peripheral Nerve in Response to Injury: Distinctive Analysis of the Nerve Repair Pathways. Cell Transpl. 2020, 29, 963689720926157. [Google Scholar] [CrossRef]
- Wagstaff, L.J.; Gomez-Sanchez, J.A.; Fazal, S.V.; Otto, G.W.; Kilpatrick, A.M.; Michael, K.; Wong, L.Y.; Ma, K.H.; Turmaine, M.; Svaren, J.; et al. Failures of Nerve Regeneration Caused by Aging or Chronic Denervation Are Rescued by Restoring Schwann Cell C-Jun. Elife 2021, 10, e62232. [Google Scholar] [CrossRef]
- Govindappa, P.K.; Talukder, M.A.H.; Gurjar, A.A.; Hegarty, J.P.; Elfar, J.C. An Effective Erythropoietin Dose Regimen Protects against Severe Nerve Injury-Induced Pathophysiological Changes with Improved Neural Gene Expression and Enhances Functional Recovery. Int. Immunopharmacol. 2020, 82, 106330. [Google Scholar] [CrossRef]
- Lee, J.; Talukder, M.H.; Karuman, Z.; Gurjar, A.; Govindappa, P.; Guddadarangaiah, J.; Manto, K.; Wandling, G.; Hegarty, J.; Waning, D.; et al. Functional Recovery and Muscle Atrophy in Pre-Clinical Models of Peripheral Nerve Transection and Gap-Grafting in Mice: Effects of 4-Aminopyridine. Neural Regen. Res. 2023, 18, 439. [Google Scholar] [CrossRef]
- Noble, M.; Tseng, K.-C.; Li, H.; Elfar, J.C. 4-Aminopyridine as a Single Agent Diagnostic and Treatment for Severe Nerve Crush Injury. Mil. Med. 2019, 184, 379–385. [Google Scholar] [CrossRef]
- Kang, H.; Lichtman, J.W. Motor Axon Regeneration and Muscle Reinnervation in Young Adult and Aged Animals. J. Neurosci. 2013, 33, 19480–19491. [Google Scholar] [CrossRef]
- Painter, M.W.; Brosius Lutz, A.; Cheng, Y.-C.; Latremoliere, A.; Duong, K.; Miller, C.M.; Posada, S.; Cobos, E.J.; Zhang, A.X.; Wagers, A.J.; et al. Diminished Schwann Cell Repair Responses Underlie Age-Associated Impaired Axonal Regeneration. Neuron 2014, 83, 331–343. [Google Scholar] [CrossRef] [PubMed]
- Stratton, J.A.; Holmes, A.; Rosin, N.L.; Sinha, S.; Vohra, M.; Burma, N.E.; Trang, T.; Midha, R.; Biernaskie, J. Macrophages Regulate Schwann Cell Maturation after Nerve Injury. Cell Rep. 2018, 24, 2561–2572.e6. [Google Scholar] [CrossRef] [PubMed]
- Stratton, J.A.; Eaton, S.; Rosin, N.L.; Jawad, S.; Holmes, A.; Yoon, G.; Midha, R.; Biernaskie, J. Macrophages and Associated Ligands in the Aged Injured Nerve: A Defective Dynamic That Contributes to Reduced Axonal Regrowth. Front. Aging Neurosci. 2020, 12, 174. [Google Scholar] [CrossRef]
- Rios, R.; Jablonka-Shariff, A.; Broberg, C.; Snyder-Warwick, A.K. Macrophage Roles in Peripheral Nervous System Injury and Pathology: Allies in Neuromuscular Junction Recovery. Mol. Cell. Neurosci. 2021, 111, 103590. [Google Scholar] [CrossRef]
- Büttner, R.; Schulz, A.; Reuter, M.; Akula, A.K.; Mindos, T.; Carlstedt, A.; Riecken, L.B.; Baader, S.L.; Bauer, R.; Morrison, H. Inflammaging Impairs Peripheral Nerve Maintenance and Regeneration. Aging Cell 2018, 17, e12833. [Google Scholar] [CrossRef] [PubMed]
- Modrak, M.; Talukder, M.A.H.; Gurgenashvili, K.; Noble, M.; Elfar, J.C. Peripheral Nerve Injury and Myelination: Potential Therapeutic Strategies. J. Neurosci. Res. 2020, 98, 780–795. [Google Scholar] [CrossRef]
- Menorca, R.M.G.; Fussell, T.S.; Elfar, J.C. Nerve Physiology. Hand Clin. 2013, 29, 317–330. [Google Scholar] [CrossRef]
