
Molecular Origins of the Mendelian Rare Diseases Reviewed by Orpha.net: A Structural Bioinformatics Investigation

 , , and
, , and 
Abstract
:
1. Introduction
2. Results
2.1. Deriving Structural Information from the Orpha.net Database
2.2. Topological Assignments of Protein Mutants Responsible for MRD
2.3. Predicting the Structural Effects of Specific Amino Acid Replacements
3. Discussion
4. Materials and Methods
4.1. Dataset of Missense Variants
4.2. Structural Analysis
4.3. Atom Depth Calculations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nguengang Wakap, S.; Lambert, D.M.; Olry, A.; Rodwell, C.; Gueydan, C.; Lanneau, V.; Murphy, D.; Le Cam, Y.; Rath, A. Estimating cumulative point prevalence of rare diseases: Analysis of the Orphanet database. Eur. J. Hum. Genet. 2020, 28, 165–173. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, T.P.; Crystal, R.G. Genetic medicines: Treatment strategies for hereditary disorders. Nat. Rev. Genet. 2006, 7, 261–276. [Google Scholar] [CrossRef] [PubMed]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Hoover, J.; et al. ClinVar: Public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 2016, 44, D862–D868. [Google Scholar] [CrossRef] [PubMed]
- Laddach, A.; Ng, J.C.F.; Fraternali, F. Pathogenic missense protein variants affect different functional pathways and proteomic features than healthy population variants. PLoS Biol. 2021, 19, e3001207. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. 2000. Available online: http://www.rcsb.org/ (accessed on 12 October 2023).
