
Tauroursodeoxycholic Acid (TUDCA) Relieves Streptozotocin (STZ)-Induced Diabetic Rat Model via Modulation of Lipotoxicity, Oxidative Stress, Inflammation, and Apoptosis


Abstract
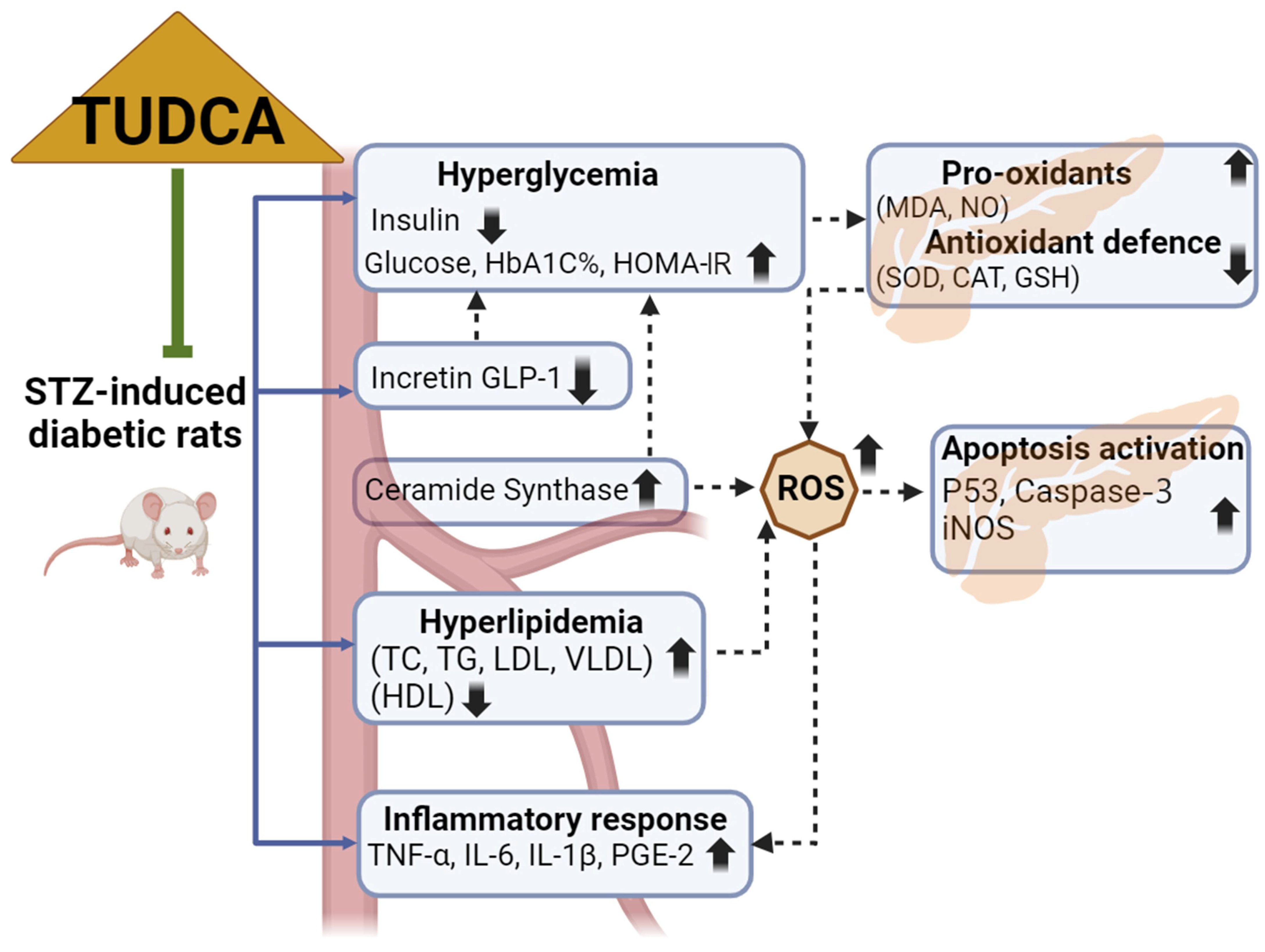
:1. Introduction
2. Results
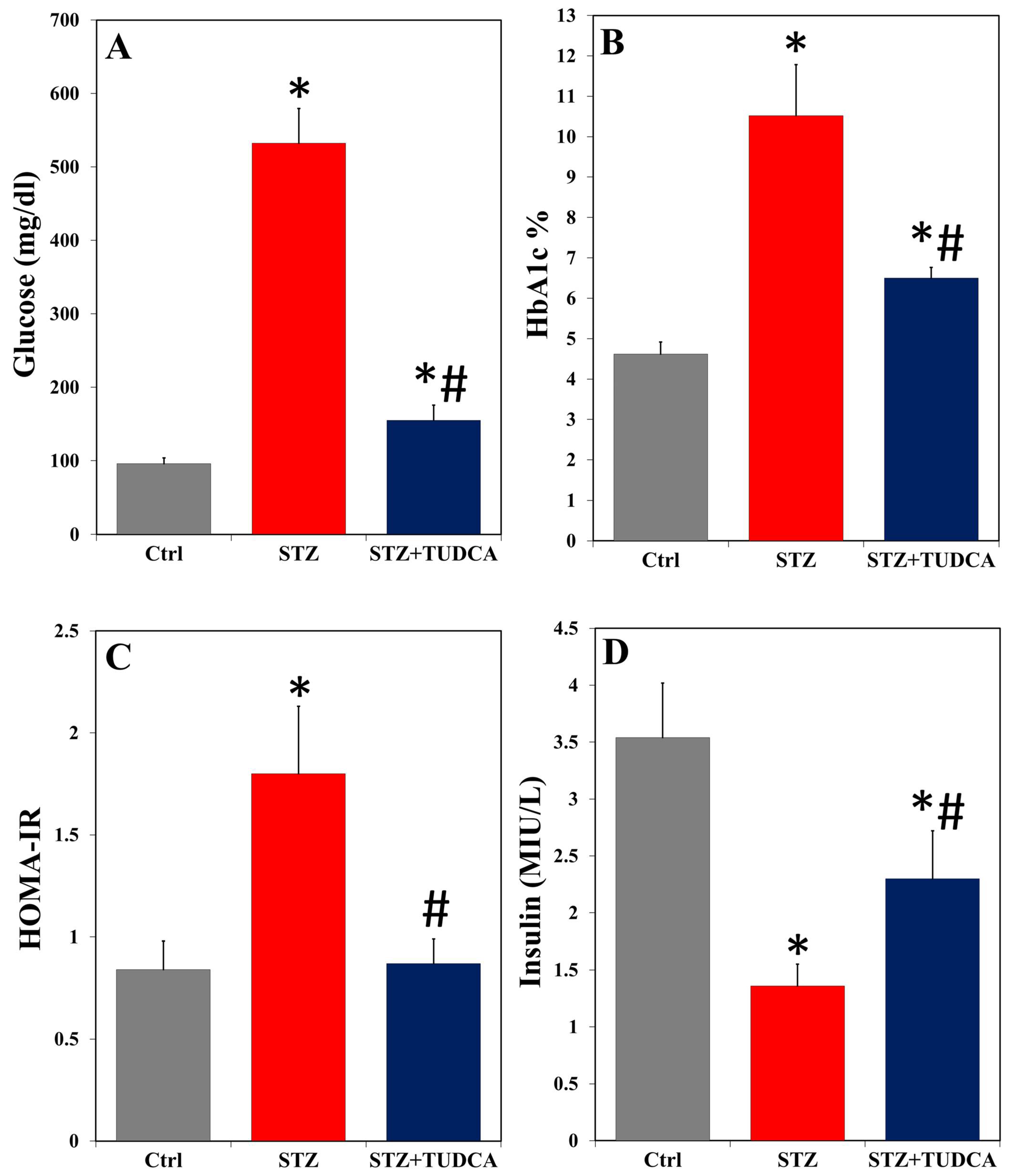
2.1. TUDCA Improves Hyperglycemia in Diabetic Rats
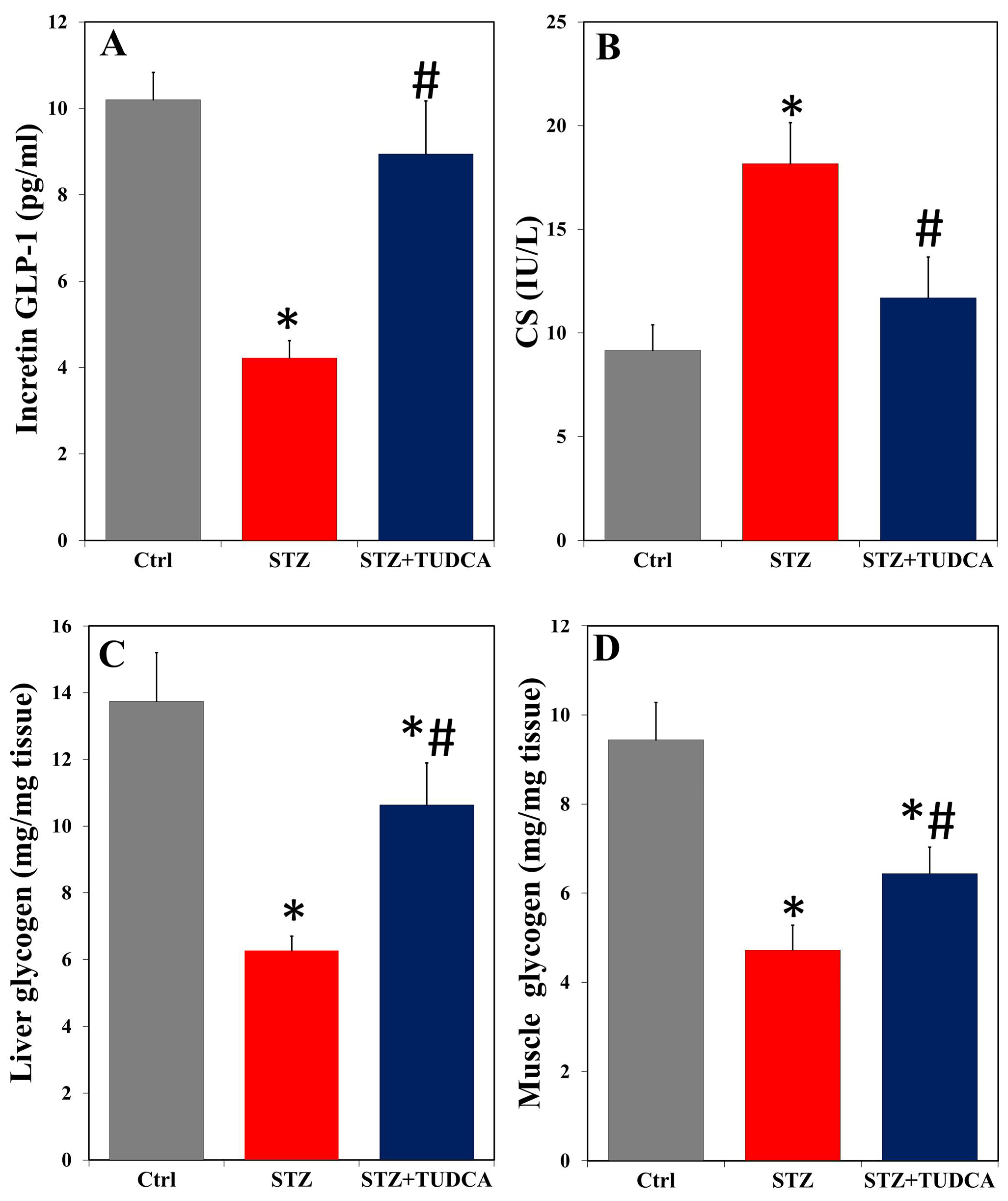
2.2. TUDCA Effects on Serum Incretin GLP-1, CS, and Liver/Muscle Glycogen in Diabetic Rats
2.3. TUDCA Effects on Dyslipidemia in Diabetic Rats
2.4. TUDCA Effects on Proinflammatory Cytokines and PGE-2 in Diabetic Rats
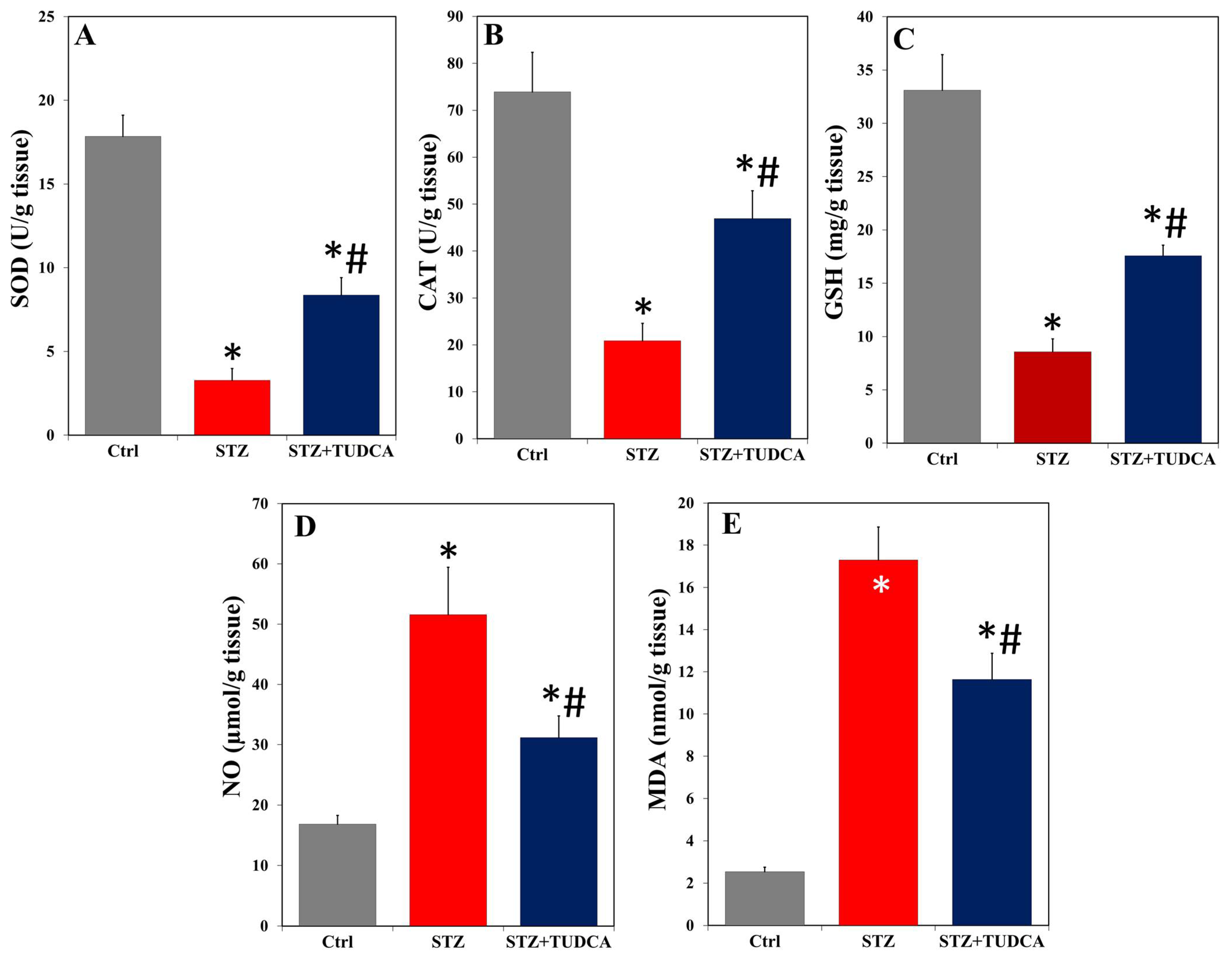
2.5. TUDCA Effects on Pancreatic Redox State in Diabetic Rats
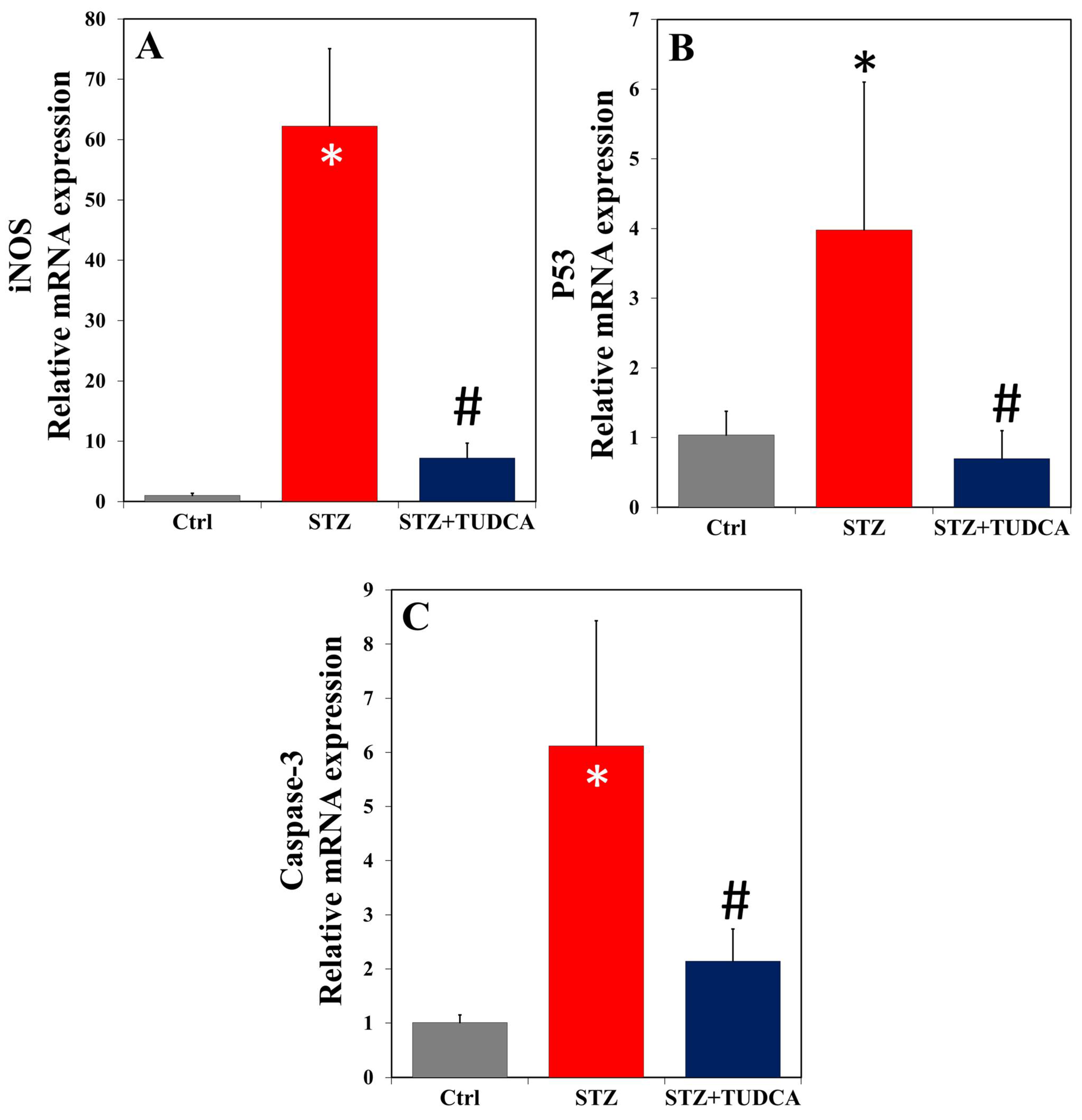
2.6. TUDCA Effects on mRNA Expression of iNOS, p53, and Caspase-3 in Diabetic Rats
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Animals
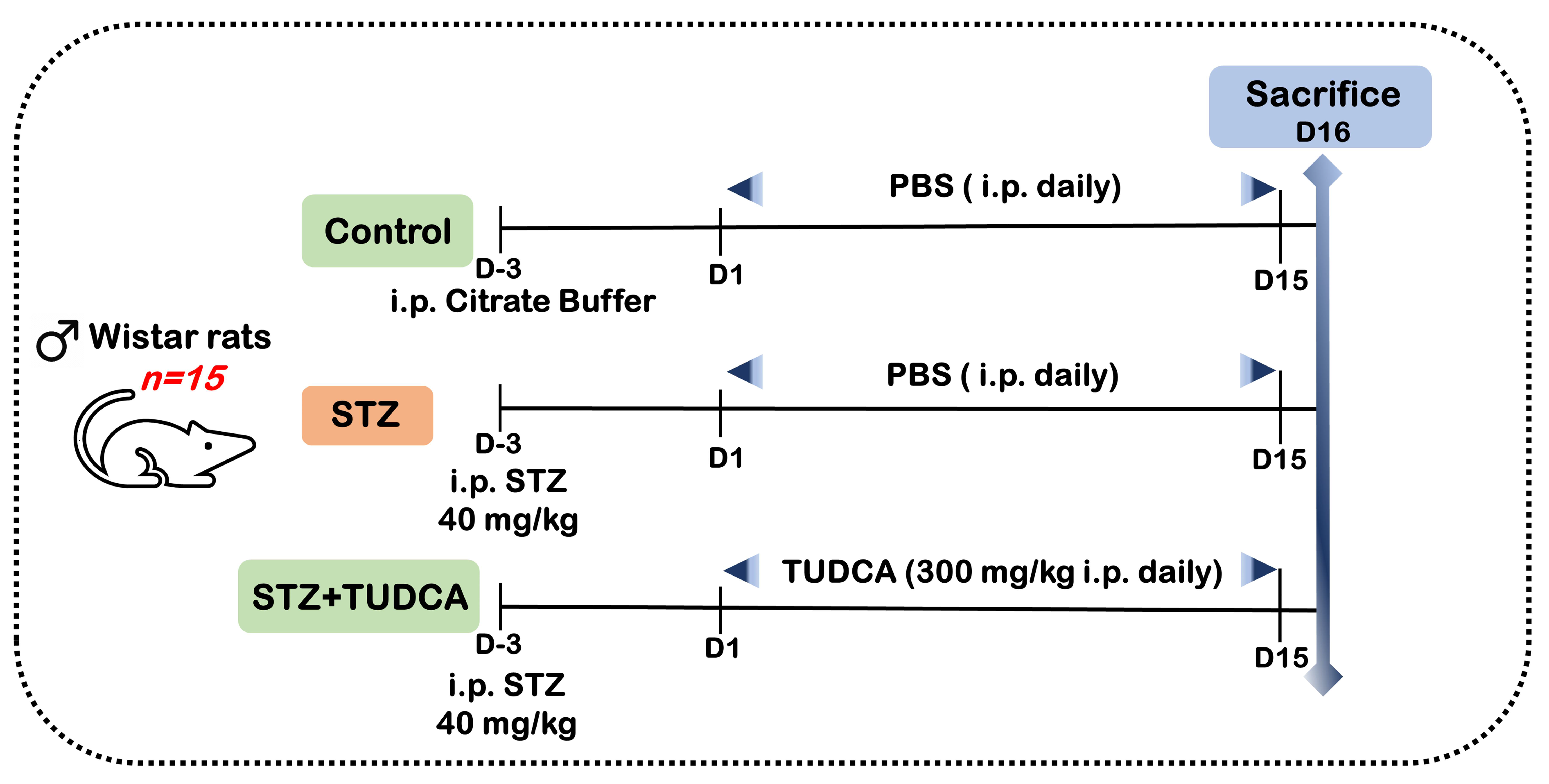
4.3. Experimental Design and Sample Collection
4.4. Evaluation of Serum Glycemic Markers
4.5. Assays for Incretin GLP-1 Level and CS Activity in Serum and Glycogen Quantification in Liver and Muscle
4.6. Quantitative Evaluation of Serum Lipids
4.7. Detection of Serum Inflammatory Response
4.8. Estimation of Oxidative Stress Biomarkers in the Pancreas
4.9. Quantitative Real-Time PCR (qRT-PCR) Assay
- ∆∆Ct = ∆Ct reference − ∆Ct target gene
- Ct = the cycle at threshold level
- reference = GAPDH.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Primer (5′->3′) | Reverse Primer (5′->3′) | GenBank Accession Number |
|---|---|---|---|
| iNOS | GACTGCACAGAATGTTCCAG | TGGCCAGATGTTCCTCTATT | NM_012611 |
| P53 | TAACAGTTCCTGCATGGGCGGC | AGGACAGGCACAAACACGCACC | NM_030989 |
| Caspase-3 | AGTTGGACCCACCTTGTGAG | AGTCTGCAGCTCCTCCACAT | NM_012922 |
| GAPDH | GGTGAAGGTCGGTGT GAACG | CTCG CTCCTGGAAGATGGTG | NM_017008 |
4.10. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Mohamed, N.A.; Ithmil, M.T.; Elkady, A.I.; Abdel Salam, S. Tauroursodeoxycholic Acid (TUDCA) Relieves Streptozotocin (STZ)-Induced Diabetic Rat Model via Modulation of Lipotoxicity, Oxidative Stress, Inflammation, and Apoptosis. Int. J. Mol. Sci. 2024, 25, 6922. [Google Scholar] [CrossRef]
- Wu, Y.; Ding, Y.; Tanaka, Y.; Zhang, W. Risk factors contributing to type 2 diabetes and recent advances in the treatment and prevention. Int. J. Med. Sci. 2014, 11, 1185–1200. [Google Scholar] [CrossRef] [PubMed]
- Farmaki, P.; Damaskos, C.; Garmpis, N.; Garmpi, A.; Savvanis, S.; Diamantis, E. Complications of the type 2 diabetes mellitus. Curr. Cardiol. Rev. 2020, 16, 249–251. [Google Scholar] [CrossRef] [PubMed]
- Oguntibeju, O.O. Type 2 diabetes mellitus, oxidative stress and inflammation: Examining the links. Int. J. Physiol. Pathophysiol. Pharmacol. 2019, 11, 45–63. Available online: http://www.ncbi.nlm.nih.gov/pmc/articles/pmc6628012/ (accessed on 29 April 2024). [PubMed]