- Jessen, K.R.; Mirsky, R. The Repair Schwann Cell and Its Function in Regenerating Nerves. J. Physiol. 2016, 594, 3521–3531. [Google Scholar] [CrossRef]
- Manole, E.; Eugenia Bastian, A.; Maria Oproiu, A.; Teodora Neagu, M.; Constantin, C.; Isvoranu, G. Schwann Cell Plasticity in Peripheral Nerve Regeneration after Injury. In Demyelination Disorders; Baloyannis, S.J., Rossi, F.H., Liu, W., Eds.; IntechOpen: London, UK, 2022; ISBN 978-1-83968-653-5. [Google Scholar]
- Nocera, G.; Jacob, C. Mechanisms of Schwann Cell Plasticity Involved in Peripheral Nerve Repair after Injury. Cell. Mol. Life Sci. 2020, 77, 3977–3989. [Google Scholar] [CrossRef]
- Fuentes-Flores, A.; Geronimo-Olvera, C.; Girardi, K.; Necuñir-Ibarra, D.; Patel, S.K.; Bons, J.; Wright, M.C.; Geschwind, D.; Hoke, A.; Gomez-Sanchez, J.A.; et al. Senescent Schwann Cells Induced by Aging and Chronic Denervation Impair Axonal Regeneration Following Peripheral Nerve Injury. EMBO Mol. Med. 2023, 15, e17907. [Google Scholar] [CrossRef]
- Wu, G.; Wen, X.; Kuang, R.; Lui, K.W.; He, B.; Li, G.; Zhu, Z. Roles of Macrophages and Their Interactions with Schwann Cells after Peripheral Nerve Injury. Cell Mol. Neurobiol. 2024, 44, 11. [Google Scholar] [CrossRef] [PubMed]
- Gitik, M.; Elberg, G.; Reichert, F.; Tal, M.; Rotshenker, S. Deletion of CD47 from Schwann Cells and Macrophages Hastens Myelin Disruption/Dismantling and Scavenging in Schwann Cells and Augments Myelin Debris Phagocytosis in Macrophages. J. Neuroinflamm. 2023, 20, 243. [Google Scholar] [CrossRef] [PubMed]
- Rolfe, A.J.; Bosco, D.B.; Broussard, E.N.; Ren, Y. In Vitro Phagocytosis of Myelin Debris by Bone Marrow-Derived Macrophages. J. Vis. Exp. 2017, 130, e56322. [Google Scholar] [CrossRef] [PubMed]
- Bauer, S.; Kerr, B.J.; Patterson, P.H. The Neuropoietic Cytokine Family in Development, Plasticity, Disease and Injury. Nat. Rev. Neurosci. 2007, 8, 221–232. [Google Scholar] [CrossRef]
- Rotshenker, S. Wallerian Degeneration: The Innate-Immune Response to Traumatic Nerve Injury. J. Neuroinflamm. 2011, 8, 109. [Google Scholar] [CrossRef]
- Neale, J.H.; Olszewski, R.T.; Zuo, D.; Janczura, K.J.; Profaci, C.P.; Lavin, K.M.; Madore, J.C.; Bzdega, T. Advances in Understanding the Peptide Neurotransmitter NAAG and Appearance of a New Member of the NAAG Neuropeptide Family. J. Neurochem. 2011, 118, 490–498. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Chae, S.S.; Koh, Y.H.; Lee, S.K.; Jo, S.A. Glutamate Carboxypeptidase II: An Amyloid Peptide-degrading Enzyme with Physiological Function in the Brain. FASEB J. 2010, 24, 4491–4502. [Google Scholar] [CrossRef] [PubMed]
- Slusher, B.S.; Robinson, M.B.; Tsai, G.; Simmons, M.L.; Richards, S.S.; Coyle, J.T. Rat Brain N-Acetylated Alpha-Linked Acidic Dipeptidase Activity. Purification and Immunologic Characterization. J. Biol. Chem. 1990, 265, 21297–21301. [Google Scholar] [CrossRef]
- Berger, U.V.; Carter, R.E.; Coyle, J.T. The Immunocytochemical Localization of N-Acetylaspartyl Glutamate, Its Hydrolysing Enzyme NAALADase, and the NMDAR-1 Receptor at a Vertebrate Neuromuscular Junction. Neuroscience 1995, 64, 847–850. [Google Scholar] [CrossRef]