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- The UniProt Consortium; Bateman, A.; Martin, M.-J.; Orchard, S.; Magrane, M.; Ahmad, S.; Alpi, E.; Bowler-Barnett, E.H.; Britto, R.; Bye-A-Jee, H.; et al. UniProt: The Universal Protein Knowledgebase in 2023. Nucleic Acids Res. 2023, 51, D523–D531. [Google Scholar] [CrossRef]
- Cheng, J.; Novati, G.; Pan, J.; Bycroft, C.; Žemgulytė, A.; Applebaum, T.; Pritzel, A.; Wong, L.H.; Zielinski, M.; Sargeant, T.; et al. Accurate proteome-wide missense variant effect prediction with AlphaMissense. Science 2023, 381, eadg7492. [Google Scholar] [CrossRef] [PubMed]
- Pavan, S.; Rommel, K.; Marquina, M.E.M.; Höhn, S.; Lanneau, V.; Rath, A. Clinical practice guidelines for rare diseases: The orphanet database. PLoS ONE 2017, 12, e0170365. [Google Scholar] [CrossRef] [PubMed]
- Bongini, P.; Niccolai, N.; Trezza, A.; Mangiavacchi, G.; Santucci, A.; Spiga, O.; Bianchini, M.; Gardini, S. Structural Bioinformatic Survey of Protein-Small Molecule Interfaces Delineates the Role of Glycine in Surface Pocket Formation. IEEE/ACM Trans. Comput. Biol. Bioinform. 2022, 19, 1881–1886. [Google Scholar] [CrossRef]
- Grantham, R. Amino Acid Difference Formula to Help Explain Protein Evolution. Science 1974, 185, 862–864. [Google Scholar] [CrossRef]
- Epstein, C.J. Non-randomness of Ammo-acid Changes in the Evolution of Homologous Proteins. Nature 1967, 215, 355–359. [Google Scholar] [CrossRef] [PubMed]
- Miyata, T.; Miyazawa, S.; Yasunaga, T. Two Types of Amino Acid Substitutions in Protein Evolution. J. Mol. Evol. 1979, 12, 219–236. [Google Scholar] [CrossRef]