- Kaneto, H.; Katakami, N.; Matsuhisa, M.; Matsuoka, T.-A. Role of reactive oxygen species in the progression of type 2 diabetes and atherosclerosis. Mediat. Inflamm. 2010, 2010, 453892. [Google Scholar] [CrossRef] [PubMed]
- Lazaridis, K.N.; Gores, G.J.; Lindor, K.D. Ursodeoxycholic acid ‘mechanisms of action and clinical use in hepatobiliary disorders’. J. Hepatol. 2001, 35, 134–146. [Google Scholar] [CrossRef] [PubMed]
- Vang, S.; Longley, K.; Steer, C.J.; Low, W.C. The Unexpected uses of urso- and tauroursodeoxycholic acid in the treatment of non-liver diseases. Glob. Adv. Health Med. 2014, 3, 58–69. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, C.M.; Solá, S.; Nan, Z.; Castro, R.E.; Ribeiro, P.S.; Low, W.C.; Steer, C.J. Tauroursodeoxycholic acid reduces apoptosis and protects against neurological injury after acute hemorrhagic stroke in rats. Proc. Natl. Acad. Sci. USA 2003, 100, 6087–6092. [Google Scholar] [CrossRef] [PubMed]
- Gaspar, J.M.; Martins, A.; Cruz, R.; Rodrigues, C.M.; Ambrósio, A.F.; Santiago, A.R. Tauroursodeoxycholic acid protects retinal neural cells from cell death induced by prolonged exposure to elevated glucose. Neuroscience 2013, 253, 380–388. [Google Scholar] [CrossRef]
- Zangerolamo, L.; Vettorazzi, J.F.; Solon, C.; Bronczek, G.A.; Engel, D.F.; Kurauti, M.A.; Soares, G.M.; Rodrigues, K.S.; Velloso, L.A.; Boschero, A.C.; et al. The bile acid TUDCA improves glucose metabolism in streptozotocin-induced Alzheimer’s disease mice model. Mol. Cell. Endocrinol. 2021, 521, 111116. [Google Scholar] [CrossRef]
- Kumar, D.; Tandon, R.K. Use of ursodeoxycholic acid in liver diseases. J. Gastroenterol. Hepatol. 2001, 16, 3–14. [Google Scholar] [CrossRef]
- Levy, C.; Lindor, K.D. Current management of primary biliary cirrhosis and primary sclerosing cholangitis. J. Hepatol. 2003, 38, 24–37. [Google Scholar] [CrossRef]
- Rodrigues, C.M.; Spellman, S.R.; Solá, S.; Grande, A.W.; Linehan-Stieers, C.; Low, W.C.; Steer, C.J. Neuroprotection by a bile acid in an acute stroke model in the rat. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2002, 22, 463–471. [Google Scholar] [CrossRef]
- Rivard, A.L.; Steer, C.J.; Kren, B.T.; Rodrigues, C.M.; Castro, R.E.; Bianco, R.W.; Low, W.C. Administration of tauroursodeoxycholic acid (TUDCA) reduces apoptosis following myocardial infarction in rat. Am. J. Chin. Med. 2007, 35, 279–295. [Google Scholar] [CrossRef]
- Çolak, A.; Kelten, B.; Sağmanligil, A.; Akdemir, O.; Karaoğlan, A.; Şahan, E.; Çelik, Ö.; Barut, Ş. Tauroursodeoxycholic acid and secondary damage after spinal cord injury in rats. J. Clin. Neurosci. Off. J. Neurosurg. Soc. Australas. 2008, 15, 665–671. [Google Scholar] [CrossRef]
