
The Role of CD4+T Cells in Nonalcoholic Steatohepatitis and Hepatocellular Carcinoma


Abstract
1. Introduction
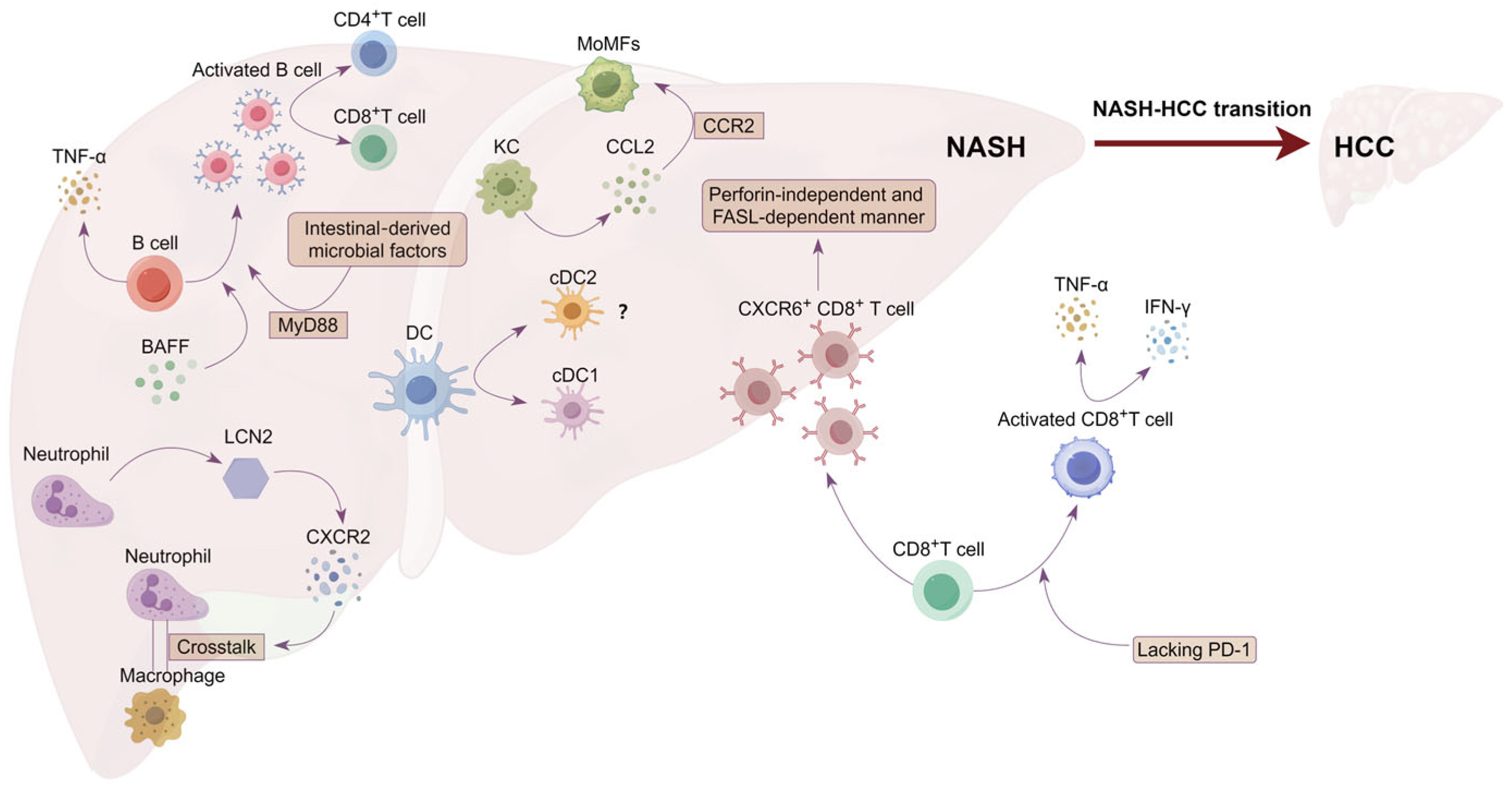
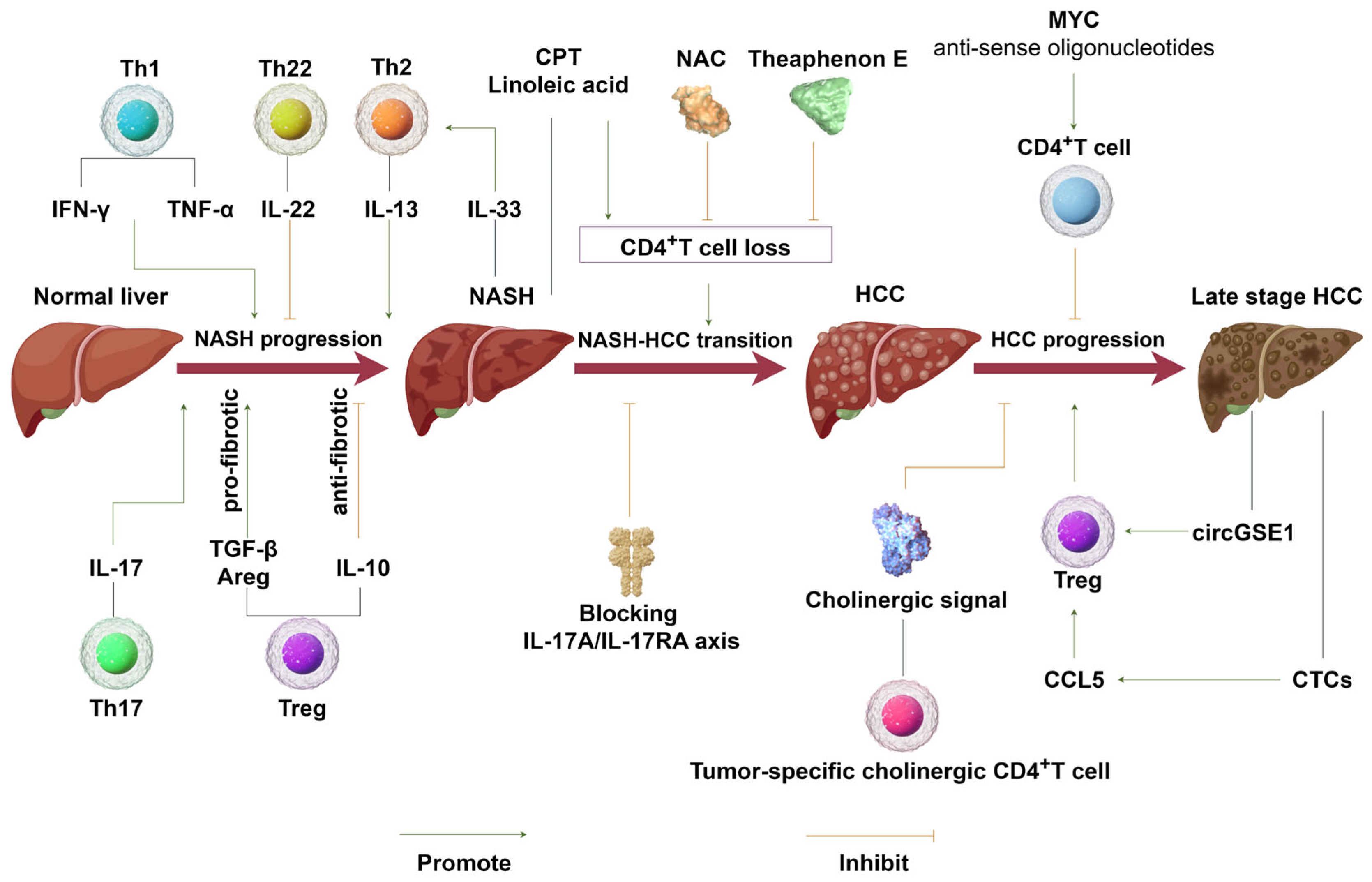
2. The Role of CD4+T Cells in NASH Progression
3. Immunomodulatory Role of CD4+T Cells in NASH–HCC Transition and HCC Progression
4. Implications of CD4+T Cells in the Treatment of NASH, NASH–HCC Transition, and HCC Immunotherapy
5. Conclusions
Funding
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.D.; Hainaut, P.; Gores, G.J.; Amadou, A.; Plymoth, A.; Roberts, L.R. A global view of hepatocellular carcinoma: Trends, risk, prevention and management. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 589–604. [Google Scholar] [CrossRef] [PubMed]
- Kanda, T.; Goto, T.; Hirotsu, Y.; Masuzaki, R.; Moriyama, M.; Omata, M. Molecular Mechanisms: Connections between Nonalcoholic Fatty Liver Disease, Steatohepatitis and Hepatocellular Carcinoma. Int. J. Mol. Sci. 2020, 21, 1525. [Google Scholar] [CrossRef] [PubMed]
- Cariou, B.; Byrne, C.D.; Loomba, R.; Sanyal, A.J. Nonalcoholic fatty liver disease as a metabolic disease in humans: A literature review. Diabetes Obes. Metab. 2021, 23, 1069–1083. [Google Scholar] [CrossRef] [PubMed]
- Rinella, M.E. Nonalcoholic fatty liver disease: A systematic review. JAMA 2015, 313, 2263–2273. [Google Scholar] [CrossRef] [PubMed]
- Koo, S.Y.; Park, E.J.; Lee, C.W. Immunological distinctions between nonalcoholic steatohepatitis and hepatocellular carcinoma. Exp. Mol. Med. 2020, 52, 1209–1219. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M. The epidemiology of nonalcoholic steatohepatitis. Clin. Liver Dis. 2018, 11, 92–94. [Google Scholar] [CrossRef]
- Gan, L.; Liu, Z.; Sun, C. Obesity linking to hepatocellular carcinoma: A global view. Biochim. Biophys. Acta. Rev. Cancer 2018, 1869, 97–102. [Google Scholar] [CrossRef]
- Hester, D.; Golabi, P.; Paik, J.; Younossi, I.; Mishra, A.; Younossi, Z.M. Among Medicare Patients With Hepatocellular Carcinoma, Non-alcoholic Fatty Liver Disease is the Most Common Etiology and Cause of Mortality. J. Clin. Gastroenterol. 2020, 54, 459–467. [Google Scholar] [CrossRef]
- EASL Clinical Practice Guidelines: Management of hepatocellular carcinoma. J. Hepatol. 2018, 69, 182–236. [CrossRef]
- Llovet, J.M.; Burroughs, A.; Bruix, J. Hepatocellular carcinoma. Lancet 2003, 362, 1907–1917. [Google Scholar] [CrossRef] [PubMed]
- Villanueva, A. Hepatocellular Carcinoma. N. Engl. J. Med. 2019, 380, 1450–1462. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.Y.; Kudo, M.; Breder, V.; Merle, P.; Kaseb, A.O.; et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N. Engl. J. Med. 2020, 382, 1894–1905. [Google Scholar] [CrossRef] [PubMed]
- Pfister, D.; Nunez, N.G.; Pinyol, R.; Govaere, O.; Pinter, M.; Szydlowska, M.; Gupta, R.; Qiu, M.; Deczkowska, A.; Weiner, A.; et al. NASH limits anti-tumour surveillance in immunotherapy-treated HCC. Nature 2021, 592, 450–456. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhang, H.; Yao, Y.; Zhang, X.; Guan, Y.; Zheng, F. CD4+ T cell activation and inflammation in NASH-related fibrosis. Front. Immunol. 2022, 13, 967410. [Google Scholar] [CrossRef] [PubMed]
- Barrow, F.; Khan, S.; Fredrickson, G.; Wang, H.; Dietsche, K.; Parthiban, P.; Robert, S.; Kaiser, T.; Winer, S.; Herman, A.; et al. Microbiota-Driven Activation of Intrahepatic B Cells Aggravates NASH Through Innate and Adaptive Signaling. Hepatology 2021, 74, 704–722. [Google Scholar] [CrossRef] [PubMed]
- Deczkowska, A.; David, E.; Ramadori, P.; Pfister, D.; Safran, M.; Li, B.; Giladi, A.; Jaitin, D.A.; Barboy, O.; Cohen, M.; et al. XCR1(+) type 1 conventional dendritic cells drive liver pathology in non-alcoholic steatohepatitis. Nat. Med. 2021, 27, 1043–1054. [Google Scholar] [CrossRef] [PubMed]
- Barrow, F.; Khan, S.; Wang, H.; Revelo, X.S. The Emerging Role of B Cells in the Pathogenesis of NAFLD. Hepatology 2021, 74, 2277–2286. [Google Scholar] [CrossRef]