- Teng, S.; Srivastava, A.K.; Schwartz, C.E.; Alexov, E.; Wang, L. Structural assessment of the effects of Amino Acid Substitutions on protein stability and protein protein interaction. Int. J. Comput. Biol. Drug Des. 2010, 3, 334–349. [Google Scholar] [CrossRef]
- Cavallo, L. POPS: A fast algorithm for solvent accessible surface areas at atomic and residue level. Nucleic Acids Res. 2003, 31, 3364–3366. [Google Scholar] [CrossRef] [PubMed]
- Vitkup, D.; Sander, C.; Church, G.M. The amino-acid mutational spectrum of human genetic disease. Genome Biol. 2003, 4, R72. [Google Scholar] [CrossRef]
- Häckel, M.; Hinz, H.-J.; Hedwig, G.R. Partial molar volumes of proteins: Amino acid side-chain contributions derived from the partial molar volumes of some tripeptides over the temperature range 10–90 °C. Biophys. Chem. 1999, 82, 35–50. [Google Scholar] [CrossRef] [PubMed]
- Harms, M.J.; Schlessman, J.L.; Sue, G.R.; García-Moreno, E.B. Arginine residues at internal positions in a protein are always charged. Proc. Natl. Acad. Sci. USA 2011, 108, 18954–18959. [Google Scholar] [CrossRef]
- Antonarakis, S.E.; Krawczak, M.; Cooper, D.N. Disease-causing mutations in the human genome. Eur. J. Pediatr. 2000, 159, S173–S178. [Google Scholar] [CrossRef]
- Branden, C.; Tooze, J. Introduction to Protein Structure, 2nd ed.; Garland Science: New York, NY, USA, 1999. [Google Scholar]
- Parrini, C.; Taddei, N.; Ramazzotti, M.; Degl’innocenti, D.; Ramponi, G.; Dobson, C.M.; Chiti, F. Glycine residues appear to be evolutionarily conserved for their ability to inhibit aggregation. Structure 2005, 13, 1143–1151. [Google Scholar] [CrossRef]