- Gao, X.; Fu, L.; Xiao, M.; Xu, C.; Sun, L.; Zhang, T.; Zheng, F.; Mei, C. The nephroprotective effect of tauroursodeoxycholic acid on ischaemia/reperfusion-induced acute kidney injury by inhibiting endoplasmic reticulum stress. Basic Clin. Pharmacol. Toxicol. 2012, 111, 14–23. [Google Scholar] [CrossRef]
- Gupta, S.; Li, S.; Abedin, M.J.; Noppakun, K.; Wang, L.; Kaur, T.; Najafian, B.; Rodrigues, C.M.; Steer, C.J. Prevention of acute kidney injury by tauroursodeoxycholic acid in rat and cell culture models. PLoS ONE 2012, 7, e48950. [Google Scholar] [CrossRef]
- Üner, A.K.; Okan, A.; Akyüz, E.; Köklü, B.; Eroğlu, E.; Yilmaz, S.; Ünalmiş, D.; Kaymak, E.; Aslan, F.Ş.; Qureshi, M.Z.; et al. Tauroursodeoxycholic acid (TUDCA) regulates inflammation and hypoxia in autonomic tissues of rats with seizures. Cell. Mol. Biol. 2023, 68, 104–111. [Google Scholar] [CrossRef]
- Arokiyaraj, S.; Balamurugan, R.; Augustian, P. Antihyperglycemic effect of Hypericum perforatum ethyl acetate extract on streptozotocin-induced diabetic rats. Asian Pac. J. Trop. Biomed. 2011, 1, 386–390. [Google Scholar] [CrossRef]
- Eliza, J.; Daisy, P.; Ignacimuthu, S.; Duraipandiyan, V. Antidiabetic and antilipidemic effect of eremanthin from Costus speciosus (Koen.) Sm., in STZ-induced diabetic rats. Chem.-Biol. Interact. 2009, 182, 67–72. [Google Scholar] [CrossRef]
- Toma, A.; Makonnen, E.; Mekonnen, Y.; Debella, A.; Adisakwattana, S. Antidiabetic activities of aqueous ethanol and n-butanol fraction of Moringa stenopetala leaves in streptozotocin-induced diabetic rats. BMC Complement. Altern. Med. 2015, 15, 242. [Google Scholar] [CrossRef] [PubMed]
- AlTamimi, L.; Zakaraya, Z.Z.; Hailat, M.; Ahmad, M.N.; Qinna, N.A.; Hamad, M.F.; Dayyih, W.A. Test of insulin resistance in nondiabetic and streptozotocin-induced diabetic rats using glycosylated hemoglobin test and other interventions. J. Adv. Pharm. Technol. Res. 2024, 15, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Simmons, R.A. Developmental origins of diabetes: The role of oxidative stress. Free. Radic. Biol. Med. 2006, 40, 917–922. [Google Scholar] [CrossRef] [PubMed]
- Ozcan, U.; Yilmaz, E.; Ozcan, L.; Furuhashi, M.; Vaillancourt, E.; Smith, R.O.; Gorgun, C.Z.; Hotamisligil, G.S. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science 2006, 313, 1137–1140. [Google Scholar] [CrossRef] [PubMed]
- Vettorazzi, J.F.; Kurauti, M.A.; Soares, G.M.; Borck, P.C.; Ferreira, S.M.; Branco, R.C.S.; Michelone, L.D.S.L.; Boschero, A.C.; Junior, J.M.C.; Carneiro, E.M. Bile acid TUDCA improves insulin clearance by increasing the expression of insulin-degrading enzyme in the liver of obese mice. Sci. Rep. 2017, 7, 14876. [Google Scholar] [CrossRef] [PubMed]