- Zhang, Z.; Bassam, B.; Thomas, A.G.; Williams, M.; Liu, J.; Nance, E.; Rojas, C.; Slusher, B.S.; Kannan, S. Maternal Inflammation Leads to Impaired Glutamate Homeostasis and Up-Regulation of Glutamate Carboxypeptidase II in Activated Microglia in the Fetal/Newborn Rabbit Brain. Neurobiol. Dis. 2016, 94, 116–128. [Google Scholar] [CrossRef]
- Tallon, C.; Sharma, A.; Zhang, Z.; Thomas, A.G.; Ng, J.; Zhu, X.; Donoghue, A.; Schulte, M.; Joe, T.R.; Kambhampati, S.P.; et al. Dendrimer-2PMPA Delays Muscle Function Loss and Denervation in a Murine Model of Amyotrophic Lateral Sclerosis. Neurotherapeutics 2022, 19, 274–288. [Google Scholar] [CrossRef] [PubMed]
- Neale, J.H.; Yamamoto, T. N-Acetylaspartylglutamate (NAAG) and Glutamate Carboxypeptidase II: An Abundant Peptide Neurotransmitter-Enzyme System with Multiple Clinical Applications. Prog. Neurobiol. 2020, 184, 101722. [Google Scholar] [CrossRef] [PubMed]
- Datta, D.; Leslie, S.N.; Woo, E.; Amancharla, N.; Elmansy, A.; Lepe, M.; Mecca, A.P.; Slusher, B.S.; Nairn, A.C.; Arnsten, A.F.T. Glutamate Carboxypeptidase II in Aging Rat Prefrontal Cortex Impairs Working Memory Performance. Front. Aging Neurosci. 2021, 13, 760270. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Xu, S.; Cui, Z.; Zhang, M.; Lin, Y.; Cai, L.; Wang, Z.; Luo, X.; Zheng, Y.; Wang, Y.; et al. Mice Lacking Glutamate Carboxypeptidase II Develop Normally, but Are Less Susceptible to Traumatic Brain Injury. J. Neurochem. 2015, 134, 340–353. [Google Scholar] [CrossRef] [PubMed]
- Ji, T.; Pang, Y.; Cheng, M.; Wang, R.; Chen, X.; Zhang, C.; Liu, M.; Zhang, J.; Zhong, C. Deletion of Glutamate Carboxypeptidase II (GCPII), but Not GCPIII, Provided Long-term Benefits in Mice with Traumatic Brain Injury. CNS Neurosci. Ther. 2023, 29, 3786–3801. [Google Scholar] [CrossRef]
- Zhang, W.; Murakawa, Y.; Wozniak, K.M.; Slusher, B.; Sima, A.A.F. The Preventive and Therapeutic Effects of GCPII (NAALADase) Inhibition on Painful and Sensory Diabetic Neuropathy. J. Neurol. Sci. 2006, 247, 217–223. [Google Scholar] [CrossRef]
- Carozzi, V.A.; Chiorazzi, A.; Canta, A.; Lapidus, R.G.; Slusher, B.S.; Wozniak, K.M.; Cavaletti, G. Glutamate Carboxypeptidase Inhibition Reduces the Severity of Chemotherapy-Induced Peripheral Neurotoxicity in Rat. Neurotox. Res. 2010, 17, 380–391. [Google Scholar] [CrossRef] [PubMed]
- Hollinger, K.R.; Alt, J.; Riehm, A.M.; Slusher, B.S.; Kaplin, A.I. Dose-Dependent Inhibition of GCPII to Prevent and Treat Cognitive Impairment in the EAE Model of Multiple Sclerosis. Brain Res. 2016, 1635, 105–112. [Google Scholar] [CrossRef]
- Endo, T.; Kadoya, K.; Suzuki, T.; Suzuki, Y.; Terkawi, M.A.; Kawamura, D.; Iwasaki, N. Mature but Not Developing Schwann Cells Promote Axon Regeneration after Peripheral Nerve Injury. NPJ Regen. Med. 2022, 7, 12. [Google Scholar] [CrossRef]
- Balakrishnan, A.; Belfiore, L.; Chu, T.-H.; Fleming, T.; Midha, R.; Biernaskie, J.; Schuurmans, C. Insights into the Role and Potential of Schwann Cells for Peripheral Nerve Repair From Studies of Development and Injury. Front. Mol. Neurosci. 2021, 13, 608442. [Google Scholar] [CrossRef]
- Kim, H.A.; Mindos, T.; Parkinson, D.B. Plastic Fantastic: Schwann Cells and Repair of the Peripheral Nervous System. Stem Cells Transl. Med. 2013, 2, 553–557. [Google Scholar] [CrossRef]