- Guarnizo, S.A.G.; Kellogg, M.K.; Miller, S.C.; Tikhonova, E.B.; Karamysheva, Z.N.; Karamyshev, A.L. Pathogenic signal peptide variants in the human genome. NAR Genom. Bioinform. 2023, 5, lqad093. [Google Scholar] [CrossRef]
- Evans, R.; O’Neill, M.; Pritzel, A.; Antropova, N.; Senior, A.; Green, T.; Žídek, A.; Bates, R.; Blackwell, S.; Yim, J.; et al. Protein complex prediction with AlphaFold-Multimer. bioRxiv 2021. [Google Scholar] [CrossRef]
- Sharo, A.G.; Zou, Y.; Adhikari, A.N.; Brenner, S.E. ClinVar and HGMD genomic variant classification accuracy has improved over time, as measured by implied disease burden. Genome Med. 2023, 15, 51. [Google Scholar] [CrossRef] [PubMed]

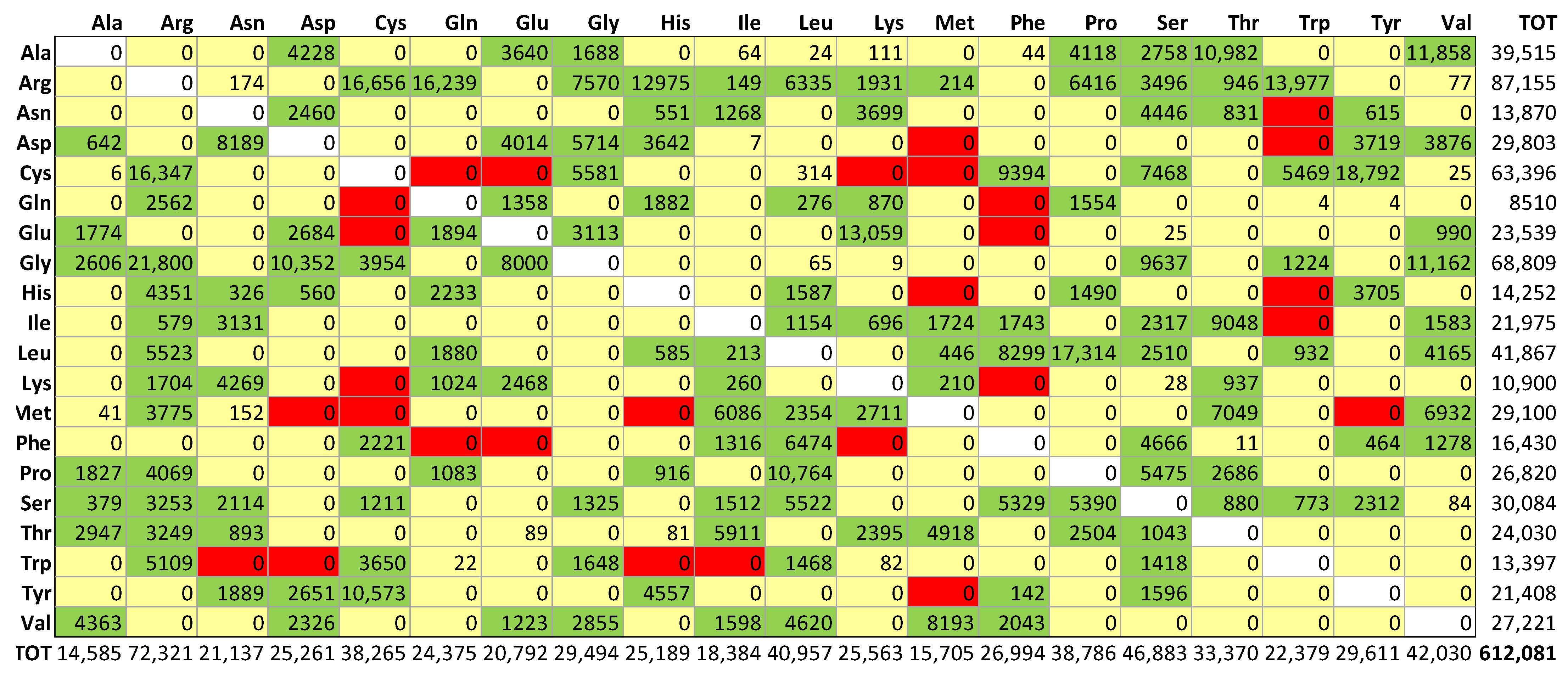
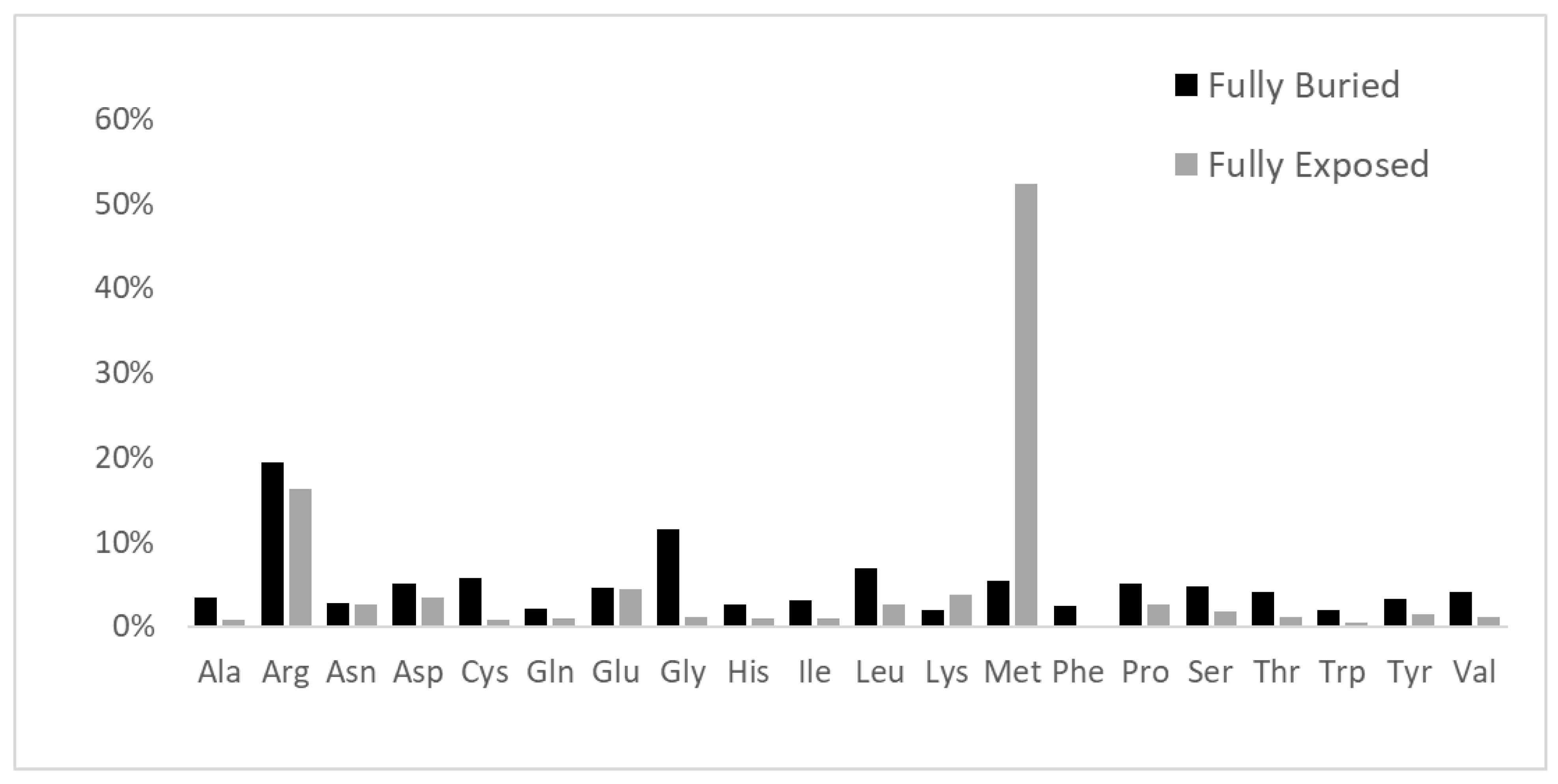
 fully buried < 0.15; (ii)
fully buried < 0.15; (ii)  internal > 0.15 and <0.24; (iii)
internal > 0.15 and <0.24; (iii)  intermediate > 0.24 and <0.60; (iv)
intermediate > 0.24 and <0.60; (iv)  external > 0.60 and <0.80; and (v)
external > 0.60 and <0.80; and (v)  fully solvent-exposed > 0.8.
fully buried < 0.15; (ii) internal > 0.15 and <0.24; (iii) intermediate > 0.24 and <0.60; (iv) external > 0.60 and <0.80; and (v) fully solvent-exposed > 0.8.
fully solvent-exposed > 0.8.
fully buried < 0.15; (ii) internal > 0.15 and <0.24; (iii) intermediate > 0.24 and <0.60; (iv) external > 0.60 and <0.80; and (v) fully solvent-exposed > 0.8.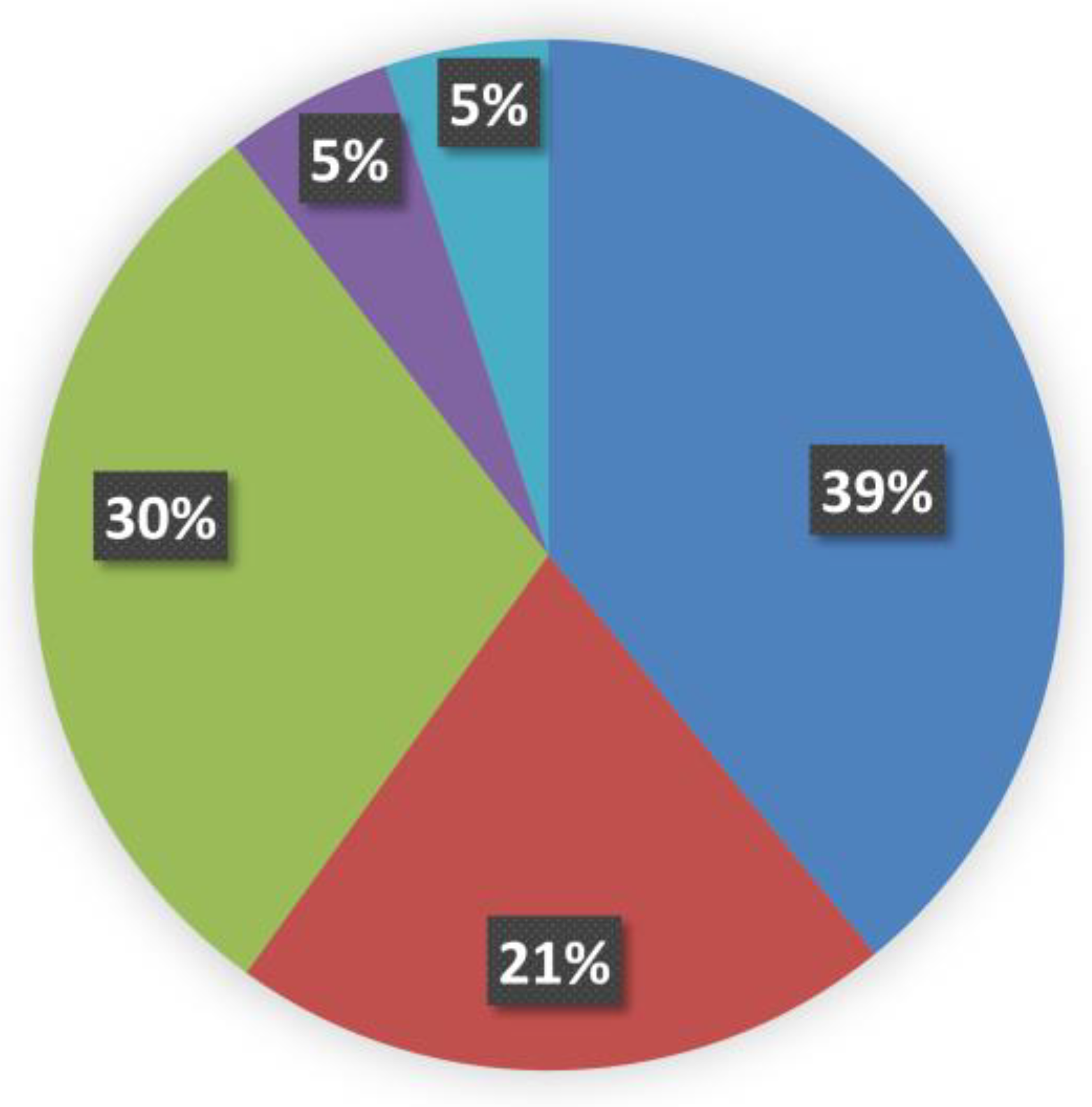