- da Silva, J.A., Jr.; Figueiredo, L.S.; Chaves, J.O.; Oliveira, K.M.; Carneiro, E.M.; Abreu, P.A.; Ribeiro, R.A. Effects of tauroursodeoxycholic acid on glucose homeostasis: Potential binding of this bile acid with the insulin receptor. Life Sci. 2021, 285, 120020. [Google Scholar] [CrossRef] [PubMed]
- Boer, G.A.; Holst, J.J. Incretin Hormones and type 2 diabetes-mechanistic insights and therapeutic approaches. Biology 2020, 9, 473. [Google Scholar] [CrossRef]
- Goldspink, D.A.; Lu, V.B.; Billing, L.J.; Larraufie, P.; Tolhurst, G.; Gribble, F.M.; Reimann, F. Mechanistic insights into the detection of free fatty and bile acids by ileal glucagon-like peptide-1 secreting cells. Mol. Metab. 2018, 7, 90–101. [Google Scholar] [CrossRef]
- Nielsen, S.; Svane, M.S.; Kuhre, R.E.; Clausen, T.R.; Kristiansen, V.B.; Rehfeld, J.F.; Holst, J.J.; Madsbad, S.; Bojsen-Moller, K.N. Chenodeoxycholic acid stimulates glucagon-like peptide-1 secretion in patients after Roux-en-Y gastric bypass. Physiol. Rep. 2017, 5, e13140. [Google Scholar] [CrossRef]
- Kuhre, R.E.; Albrechtsen, N.J.W.; Larsen, O.; Jepsen, S.L.; Balk-Møller, E.; Andersen, D.B.; Deacon, C.F.; Schoonjans, K.; Reimann, F.; Gribble, F.M.; et al. Bile acids are important direct and indirect regulators of the secretion of appetite- and metabolism-regulating hormones from the gut and pancreas. Mol. Metab. 2018, 11, 84–95. [Google Scholar] [CrossRef]
- El-Kordy, E.A.; Alshahrani, A.M. Effect of genistein, a natural soy isoflavone, on pancreatic β-cells of streptozotocin-induced diabetic rats: Histological and immunohistochemical study. J. Microsc. Ultrastruct. 2015, 3, 108–119. [Google Scholar] [CrossRef]
- Yadav, S.K.; Nagori, B.P.; Desai, P.K. Pharmacological characterization of different fractions of Calotropis procera (Asclepiadaceae) in streptozotocin induced experimental model of diabetic neuropathy. J. Ethnopharmacol. 2014, 152, 349–357. [Google Scholar] [CrossRef] [PubMed]
- Altura, B.M.; Shah, N.C.; Shah, G.; Zhang, A.; Li, W.; Zheng, T.; Perez-Albela, J.L.; Altura, B.T. Short-term magnesium deficiency upregulates ceramide synthase in cardiovascular tissues and cells: Cross-talk among cytokines, Mg2+, NF-κB, and de novo ceramide. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H319–H332. [Google Scholar] [CrossRef]
- Mandal, N.; Grambergs, R.; Mondal, K.; Basu, S.K.; Tahia, F.; Dagogo-Jack, S. Role of ceramides in the pathogenesis of diabetes mellitus and its complications. J. Diabetes Its Complicat. 2021, 35, 107734. [Google Scholar] [CrossRef] [PubMed]
- Boslem, E.; Meikle, P.J.; Biden, T.J. Roles of ceramide and sphingolipids in pancreatic β-cell function and dysfunction. Islets 2012, 4, 177–187. [Google Scholar] [CrossRef] [PubMed]