- Pitzalis, C.; Jones, G.W.; Bombardieri, M.; Jones, S.A. Ectopic lymphoid-like structures in infection, cancer and autoimmunity. Nat. Rev. Immunol. 2014, 14, 447–462. [Google Scholar] [CrossRef]
- Sutti, S.; Albano, E. Adaptive immunity: An emerging player in the progression of NAFLD. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Dudek, M.; Pfister, D.; Donakonda, S.; Filpe, P.; Schneider, A.; Laschinger, M.; Hartmann, D.; Huser, N.; Meiser, P.; Bayerl, F.; et al. Auto-aggressive CXCR6(+) CD8 T cells cause liver immune pathology in NASH. Nature 2021, 592, 444–449. [Google Scholar] [CrossRef] [PubMed]
- Heier, E.C.; Meier, A.; Julich-Haertel, H.; Djudjaj, S.; Rau, M.; Tschernig, T.; Geier, A.; Boor, P.; Lammert, F.; Lukacs-Kornek, V. Murine CD103(+) dendritic cells protect against steatosis progression towards steatohepatitis. J. Hepatol. 2017, 66, 1241–1250. [Google Scholar] [CrossRef] [PubMed]
- Hirsova, P.; Bamidele, A.O.; Wang, H.; Povero, D.; Revelo, X.S. Emerging Roles of T Cells in the Pathogenesis of Nonalcoholic Steatohepatitis and Hepatocellular Carcinoma. Front. Endocrinol. 2021, 12, 760860. [Google Scholar] [CrossRef] [PubMed]
- Keene, J.A.; Forman, J. Helper activity is required for the in vivo generation of cytotoxic T lymphocytes. J. Exp. Med. 1982, 155, 768–782. [Google Scholar] [CrossRef] [PubMed]
- Antony, P.A.; Piccirillo, C.A.; Akpinarli, A.; Finkelstein, S.E.; Speiss, P.J.; Surman, D.R.; Palmer, D.C.; Chan, C.C.; Klebanoff, C.A.; Overwijk, W.W.; et al. CD8+ T cell immunity against a tumor/self-antigen is augmented by CD4+ T helper cells and hindered by naturally occurring T regulatory cells. J. Immunol. 2005, 174, 2591–2601. [Google Scholar] [CrossRef] [PubMed]
- Bos, R.; Sherman, L.A. CD4+ T-cell help in the tumor milieu is required for recruitment and cytolytic function of CD8+ T lymphocytes. Cancer Res. 2010, 70, 8368–8377. [Google Scholar] [CrossRef] [PubMed]
- Tay, R.E.; Richardson, E.K.; Toh, H.C. Revisiting the role of CD4+ T cells in cancer immunotherapy-new insights into old paradigms. Cancer Gene Ther. 2021, 28, 5–17. [Google Scholar] [CrossRef]
- Linnemann, C.; van Buuren, M.M.; Bies, L.; Verdegaal, E.M.; Schotte, R.; Calis, J.J.; Behjati, S.; Velds, A.; Hilkmann, H.; Atmioui, D.E.; et al. High-throughput epitope discovery reveals frequent recognition of neo-antigens by CD4+ T cells in human melanoma. Nat. Med. 2015, 21, 81–85. [Google Scholar] [CrossRef]
- Kreiter, S.; Vormehr, M.; van de Roemer, N.; Diken, M.; Löwer, M.; Diekmann, J.; Boegel, S.; Schrörs, B.; Vascotto, F.; Castle, J.C.; et al. Mutant MHC class II epitopes drive therapeutic immune responses to cancer. Nature 2015, 520, 692–696. [Google Scholar] [CrossRef]
- Seung, E.; Xing, Z.; Wu, L.; Rao, E.; Cortez-Retamozo, V.; Ospina, B.; Chen, L.; Beil, C.; Song, Z.; Zhang, B.; et al. A trispecific antibody targeting HER2 and T cells inhibits breast cancer growth via CD4 cells. Nature 2022, 603, 328–334. [Google Scholar] [CrossRef] [PubMed]
- Oh, D.Y.; Kwek, S.S.; Raju, S.S.; Li, T.; McCarthy, E.; Chow, E.; Aran, D.; Ilano, A.; Pai, C.S.; Rancan, C.; et al. Intratumoral CD4+ T Cells Mediate Anti-tumor Cytotoxicity in Human Bladder Cancer. Cell 2020, 181, 1612–1625.e1613. [Google Scholar] [CrossRef] [PubMed]
- Speiser, D.E.; Chijioke, O.; Schaeuble, K.; Münz, C. CD4+ T cells in cancer. Nat. Cancer 2023, 4, 317–329. [Google Scholar] [CrossRef] [PubMed]
- Saravia, J.; Chapman, N.M.; Chi, H. Helper T cell differentiation. Cell. Mol. Immunol. 2019, 16, 634–643. [Google Scholar] [CrossRef] [PubMed]
- Her, Z.; Tan, J.H.L.; Lim, Y.S.; Tan, S.Y.; Chan, X.Y.; Tan, W.W.S.; Liu, M.; Yong, K.S.M.; Lai, F.; Ceccarello, E.; et al. CD4+ T Cells Mediate the Development of Liver Fibrosis in High Fat Diet-Induced NAFLD in Humanized Mice. Front. Immunol. 2020, 11, 580968. [Google Scholar] [CrossRef] [PubMed]
- Rau, M.; Schilling, A.K.; Meertens, J.; Hering, I.; Weiss, J.; Jurowich, C.; Kudlich, T.; Hermanns, H.M.; Bantel, H.; Beyersdorf, N.; et al. Progression from Nonalcoholic Fatty Liver to Nonalcoholic Steatohepatitis Is Marked by a Higher Frequency of Th17 Cells in the Liver and an Increased Th17/Resting Regulatory T Cell Ratio in Peripheral Blood and in the Liver. J. Immunol. 2016, 196, 97–105. [Google Scholar] [CrossRef] [PubMed]