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Electrically Charged Side Chains | Polar Uncharged Side Chains | Hydrophobic Side Chains | Special Cases |
|---|---|---|---|
| Positive: Arg, His, Lys Negative: Asp, Glu | Small size: Ser, Thr Large size: Asn, Gln, Tyr | Small size: Ala, Val Medium size: Ile, Leu, Met Large size: Phe, Trp | Cys; Gly; Pro |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Visibelli, A.; Finetti, R.; Niccolai, N.; Spiga, O.; Santucci, A. Molecular Origins of the Mendelian Rare Diseases Reviewed by Orpha.net: A Structural Bioinformatics Investigation. Int. J. Mol. Sci. 2024, 25, 6953. https://doi.org/10.3390/ijms25136953
Visibelli A, Finetti R, Niccolai N, Spiga O, Santucci A. Molecular Origins of the Mendelian Rare Diseases Reviewed by Orpha.net: A Structural Bioinformatics Investigation. International Journal of Molecular Sciences. 2024; 25(13):6953. https://doi.org/10.3390/ijms25136953
Chicago/Turabian StyleVisibelli, Anna, Rebecca Finetti, Neri Niccolai, Ottavia Spiga, and Annalisa Santucci. 2024. "Molecular Origins of the Mendelian Rare Diseases Reviewed by Orpha.net: A Structural Bioinformatics Investigation" International Journal of Molecular Sciences 25, no. 13: 6953. https://doi.org/10.3390/ijms25136953
APA StyleVisibelli, A., Finetti, R., Niccolai, N., Spiga, O., & Santucci, A. (2024). Molecular Origins of the Mendelian Rare Diseases Reviewed by Orpha.net: A Structural Bioinformatics Investigation. International Journal of Molecular Sciences, 25(13), 6953. https://doi.org/10.3390/ijms25136953