- Galadari, S.; Rahman, A.; Pallichankandy, S.; Galadari, A.; Thayyullathil, F. Role of ceramide in diabetes mellitus: Evidence and mechanisms. Lipids Health Dis. 2013, 12, 98. [Google Scholar] [CrossRef]
- Huang, W.C.; Chen, C.L.; Lin, Y.S.; Lin, C.F. Apoptotic sphingolipid ceramide in cancer therapy. J. Lipids 2011, 2011, 565316. [Google Scholar] [CrossRef]
- Novgorodov, S.A.; Gudz, T.I. Ceramide and mitochondria in ischemia/reperfusion. J. Cardiovasc. Pharmacol. 2009, 53, 198–208. [Google Scholar] [CrossRef] [PubMed]
- Oh, Y.S.; Bae, G.D.; Baek, D.J.; Park, E.Y.; Jun, H.S. Fatty acid-induced lipotoxicity in pancreatic beta-cells during development of type 2 diabetes. Front. Endocrinol. 2018, 9, 384. [Google Scholar] [CrossRef]
- Liu, Z.; Xia, Y.; Li, B.; Xu, H.; Wang, C.; Liu, Y.; Li, Y.; Li, C.; Gao, N.; Li, L. Induction of ER stress-mediated apoptosis by ceramide via disruption of ER Ca2+ homeostasis in human adenoid cystic carcinoma cells. Cell Biosci. 2014, 4, 71. [Google Scholar] [CrossRef]
- Arabshomali, A.; Bazzazzadehgan, S.; Mahdi, F.; Shariat-Madar, Z. Potential benefits of antioxidant phytochemicals in type 2 diabetes. Molecules 2023, 28, 7209. [Google Scholar] [CrossRef] [PubMed]
- Cui, N.; Zhang, W.; Su, F.; Zhang, Z.; Qiao, W.; Sun, Y.; Yang, B.; Kuang, H.; Wang, Q. Metabolomics and lipidomics study unveils the impact of tauroursodeoxycholic acid on hyperlipidemic mice. Molecules 2023, 28, 6352. [Google Scholar] [CrossRef] [PubMed]
- Balasubashini, M.S.; Rukkumani, R.; Viswanathan, P.; Menon, V.P. Ferulic acid alleviates lipid peroxidation in diabetic rats. Phytother. Res. 2004, 18, 310–314. [Google Scholar] [CrossRef] [PubMed]
- Pillon, N.J.; Vella, R.E.; Soulère, L.; Becchi, M.; Lagarde, M.; Soulage, C.O. Structural and functional changes in human insulin induced by the lipid peroxidation byproducts 4-hydroxy-2-nonenal and 4-hydroxy-2-hexenal. Chem. Res. Toxicol. 2011, 24, 752–762. [Google Scholar] [CrossRef] [PubMed]
- Tangvarasittichai, S. Oxidative stress, insulin resistance, dyslipidemia and type 2 diabetes mellitus. World J. Diabetes 2015, 6, 456–480. [Google Scholar] [CrossRef] [PubMed]
- Caturano, A.; D’Angelo, M.; Mormone, A.; Russo, V.; Mollica, M.P.; Salvatore, T.; Galiero, R.; Rinaldi, L.; Vetrano, E.; Marfella, R.; et al. Oxidative stress in type 2 diabetes: Impacts from pathogenesis to lifestyle modifications. Curr. Issues Mol. Biol. 2023, 45, 6651–6666. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, B.K.; Pandey, K.B.; Abidi, A.B.; Rizvi, S.I. Markers of oxidative stress during diabetes mellitus. J. Biomark. 2013, 2013, 378790. [Google Scholar] [CrossRef] [PubMed]
- Góth, L.; Eaton, J.W. Hereditary catalase deficiencies and increased risk of diabetes. Lancet 2000, 356, 1820–1821. [Google Scholar] [CrossRef] [PubMed]