- O’Garra, A.; Robinson, D. Development and function of T helper 1 cells. Adv. Immunol. 2004, 83, 133–162. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Kodumudi, K.N.; Ramamoorthi, G.; Basu, A.; Snyder, C.; Wiener, D.; Pilon-Thomas, S.; Grover, P.; Zhang, H.; Greene, M.I.; et al. Th1 cytokine interferon gamma improves response in HER2 breast cancer by modulating the ubiquitin proteasomal pathway. Mol. Ther. J. Am. Soc. Gene Ther. 2021, 29, 1541–1556. [Google Scholar] [CrossRef]
- Pacifico, L.; Di Renzo, L.; Anania, C.; Osborn, J.F.; Ippoliti, F.; Schiavo, E.; Chiesa, C. Increased T-helper interferon-gamma-secreting cells in obese children. Eur. J. Endocrinol. 2006, 154, 691–697. [Google Scholar] [CrossRef]
- Leung, O.M.; Li, J.; Li, X.; Chan, V.W.; Yang, K.Y.; Ku, M.; Ji, L.; Sun, H.; Waldmann, H.; Tian, X.Y.; et al. Regulatory T Cells Promote Apelin-Mediated Sprouting Angiogenesis in Type 2 Diabetes. Cell Rep. 2018, 24, 1610–1626. [Google Scholar] [CrossRef]
- Inzaugarat, M.E.; Ferreyra Solari, N.E.; Billordo, L.A.; Abecasis, R.; Gadano, A.C.; Cherñavsky, A.C. Altered phenotype and functionality of circulating immune cells characterize adult patients with nonalcoholic steatohepatitis. J. Clin. Immunol. 2011, 31, 1120–1130. [Google Scholar] [CrossRef] [PubMed]
- Bertola, A.; Bonnafous, S.; Anty, R.; Patouraux, S.; Saint-Paul, M.C.; Iannelli, A.; Gugenheim, J.; Barr, J.; Mato, J.M.; Le Marchand-Brustel, Y.; et al. Hepatic expression patterns of inflammatory and immune response genes associated with obesity and NASH in morbidly obese patients. PLoS ONE 2010, 5, e13577. [Google Scholar] [CrossRef] [PubMed]
- Kremer, M.; Hines, I.N.; Milton, R.J.; Wheeler, M.D. Favored T helper 1 response in a mouse model of hepatosteatosis is associated with enhanced T cell-mediated hepatitis. Hepatology 2006, 44, 216–227. [Google Scholar] [CrossRef] [PubMed]
- Rolla, S.; Alchera, E.; Imarisio, C.; Bardina, V.; Valente, G.; Cappello, P.; Mombello, C.; Follenzi, A.; Novelli, F.; Carini, R. The balance between IL-17 and IL-22 produced by liver-infiltrating T-helper cells critically controls NASH development in mice. Clin. Sci. 2016, 130, 193–203. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.Y.; Takahara, T.; Kawai, K.; Fujino, M.; Sugiyama, T.; Tsuneyama, K.; Tsukada, K.; Nakae, S.; Zhong, L.; Li, X.K. IFN-γ deficiency attenuates hepatic inflammation and fibrosis in a steatohepatitis model induced by a methionine- and choline-deficient high-fat diet. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 305, G891–G899. [Google Scholar] [CrossRef] [PubMed]
- Horras, C.J.; Lamb, C.L.; Mitchell, K.A. Regulation of hepatocyte fate by interferon-gamma. Cytokine Growth Factor Rev. 2011, 22, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Tura, B.J.; Bunyan, K.E.; Harrison, D.J. The effect of IFNgamma on the hepatocyte: Cell cycle and apoptosis. Int. J. Exp. Pathol. 2001, 82, 317–326. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.; Park, O.; Horiguchi, N.; Kulkarni, S.; Jeong, W.I.; Sun, H.Y.; Radaeva, S.; Gao, B. STAT1 contributes to dsRNA inhibition of liver regeneration after partial hepatectomy in mice. Hepatology 2006, 44, 955–966. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Shen, J.; Man, K.; Chu, E.S.; Yau, T.O.; Sung, J.C.; Go, M.Y.; Deng, J.; Lu, L.; Wong, V.W.; et al. CXCL10 plays a key role as an inflammatory mediator and a non-invasive biomarker of non-alcoholic steatohepatitis. J. Hepatol. 2014, 61, 1365–1375. [Google Scholar] [CrossRef]
- Lu, S.; Wang, Y.; Liu, J. Tumor necrosis factor-alpha signaling in nonalcoholic steatohepatitis and targeted therapies. J. Genet. Genom. 2022, 49, 269–278. [Google Scholar] [CrossRef]