- Gomez-Sanchez, J.A.; Pilch, K.S.; Van Der Lans, M.; Fazal, S.V.; Benito, C.; Wagstaff, L.J.; Mirsky, R.; Jessen, K.R. After Nerve Injury, Lineage Tracing Shows That Myelin and Remak Schwann Cells Elongate Extensively and Branch to Form Repair Schwann Cells, Which Shorten Radically on Remyelination. J. Neurosci. 2017, 37, 9086–9099. [Google Scholar] [CrossRef]
- Arthur-Farraj, P.J.; Latouche, M.; Wilton, D.K.; Quintes, S.; Chabrol, E.; Banerjee, A.; Woodhoo, A.; Jenkins, B.; Rahman, M.; Turmaine, M.; et al. C-Jun Reprograms Schwann Cells of Injured Nerves to Generate a Repair Cell Essential for Regeneration. Neuron 2012, 75, 633–647. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Sanchez, J.A.; Carty, L.; Iruarrizaga-Lejarreta, M.; Palomo-Irigoyen, M.; Varela-Rey, M.; Griffith, M.; Hantke, J.; Macias-Camara, N.; Azkargorta, M.; Aurrekoetxea, I.; et al. Schwann Cell Autophagy, Myelinophagy, Initiates Myelin Clearance from Injured Nerves. J. Cell Biol. 2015, 210, 153–168. [Google Scholar] [CrossRef]
- Cattin, A.-L.; Burden, J.J.; Van Emmenis, L.; Mackenzie, F.E.; Hoving, J.J.A.; Garcia Calavia, N.; Guo, Y.; McLaughlin, M.; Rosenberg, L.H.; Quereda, V.; et al. Macrophage-Induced Blood Vessels Guide Schwann Cell-Mediated Regeneration of Peripheral Nerves. Cell 2015, 162, 1127–1139. [Google Scholar] [CrossRef] [PubMed]
- Ha, D.; Bing, S.J.; Ahn, G.; Kim, J.; Cho, J.; Kim, A.; Herath, K.H.I.N.M.; Yu, H.S.; Jo, S.A.; Cho, I.; et al. Blocking Glutamate Carboxypeptidase II Inhibits Glutamate Excitotoxicity and Regulates Immune Responses in Experimental Autoimmune Encephalomyelitis. FEBS J. 2016, 283, 3438–3456. [Google Scholar] [CrossRef]
- Scheib, J.L.; Höke, A. An Attenuated Immune Response by Schwann Cells and Macrophages Inhibits Nerve Regeneration in Aged Rats. Neurobiol. Aging 2016, 45, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Jessen, K.R.; Mirsky, R. The Success and Failure of the Schwann Cell Response to Nerve Injury. Front. Cell. Neurosci. 2019, 13, 33. [Google Scholar] [CrossRef]
- Saitoh, F.; Araki, T. Proteasomal Degradation of Glutamine Synthetase Regulates Schwann Cell Differentiation. J. Neurosci. 2010, 30, 1204–1212. [Google Scholar] [CrossRef]
- Fu, Y.; Sun, W.; Shi, Y.; Shi, R.; Cheng, J.-X. Glutamate Excitotoxicity Inflicts Paranodal Myelin Splitting and Retraction. PLoS ONE 2009, 4, e6705. [Google Scholar] [CrossRef]
- Farah, M.H.; Pan, B.H.; Hoffman, P.N.; Ferraris, D.; Tsukamoto, T.; Nguyen, T.; Wong, P.C.; Price, D.L.; Slusher, B.S.; Griffin, J.W. Reduced BACE1 Activity Enhances Clearance of Myelin Debris and Regeneration of Axons in the Injured Peripheral Nervous System. J. Neurosci. 2011, 31, 5744–5754. [Google Scholar] [CrossRef] [PubMed]
- Rojas, C.; Frazier, S.T.; Flanary, J.; Slusher, B.S. Kinetics and Inhibition of Glutamate Carboxypeptidase II Using a Microplate Assay. Anal. Biochem. 2002, 310, 50–54. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Ahn, B.; Macpherson, P.C.D.; Ranjit, R.; Claflin, D.R.; Van Remmen, H.; Brooks, S.V. Transgenic Expression of SOD1 Specifically in Neurons of Sod1 Deficient Mice Prevents Defects in Muscle Mitochondrial Function and Calcium Handling. Free Radic. Biol. Med. 2021, 165, 299–311. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Tallon, C.; Zhu, X.; Huizar, K.D.J.; Picciolini, S.; Thomas, A.G.; Tenora, L.; Liyanage, W.; Rodà, F.; Gualerzi, A.; et al. Microglial-Targeted nSMase2 Inhibitor Fails to Reduce Tau Propagation in PS19 Mice. Pharmaceutics 2023, 15, 2364. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Claflin, D.R.; Huang, M.; Davis, C.S.; Macpherson, P.C.D.; Richardson, A.; Van Remmen, H.; Brooks, S.V. Deletion of Neuronal CuZnSOD Accelerates Age-Associated Muscle Mitochondria and Calcium Handling Dysfunction That Is Independent of Denervation and Precedes Sarcopenia. Int. J. Mol. Sci. 2021, 22, 10735. [Google Scholar] [CrossRef] [PubMed]
- Numata-Uematasu, Y.; Wakatsuki, S.; Kobayashi-Ujiie, Y.; Sakai, K.; Ichinohe, N.; Araki, T. In Vitro Myelination Using Explant Culture of Dorsal Root Ganglia: An Efficient Tool for Analyzing Peripheral Nerve Differentiation and Disease Modeling. PLoS ONE 2023, 18, e0285897. [Google Scholar] [CrossRef]
- Taveggia, C.; Bolino, A. DRG Neuron/Schwann Cells Myelinating Cocultures. In Myelin; Woodhoo, A., Ed.; Methods in Molecular Biology; Springer: New York, NY, USA, 2018; Volume 1791, pp. 115–129. ISBN 978-1-4939-7861-8. [Google Scholar]
- Sango, K. Novel Neuron-Schwann Cell Co-Culture Models to Study Peripheral Nerve Degeneration and Regeneration. Neural Regen. Res. 2023, 18, 1733. [Google Scholar] [CrossRef]
- Stoll, G.; Griffin, J.W.; Li, C.Y.; Trapp, B.D. Wallerian Degeneration in the Peripheral Nervous System: Participation of Both Schwann Cells and Macrophages in Myelin Degradation. J. Neurocytol. 1989, 18, 671–683. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | Primary Antibody | Secondary Antibody |
|---|---|---|
| GCPII | GCP-04 (Goat anti-Mouse, 1:250) (Novus, NBP1-45057, Centennial, CO, USA) | 647 nm, anti-mouse IgG1 (1:1000) (Invitrogen, A66784, Waltham, MA, USA) |
| Repair Schwann cells | GFAP (Goat anti-Chicken, 1:250) (Abcam, Ab4674, Waltham, MA, USA) | 488 nm, anti-chicken IgG (H + L) (1:1000) (Invitrogen, A11039, Waltham, MA, USA) |
| Myelin Schwann cells | S100 (Goat anti-Rabbit, 1:25) (DAKO, GA50461-2, Santa Clara, CA, USA) | 546 nm, anti-rabbit IgG (H + L) (1:1000) (Invitrogen, A11035, Waltham, MA, USA) |
| Macrophages | CD68 (Goat anti-Rat, 1:250) (Invitrogen, PA5-109344, Waltham, MA, USA) | 405 nm, anti-rat IgG (H + L) (1:1000) (Invitrogen, A48261, Waltham, MA, USA) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Su, Y.; Huang, M.; Thomas, A.G.; Maragakis, J.; Huizar, K.D.J.; Zheng, Y.; Wu, Y.; Farah, M.H.; Slusher, B.S. GCPII Inhibition Promotes Remyelination after Peripheral Nerve Injury in Aged Mice. Int. J. Mol. Sci. 2024, 25, 6893. https://doi.org/10.3390/ijms25136893
Su Y, Huang M, Thomas AG, Maragakis J, Huizar KDJ, Zheng Y, Wu Y, Farah MH, Slusher BS. GCPII Inhibition Promotes Remyelination after Peripheral Nerve Injury in Aged Mice. International Journal of Molecular Sciences. 2024; 25(13):6893. https://doi.org/10.3390/ijms25136893
Chicago/Turabian StyleSu, Yu, Meixiang Huang, Ajit G. Thomas, John Maragakis, Kaitlyn D. J. Huizar, Yuxin Zheng, Ying Wu, Mohamed H. Farah, and Barbara S. Slusher. 2024. "GCPII Inhibition Promotes Remyelination after Peripheral Nerve Injury in Aged Mice" International Journal of Molecular Sciences 25, no. 13: 6893. https://doi.org/10.3390/ijms25136893