- Tuell, D.; Ford, G.; Los, E.; Stone, W. The Role of glutathione and its precursors in type 2 diabetes. Antioxidants 2024, 13, 184. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, Y. Tauroursodeoxycholic acid alleviates H2O2-induced oxidative stress and apoptosis via suppressing endoplasmic reticulum stress in neonatal rat cardiomyocytes. Dose-Response 2018, 9, 1559325818782631. [Google Scholar] [CrossRef]
- Alhasani, R.H.; Almarhoun, M.; Zhou, X.; Reilly, J.; Patterson, S.; Zeng, Z.; Shu, X. Tauroursodeoxycholic acid protects retinal pigment epithelial cells from oxidative injury and endoplasmic reticulum stress in vitro. Biomedicines 2020, 8, 367. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Luan, J.; Huang, T.; Deng, T.; Li, X.; Xiao, Z.; Zhan, J.; Luo, D.; Hou, Y.; Xu, L.; et al. Tauroursodeoxycholic acid alleviates secondary injury in spinal cord injury mice by reducing oxidative stress, apoptosis, and inflammatory response. J. Neuroinflamm. 2021, 18, 216. [Google Scholar] [CrossRef] [PubMed]
- Moreira, S.; Fonseca, I.; Nunes, M.J.; Rosa, A.; Lemos, L.; Rodrigues, E.; Carvalho, A.N.; Outeiro, T.F.; Rodrigues, C.M.P.; Gama, M.J.; et al. Nrf2 activation by tauroursodeoxycholic acid in experimental models of Parkinson’s disease. Exp. Neurol. 2017, 295, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Keane, K.N.; Cruzat, V.F.; Carlessi, R.; de Bittencourt, P.I.H.; Newsholme, P. Molecular events linking oxidative stress and inflammation to insulin resistance and β-cell dysfunction. Oxidative Med. Cell. Longev. 2015, 2015, 181643. [Google Scholar] [CrossRef]
- Busa, P.; Kuthati, Y.; Huang, N.; Wong, C.S. New Advances on pathophysiology of diabetes neuropathy and pain management: Potential role of melatonin and DPP-4 inhibitors. Front. Pharmacol. 2022, 13, 864088. [Google Scholar] [CrossRef]
- Kim, S.J.; Ko, W.K.; Jo, M.J.; Arai, Y.; Choi, H.; Kumar, H.; Han, I.B.; Sohn, S. Anti-inflammatory effect of Tauroursodeoxycholic acid in RAW 264.7 macrophages, Bone marrow-derived macrophages, BV2 microglial cells, and spinal cord injury. Sci. Rep. 2018, 8, 3176. [Google Scholar] [CrossRef] [PubMed]
- Yanguas-Casás, N.; Barreda-Manso, M.A.; Nieto-Sampedro, M.; Romero-Ramírez, L. Tauroursodeoxycholic acid reduces glial cell activation in an animal model of acute neuroinflammation. J. Neuroinflamm. 2014, 11, 50. [Google Scholar] [CrossRef] [PubMed]
- Dludla, P.V.; Mabhida, S.E.; Ziqubu, K.; Nkambule, B.B.; Mazibuko-Mbeje, S.E.; Hanser, S.; Basson, A.K.; Pheiffer, C.; Kengne, A.P. Pancreatic β-cell dysfunction in type 2 diabetes: Implications of inflammation and oxidative stress. World J. Diabetes 2023, 14, 130–146. [Google Scholar] [CrossRef]