- Paredes-Turrubiarte, G.; Gonzalez-Chavez, A.; Perez-Tamayo, R.; Salazar-Vazquez, B.Y.; Hernandez, V.S.; Garibay-Nieto, N.; Fragoso, J.M.; Escobedo, G. Severity of non-alcoholic fatty liver disease is associated with high systemic levels of tumor necrosis factor alpha and low serum interleukin 10 in morbidly obese patients. Clin. Exp. Med. 2016, 16, 193–202. [Google Scholar] [CrossRef]
- Zahran, W.E.; Salah El-Dien, K.A.; Kamel, P.G.; El-Sawaby, A.S. Efficacy of Tumor Necrosis Factor and Interleukin-10 Analysis in the Follow-up of Nonalcoholic Fatty Liver Disease Progression. Indian J. Clin. Biochem. 2013, 28, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Alaaeddine, N.; Sidaoui, J.; Hilal, G.; Serhal, R.; Abedelrahman, A.; Khoury, S. TNF-alpha messenger ribonucleic acid (mRNA) in patients with nonalcoholic steatohepatitis. Eur. Cytokine Netw. 2012, 23, 107–111. [Google Scholar] [CrossRef]
- Tasci, I.; Dogru, T.; Ercin, C.N.; Erdem, G.; Sonmez, A. Adipokines and cytokines in non-alcoholic fatty liver disease. Aliment. Pharmacol. Ther. 2008, 28, 266–267. [Google Scholar] [CrossRef] [PubMed]
- Bahcecioglu, I.H.; Yalniz, M.; Ataseven, H.; Ilhan, N.; Ozercan, I.H.; Seckin, D.; Sahin, K. Levels of serum hyaluronic acid, TNF-alpha and IL-8 in patients with nonalcoholic steatohepatitis. Hepato-Gastroenterology 2005, 52, 1549–1553. [Google Scholar]
- Crespo, J.; Cayon, A.; Fernandez-Gil, P.; Hernandez-Guerra, M.; Mayorga, M.; Dominguez-Diez, A.; Fernandez-Escalante, J.C.; Pons-Romero, F. Gene expression of tumor necrosis factor alpha and TNF-receptors, p55 and p75, in nonalcoholic steatohepatitis patients. Hepatology 2001, 34, 1158–1163. [Google Scholar] [CrossRef] [PubMed]
- Wandrer, F.; Liebig, S.; Marhenke, S.; Vogel, A.; John, K.; Manns, M.P.; Teufel, A.; Itzel, T.; Longerich, T.; Maier, O.; et al. TNF-Receptor-1 inhibition reduces liver steatosis, hepatocellular injury and fibrosis in NAFLD mice. Cell Death Dis. 2020, 11, 212. [Google Scholar] [CrossRef] [PubMed]
- Fagerberg, L.; Hallstrom, B.M.; Oksvold, P.; Kampf, C.; Djureinovic, D.; Odeberg, J.; Habuka, M.; Tahmasebpoor, S.; Danielsson, A.; Edlund, K.; et al. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol. Cell Proteom. 2014, 13, 397–406. [Google Scholar] [CrossRef]
- Nakayama, T.; Hirahara, K.; Onodera, A.; Endo, Y.; Hosokawa, H.; Shinoda, K.; Tumes, D.J.; Okamoto, Y. Th2 Cells in Health and Disease. Annu. Rev. Immunol. 2017, 35, 53–84. [Google Scholar] [CrossRef]
- Kokubo, K.; Onodera, A.; Kiuchi, M.; Tsuji, K.; Hirahara, K.; Nakayama, T. Conventional and pathogenic Th2 cells in inflammation, tissue repair, and fibrosis. Front. Immunol. 2022, 13, 945063. [Google Scholar] [CrossRef]
- Gieseck, R.L., 3rd; Wilson, M.S.; Wynn, T.A. Type 2 immunity in tissue repair and fibrosis. Nat. Rev. Immunol. 2018, 18, 62–76. [Google Scholar] [CrossRef] [PubMed]
- Shimamura, T.; Fujisawa, T.; Husain, S.R.; Kioi, M.; Nakajima, A.; Puri, R.K. Novel role of IL-13 in fibrosis induced by nonalcoholic steatohepatitis and its amelioration by IL-13R-directed cytotoxin in a rat model. J. Immunol. 2008, 181, 4656–4665. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Liu, Y.; Yang, M.; Guo, X.; Zhang, M.; Li, H.; Li, J.; Zhao, J. IL-33 treatment attenuated diet-induced hepatic steatosis but aggravated hepatic fibrosis. Oncotarget 2016, 7, 33649–33661. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, J.; Owyang, A.; Oldham, E.; Song, Y.; Murphy, E.; McClanahan, T.K.; Zurawski, G.; Moshrefi, M.; Qin, J.; Li, X.; et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity 2005, 23, 479–490. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Zheng, C.; Xiang, Y.; Malik, S.; Su, D.; Xu, G.; Zhang, M. The involvement of TH17 cells in the pathogenesis of IBD. Cytokine Growth Factor Rev. 2023, 69, 28–42. [Google Scholar] [CrossRef] [PubMed]
- Luckheeram, R.V.; Zhou, R.; Verma, A.D.; Xia, B. CD4+T cells: Differentiation and functions. Clin. Dev. Immunol. 2012, 2012, 925135. [Google Scholar] [CrossRef]
- Van Herck, M.A.; Weyler, J.; Kwanten, W.J.; Dirinck, E.L.; De Winter, B.Y.; Francque, S.M.; Vonghia, L. The Differential Roles of T Cells in Non-alcoholic Fatty Liver Disease and Obesity. Front. Immunol. 2019, 10, 82. [Google Scholar] [CrossRef] [PubMed]
- Bunte, K.; Beikler, T. Th17 Cells and the IL-23/IL-17 Axis in the Pathogenesis of Periodontitis and Immune-Mediated Inflammatory Diseases. Int. J. Mol. Sci. 2019, 20, 3394. [Google Scholar] [CrossRef]
- Yasuda, K.; Takeuchi, Y.; Hirota, K. The pathogenicity of Th17 cells in autoimmune diseases. Semin. Immunopathol. 2019, 41, 283–297. [Google Scholar] [CrossRef]
- Nie, Y.J.; Wu, S.H.; Xuan, Y.H.; Yan, G. Role of IL-17 family cytokines in the progression of IPF from inflammation to fibrosis. Mil. Med. Res. 2022, 9, 21. [Google Scholar] [CrossRef] [PubMed]
- Giles, D.A.; Moreno-Fernandez, M.E.; Stankiewicz, T.E.; Cappelletti, M.; Huppert, S.S.; Iwakura, Y.; Dong, C.; Shanmukhappa, S.K.; Divanovic, S. Regulation of Inflammation by IL-17A and IL-17F Modulates Non-Alcoholic Fatty Liver Disease Pathogenesis. PLoS ONE 2016, 11, e0149783. [Google Scholar] [CrossRef] [PubMed]
- Mendez-Sanchez, N.; Valencia-Rodriguez, A.; Coronel-Castillo, C.; Vera-Barajas, A.; Contreras-Carmona, J.; Ponciano-Rodriguez, G.; Zamora-Valdes, D. The cellular pathways of liver fibrosis in non-alcoholic steatohepatitis. Ann. Transl. Med. 2020, 8, 400. [Google Scholar] [CrossRef] [PubMed]
- Harley, I.T.; Stankiewicz, T.E.; Giles, D.A.; Softic, S.; Flick, L.M.; Cappelletti, M.; Sheridan, R.; Xanthakos, S.A.; Steinbrecher, K.A.; Sartor, R.B.; et al. IL-17 signaling accelerates the progression of nonalcoholic fatty liver disease in mice. Hepatology 2014, 59, 1830–1839. [Google Scholar] [CrossRef] [PubMed]
- Fabre, T.; Kared, H.; Friedman, S.L.; Shoukry, N.H. IL-17A enhances the expression of profibrotic genes through upregulation of the TGF-β receptor on hepatic stellate cells in a JNK-dependent manner. J. Immunol. 2014, 193, 3925–3933. [Google Scholar] [CrossRef] [PubMed]
- Trifari, S.; Kaplan, C.D.; Tran, E.H.; Crellin, N.K.; Spits, H. Identification of a human helper T cell population that has abundant production of interleukin 22 and is distinct from T(H)-17, T(H)1 and T(H)2 cells. Nat. Immunol. 2009, 10, 864–871. [Google Scholar] [CrossRef] [PubMed]
- Plank, M.W.; Kaiko, G.E.; Maltby, S.; Weaver, J.; Tay, H.L.; Shen, W.; Wilson, M.S.; Durum, S.K.; Foster, P.S. Th22 Cells Form a Distinct Th Lineage from Th17 Cells In Vitro with Unique Transcriptional Properties and Tbet-Dependent Th1 Plasticity. J. Immunol. 2017, 198, 2182–2190. [Google Scholar] [CrossRef] [PubMed]
- Keir, M.; Yi, Y.; Lu, T.; Ghilardi, N. The role of IL-22 in intestinal health and disease. J. Exp. Med. 2020, 217, e20192195. [Google Scholar] [CrossRef]
- Wang, X.; Ota, N.; Manzanillo, P.; Kates, L.; Zavala-Solorio, J.; Eidenschenk, C.; Zhang, J.; Lesch, J.; Lee, W.P.; Ross, J.; et al. Interleukin-22 alleviates metabolic disorders and restores mucosal immunity in diabetes. Nature 2014, 514, 237–241. [Google Scholar] [CrossRef]
- Zenewicz, L.A.; Yancopoulos, G.D.; Valenzuela, D.M.; Murphy, A.J.; Karow, M.; Flavell, R.A. Interleukin-22 but not interleukin-17 provides protection to hepatocytes during acute liver inflammation. Immunity 2007, 27, 647–659. [Google Scholar] [CrossRef]
- Yang, L.; Zhang, Y.; Wang, L.; Fan, F.; Zhu, L.; Li, Z.; Ruan, X.; Huang, H.; Wang, Z.; Huang, Z.; et al. Amelioration of high fat diet induced liver lipogenesis and hepatic steatosis by interleukin-22. J. Hepatol. 2010, 53, 339–347. [Google Scholar] [CrossRef]
- Hamaguchi, M.; Okamura, T.; Fukuda, T.; Nishida, K.; Yoshimura, Y.; Hashimoto, Y.; Ushigome, E.; Nakanishi, N.; Majima, S.; Asano, M.; et al. Group 3 Innate Lymphoid Cells Protect Steatohepatitis From High-Fat Diet Induced Toxicity. Front. Immunol. 2021, 12, 648754. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.; He, Y.; Xiang, X.; Seo, W.; Kim, S.J.; Ma, J.; Ren, T.; Park, S.H.; Zhou, Z.; Feng, D.; et al. Interleukin-22 Ameliorates Neutrophil-Driven Nonalcoholic Steatohepatitis Through Multiple Targets. Hepatology 2020, 72, 412–429. [Google Scholar] [CrossRef] [PubMed]