- Peng, J.; Li, X.; Zhang, D.; Chen, J.K.; Su, Y.; Smith, S.B.; Dong, Z. Hyperglycemia, p53, and mitochondrial pathway of apoptosis are involved in the susceptibility of diabetic models to ischemic acute kidney injury. Kidney Int. 2015, 87, 137–150. [Google Scholar] [CrossRef]
- Kung, C.P.; Murphy, M.E. The role of the p53 tumor suppressor in metabolism and diabetes. J. Endocrinol. 2016, 231, R61–R75. Available online: https://pubmed.ncbi.nlm.nih.gov/?term=Murphy+ME&cauthor_id=27613337 (accessed on 29 April 2024). [CrossRef]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2016, 1863, 2977–2992. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-M.; Bombeck, C.A.; Billiar, T.R. Nitric oxide as a bifunctional regulator of apoptosis. Circ. Res. 1999, 84, 253–256. [Google Scholar] [CrossRef] [PubMed]
- Khalaf, K.; Tornese, P.; Cocco, A.; Albanese, A. Tauroursodeoxycholic acid: A potential therapeutic tool in neurodegenerative diseases. Transl. Neurodegener. 2022, 11, 33. [Google Scholar] [CrossRef] [PubMed]
- Song, G.; Weng, F.; Zou, B.; Zhao, J.; Jin, J.; Yan, D.; Huang, K.; Sun, X.; Liu, C.; Hu, Y.; et al. Potential therapeutic action of tauroursodeoxycholic acid against cholestatic liver injury via hepatic Fxr/Nrf2 and CHOP-DR5-caspase-8 pathway. Clin. Sci. 2023, 137, 561–577. [Google Scholar] [CrossRef] [PubMed]
- Amaral, J.D.; Viana, R.J.S.; Ramalho, R.M.; Steer, C.J.; Rodrigues, C.M.P. Bile acids: Regulation of apoptosis by ursodeoxycholic acid. J. Lipid Res. 2009, 50, 1721–1734. [Google Scholar] [CrossRef]
- Aloud, A.A.; Veeramani, C.; Govindasamy, C.; Alsaif, M.A.; El Newehy, A.S.; Al-Numair, K.S. Galangin, a dietary flavonoid, improves antioxidant status and reduces hyperglycemia-mediated oxidative stress in streptozotocin-induced diabetic rats. Redox Rep. Commun. Free. Radic. Res. 2017, 22, 290–300. [Google Scholar] [CrossRef]
- Trinder, P. Determination of blood glucose using an oxidase-peroxidase system with a non-carcinogenic chromogen. J. Clin. Pathol. 1969, 22, 158–161. [Google Scholar] [CrossRef]
- Matthews, D.R.; Hosker, J.P.; Rudenski, A.S.; Naylor, B.A.; Treacher, D.F.; Turner, R.C. Homeostasis model assessment: Insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia 1985, 28, 412–419. [Google Scholar] [CrossRef]
- Nathan, D.M.; Singer, D.E.; Hurxthal, K.; Goodson, J.D. The clinical information value of the glycosylated hemoglobin assay. N. Engl. J. Med. 1984, 310, 341–346. [Google Scholar] [CrossRef]
- Huijing, F. A rapid enzymic method for glycogen estimation in very small tissue samples. Clin. Chim. Acta Int. J. Clin. Chem. 1970, 30, 567–572. [Google Scholar] [CrossRef]
- Allain, C.C.; Poon, L.S.; Chan, C.S.; Richmond, W.F.P.C.; Fu, P.C. Enzymatic determination of total serum cholesterol. Clin. Chem. 1974, 20, 470–475. [Google Scholar] [CrossRef] [PubMed]
- Bucolo, G.; David, H. Quantitative determination of serum triglycerides by the use of enzymes. Clin. Chem. 1973, 19, 476–482. [Google Scholar] [CrossRef] [PubMed]
- Lopes-Virella, M.F.; Stone, P.; Ellis, S.; Colwell, J.A. Cholesterol determination in high density lipoproteins separated by three different methods. Clin. Chem. 1977, 23, 882–884. [Google Scholar] [CrossRef] [PubMed]
- Friedewald, W.T.; Levy, R.I.; Fredrickson, D.S. Estimation of the concentration of low-density lipoprotein cholesterol in plasma, without use of the preparative ultracentrifuge. Clin. Chem. 1972, 18, 499–502. [Google Scholar] [CrossRef]
- Nishikimi, M.; Appaji Rao, N.; Yagi, K. The occurrence of superoxide anion in the reaction of reduced phenazine methosulfate and molecular oxygen. Biochem. Biophys. Res. Commun. 1972, 46, 849–854. [Google Scholar] [CrossRef] [PubMed]
- Aebi, H. Catalase in vitro. Methods Enzymol. 1984, 105, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Beutler, E.; Duron, O.; Kelly, B.M. Improved method for the determination of blood glutathione. J. Lab. Clin. Med. 1963, 61, 882–888. [Google Scholar]
- Montgomery, H.; Dymock, J. Determination of Nitrite in Water, Royal Soc Chemistry Thomas Graham House; Science Park, Milton Rd: Cambridge, UK, 1961. [Google Scholar]
- Ohkawa, H.; Ohishi, N.; Yagi, K. Assay for lipid peroxides in animal tissues by thiobarbituric acid reaction. Anal. Biochem. 1979, 95, 351–358. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mohamed, N.A.; Ithmil, M.T.; Elkady, A.I.; Abdel Salam, S. Tauroursodeoxycholic Acid (TUDCA) Relieves Streptozotocin (STZ)-Induced Diabetic Rat Model via Modulation of Lipotoxicity, Oxidative Stress, Inflammation, and Apoptosis. Int. J. Mol. Sci. 2024, 25, 6922. https://doi.org/10.3390/ijms25136922
Mohamed NA, Ithmil MT, Elkady AI, Abdel Salam S. Tauroursodeoxycholic Acid (TUDCA) Relieves Streptozotocin (STZ)-Induced Diabetic Rat Model via Modulation of Lipotoxicity, Oxidative Stress, Inflammation, and Apoptosis. International Journal of Molecular Sciences. 2024; 25(13):6922. https://doi.org/10.3390/ijms25136922
Chicago/Turabian StyleMohamed, Nema A., Mohammed T. Ithmil, Ayman I. Elkady, and Sherine Abdel Salam. 2024. "Tauroursodeoxycholic Acid (TUDCA) Relieves Streptozotocin (STZ)-Induced Diabetic Rat Model via Modulation of Lipotoxicity, Oxidative Stress, Inflammation, and Apoptosis" International Journal of Molecular Sciences 25, no. 13: 6922. https://doi.org/10.3390/ijms25136922
APA StyleMohamed, N. A., Ithmil, M. T., Elkady, A. I., & Abdel Salam, S. (2024). Tauroursodeoxycholic Acid (TUDCA) Relieves Streptozotocin (STZ)-Induced Diabetic Rat Model via Modulation of Lipotoxicity, Oxidative Stress, Inflammation, and Apoptosis. International Journal of Molecular Sciences, 25(13), 6922. https://doi.org/10.3390/ijms25136922