- Ohkura, N.; Sakaguchi, S. Transcriptional and epigenetic basis of Treg cell development and function: Its genetic anomalies or variations in autoimmune diseases. Cell Res. 2020, 30, 465–474. [Google Scholar] [CrossRef] [PubMed]
- Scheinecker, C.; Göschl, L.; Bonelli, M. Treg cells in health and autoimmune diseases: New insights from single cell analysis. J. Autoimmun. 2020, 110, 102376. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Hua, J.; Mohamood, A.R.; Hamad, A.R.; Ravi, R.; Li, Z. A high-fat diet and regulatory T cells influence susceptibility to endotoxin-induced liver injury. Hepatology 2007, 46, 1519–1529. [Google Scholar] [CrossRef] [PubMed]
- Roh, Y.S.; Kim, J.W.; Park, S.; Shon, C.; Kim, S.; Eo, S.K.; Kwon, J.K.; Lim, C.W.; Kim, B. Toll-Like Receptor-7 Signaling Promotes Nonalcoholic Steatohepatitis by Inhibiting Regulatory T Cells in Mice. Am. J. Pathol. 2018, 188, 2574–2588. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Li, L.; Feng, K.; Fan, D.; Xue, W.; Lu, J. Repair Treg Cells in Tissue Injury. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2017, 43, 2155–2169. [Google Scholar] [CrossRef]
- Katz, S.C.; Ryan, K.; Ahmed, N.; Plitas, G.; Chaudhry, U.I.; Kingham, T.P.; Naheed, S.; Nguyen, C.; Somasundar, P.; Espat, N.J.; et al. Obstructive jaundice expands intrahepatic regulatory T cells, which impair liver T lymphocyte function but modulate liver cholestasis and fibrosis. J. Immunol. 2011, 187, 1150–1156. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yang, K.Y.; Tam, R.C.Y.; Chan, V.W.; Lan, H.Y.; Hori, S.; Zhou, B.; Lui, K.O. Regulatory T-cells regulate neonatal heart regeneration by potentiating cardiomyocyte proliferation in a paracrine manner. Theranostics 2019, 9, 4324–4341. [Google Scholar] [CrossRef]
- Wu, J.; Ren, B.; Wang, D.; Lin, H. Regulatory T cells in skeletal muscle repair and regeneration: Recent insights. Cell Death Dis. 2022, 13, 680. [Google Scholar] [CrossRef]
- Savage, T.M.; Fortson, K.T.; de Los Santos-Alexis, K.; Oliveras-Alsina, A.; Rouanne, M.; Rae, S.S.; Gamarra, J.R.; Shayya, H.; Kornberg, A.; Cavero, R.; et al. Amphiregulin from regulatory T cells promotes liver fibrosis and insulin resistance in non-alcoholic steatohepatitis. Immunity 2024, 57, 303–318.e306. [Google Scholar] [CrossRef]
- Ma, C.; Kesarwala, A.H.; Eggert, T.; Medina-Echeverz, J.; Kleiner, D.E.; Jin, P.; Stroncek, D.F.; Terabe, M.; Kapoor, V.; ElGindi, M.; et al. NAFLD causes selective CD4+ T lymphocyte loss and promotes hepatocarcinogenesis. Nature 2016, 531, 253–257. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, B.; Brown, Z.J.; Diggs, L.P.; Vormehr, M.; Ma, C.; Subramanyam, V.; Rosato, U.; Ruf, B.; Walz, J.S.; McVey, J.C.; et al. Steatohepatitis Impairs T-cell-Directed Immunotherapies Against Liver Tumors in Mice. Gastroenterology 2021, 160, 331–345.e336. [Google Scholar] [CrossRef] [PubMed]
- Lei, Z.; Yu, J.; Wu, Y.; Shen, J.; Lin, S.; Xue, W.; Mao, C.; Tang, R.; Sun, H.; Qi, X.; et al. CD1d protects against hepatocyte apoptosis in non-alcoholic steatohepatitis. J. Hepatol. 2024, 80, 194–208. [Google Scholar] [CrossRef] [PubMed]
- Baboota, R.K.; Rawshani, A.; Bonnet, L.; Li, X.; Yang, H.; Mardinoglu, A.; Tchkonia, T.; Kirkland, J.L.; Hoffmann, A.; Dietrich, A.; et al. BMP4 and Gremlin 1 regulate hepatic cell senescence during clinical progression of NAFLD/NASH. Nat. Metab. 2022, 4, 1007–1021. [Google Scholar] [CrossRef] [PubMed]
- Kang, T.W.; Yevsa, T.; Woller, N.; Hoenicke, L.; Wuestefeld, T.; Dauch, D.; Hohmeyer, A.; Gereke, M.; Rudalska, R.; Potapova, A.; et al. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature 2011, 479, 547–551. [Google Scholar] [CrossRef]
- Hirsova, P.; Gores, G.J. Fatty liver progression and carcinogenesis: Beware of pathogenic T cells. Med 2021, 2, 453–455. [Google Scholar] [CrossRef] [PubMed]
- Shalapour, S.; Lin, X.J.; Bastian, I.N.; Brain, J.; Burt, A.D.; Aksenov, A.A.; Vrbanac, A.F.; Li, W.; Perkins, A.; Matsutani, T.; et al. Inflammation-induced IgA+ cells dismantle anti-liver cancer immunity. Nature 2017, 551, 340–345. [Google Scholar] [CrossRef] [PubMed]
- Gomes, A.L.; Teijeiro, A.; Buren, S.; Tummala, K.S.; Yilmaz, M.; Waisman, A.; Theurillat, J.P.; Perna, C.; Djouder, N. Metabolic Inflammation-Associated IL-17A Causes Non-alcoholic Steatohepatitis and Hepatocellular Carcinoma. Cancer Cell 2016, 30, 161–175. [Google Scholar] [CrossRef]
- Zheng, Q.; Martin, R.C.; Shi, X.; Pandit, H.; Yu, Y.; Liu, X.; Guo, W.; Tan, M.; Bai, O.; Meng, X.; et al. Lack of FGF21 promotes NASH-HCC transition via hepatocyte-TLR4-IL-17A signaling. Theranostics 2020, 10, 9923–9936. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, H.; Wang, Y.; Brown, Z.J.; Xia, Y.; Huang, Z.; Shen, C.; Hu, Z.; Beane, J.; Ansa-Addo, E.A.; et al. Regulatory T-cell and neutrophil extracellular trap interaction contributes to carcinogenesis in non-alcoholic steatohepatitis. J. Hepatol. 2021, 75, 1271–1283. [Google Scholar] [CrossRef] [PubMed]
- Zheng, B.; Wang, D.; Qiu, X.; Luo, G.; Wu, T.; Yang, S.; Li, Z.; Zhu, Y.; Wang, S.; Wu, R.; et al. Trajectory and Functional Analysis of PD-1(high) CD4+CD8+ T Cells in Hepatocellular Carcinoma by Single-Cell Cytometry and Transcriptome Sequencing. Adv. Sci. 2020, 7, 2000224. [Google Scholar] [CrossRef] [PubMed]
- Chaoul, N.; Mancarella, S.; Lupo, L.; Giannelli, G.; Dituri, F. Impaired Anti-Tumor T cell Response in Hepatocellular Carcinoma. Cancers 2020, 12, 627. [Google Scholar] [CrossRef]
- Rakhra, K.; Bachireddy, P.; Zabuawala, T.; Zeiser, R.; Xu, L.; Kopelman, A.; Fan, A.C.; Yang, Q.; Braunstein, L.; Crosby, E.; et al. CD4+ T cells contribute to the remodeling of the microenvironment required for sustained tumor regression upon oncogene inactivation. Cancer Cell 2010, 18, 485–498. [Google Scholar] [CrossRef] [PubMed]
- Dhanasekaran, R.; Park, J.; Yevtodiyenko, A.; Bellovin, D.I.; Adam, S.J.; Kd, A.R.; Gabay, M.; Fernando, H.; Arzeno, J.; Arjunan, V.; et al. MYC ASO Impedes Tumorigenesis and Elicits Oncogene Addiction in Autochthonous Transgenic Mouse Models of HCC and RCC. Mol. Ther. Nucleic Acids 2020, 21, 850–859. [Google Scholar] [CrossRef]
- Zheng, C.; Snow, B.E.; Elia, A.J.; Nechanitzky, R.; Dominguez-Brauer, C.; Liu, S.; Tong, Y.; Cox, M.A.; Focaccia, E.; Wakeham, A.C.; et al. Tumor-specific cholinergic CD4+ T lymphocytes guide immunosurveillance of hepatocellular carcinoma. Nat. Cancer 2023, 4, 1437–1454. [Google Scholar] [CrossRef]
- Huang, M.; Huang, X.; Huang, N. Exosomal circGSE1 promotes immune escape of hepatocellular carcinoma by inducing the expansion of regulatory T cells. Cancer Sci. 2022, 113, 1968–1983. [Google Scholar] [CrossRef]
- Sun, Y.F.; Wu, L.; Liu, S.P.; Jiang, M.M.; Hu, B.; Zhou, K.Q.; Guo, W.; Xu, Y.; Zhong, Y.; Zhou, X.R.; et al. Dissecting spatial heterogeneity and the immune-evasion mechanism of CTCs by single-cell RNA-seq in hepatocellular carcinoma. Nat. Commun. 2021, 12, 4091. [Google Scholar] [CrossRef]
- Qi, X.; Yun, C.; Sun, L.; Xia, J.; Wu, Q.; Wang, Y.; Wang, L.; Zhang, Y.; Liang, X.; Wang, L.; et al. Gut microbiota-bile acid-interleukin-22 axis orchestrates polycystic ovary syndrome. Nat. Med. 2019, 25, 1225–1233. [Google Scholar] [CrossRef]
- Verrastro, O.; Panunzi, S.; Castagneto-Gissey, L.; De Gaetano, A.; Lembo, E.; Capristo, E.; Guidone, C.; Angelini, G.; Pennestrì, F.; Sessa, L.; et al. Bariatric-metabolic surgery versus lifestyle intervention plus best medical care in non-alcoholic steatohepatitis (BRAVES): A multicentre, open-label, randomised trial. Lancet 2023, 401, 1786–1797. [Google Scholar] [CrossRef]
- Rai, R.P.; Liu, Y.; Iyer, S.S.; Liu, S.; Gupta, B.; Desai, C.; Kumar, P.; Smith, T.; Singhi, A.D.; Nusrat, A.; et al. Blocking integrin α(4)β(7)-mediated CD4 T cell recruitment to the intestine and liver protects mice from western diet-induced non-alcoholic steatohepatitis. J. Hepatol. 2020, 73, 1013–1022. [Google Scholar] [CrossRef] [PubMed]
- Jian, C.; Fu, J.; Cheng, X.; Shen, L.J.; Ji, Y.X.; Wang, X.; Pan, S.; Tian, H.; Tian, S.; Liao, R.; et al. Low-Dose Sorafenib Acts as a Mitochondrial Uncoupler and Ameliorates Nonalcoholic Steatohepatitis. Cell Metab. 2020, 31, 892–908.e811. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.; Tanaka, T.; Kai, K.; Matsufuji, S.; Ito, K.; Kitajima, Y.; Manabe, T.; Noshiro, H. Suppression of NASH-Related HCC by Farnesyltransferase Inhibitor through Inhibition of Inflammation and Hypoxia-Inducible Factor-1α Expression. Int. J. Mol. Sci. 2023, 24, 11546. [Google Scholar] [CrossRef] [PubMed]
- Brown, Z.J.; Fu, Q.; Ma, C.; Kruhlak, M.; Zhang, H.; Luo, J.; Heinrich, B.; Yu, S.J.; Zhang, Q.; Wilson, A.; et al. Carnitine palmitoyltransferase gene upregulation by linoleic acid induces CD4+ T cell apoptosis promoting HCC development. Cell Death Dis. 2018, 9, 620. [Google Scholar] [CrossRef] [PubMed]
- Coia, H.; Ma, N.; Hou, Y.; Permaul, E.; Berry, D.L.; Cruz, M.I.; Pannkuk, E.; Girgis, M.; Zhu, Z.; Lee, Y.; et al. Theaphenon E prevents fatty liver disease and increases CD4+ T cell survival in mice fed a high-fat diet. Clin. Nutr. 2021, 40, 110–119. [Google Scholar] [CrossRef] [PubMed]
- Abou-Alfa, G.K.; Lau, G.; Kudo, M.; Chan, S.L.; Kelley, R.K.; Furuse, J.; Sukeepaisarnjaroen, W.; Kang, Y.K.; Van Dao, T.; De Toni, E.N.; et al. Tremelimumab plus Durvalumab in Unresectable Hepatocellular Carcinoma. NEJM Evid. 2022, 1, EVIDoa2100070. [Google Scholar] [CrossRef] [PubMed]
- Fu, Q.; Zheng, Y.; Fang, W.; Zhao, Q.; Zhao, P.; Liu, L.; Zhai, Y.; Tong, Z.; Zhang, H.; Lin, M.; et al. RUNX-3-expressing CAR T cells targeting glypican-3 in patients with heavily pretreated advanced hepatocellular carcinoma: A phase I trial. EClinicalMedicine 2023, 63, 102175. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Shi, D.; Chi, J.; Cui, D.; Tang, X.; Lin, Y.; Wang, S.; Li, Z.; Jin, H.; Zhai, B. Combined local therapy and CAR-GPC3 T-cell therapy in advanced hepatocellular carcinoma: A proof-of-concept treatment strategy. Cancer Commun. 2023, 43, 1064–1068. [Google Scholar] [CrossRef]
- Zhou, M.; Liu, B.; Shen, J. Immunotherapy for hepatocellular carcinoma. Clin. Exp. Med. 2023, 23, 569–577. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Xu, C. Immune checkpoint signaling and cancer immunotherapy. Cell Res. 2020, 30, 660–669. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Process | Subset | Effect | Mechanism | Model | References |
|---|---|---|---|---|---|
| NASH progression | Th1 | Promote | IFN-γ and TNF-α | NASH and NAFLD patient samples, MCD, MCD-HF | [36,44,50] |
| Th2 | Promote | IL-33 | NASH and NAFLD patient samples | [36,62] | |
| Th17 | Promote | IL-17 | MCDD, HFD | [71,72,73] | |
| Th22 | Inhibit | IL-22 | HFD | [80,82,109] | |
| Treg | Bidirectional | TGF-β and Areg (Promote) IL-22 (Inhibit) | MCD, CDAA-HFD | [88,90,91] | |
| NASH–HCC transition | effector CD4+T | Inhibit | Immunological surveillance | MYC-ON MCD | [15,92] |
| Th17 | Promote | IL-17A/IL-17RA axis | --- | [99,100] | |
| Treg | Promote | Immunosuppressive | --- | [101] | |
| HCC progression | ChAT-CD4+T | Inhibit | Cholinergic signal | --- | [105] |
| Treg | Promote | miR-324-5p/TGFBR1/Smad3 axis | --- | [106,107] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miao, Y.; Li, Z.; Feng, J.; Lei, X.; Shan, J.; Qian, C.; Li, J. The Role of CD4+T Cells in Nonalcoholic Steatohepatitis and Hepatocellular Carcinoma. Int. J. Mol. Sci. 2024, 25, 6895. https://doi.org/10.3390/ijms25136895
Miao Y, Li Z, Feng J, Lei X, Shan J, Qian C, Li J. The Role of CD4+T Cells in Nonalcoholic Steatohepatitis and Hepatocellular Carcinoma. International Journal of Molecular Sciences. 2024; 25(13):6895. https://doi.org/10.3390/ijms25136895
Chicago/Turabian StyleMiao, Yadi, Ziyong Li, Juan Feng, Xia Lei, Juanjuan Shan, Cheng Qian, and Jiatao Li. 2024. "The Role of CD4+T Cells in Nonalcoholic Steatohepatitis and Hepatocellular Carcinoma" International Journal of Molecular Sciences 25, no. 13: 6895. https://doi.org/10.3390/ijms25136895
APA StyleMiao, Y., Li, Z., Feng, J., Lei, X., Shan, J., Qian, C., & Li, J. (2024). The Role of CD4+T Cells in Nonalcoholic Steatohepatitis and Hepatocellular Carcinoma. International Journal of Molecular Sciences, 25(13), 6895. https://doi.org/10.3390/ijms25136895