Abstract
Alkoxyalkylation and hydroxyalkylation methods utilizing oxo-compound derivatives such as aldehydes, acetals or acetylenes and various alcohols or water are widely used tools in preparative organic chemistry to synthesize bioactive compounds, biosensors, supramolecular compounds and petrochemicals. The syntheses of such molecules of broad relevance are facilitated by acid, base or heterogenous catalysis. However, degradation of the N-analogous Mannich bases are reported to yield alkoxyalkyl derivatives via the retro-Mannich reaction. The mutual derivative of all mentioned species are quinone methides, which are reported to form under both alkoxy- and aminoalkylative conditions and via the degradation of the Mannich-products. The aim of this review is to summarize the alkoxyalkylation (most commonly alkoxymethylation) of electron-rich arenes sorted by the methods of alkoxyalkylation (direct or via retro-Mannich reaction) and the substrate arenes, such as phenolic and derived carbocycles, heterocycles and the widely examined indole derivatives.
1. Introduction
In the field of organic chemistry, C–C coupling reactions are a unique and versatile means of molecular fine-tuning. Coupling a carbon atom bearing active hydrogen with an electron-deficient carbon (e.g., carbonyl) atom has been known to literature. Various methods of synthesizing benzylic-type ethers and alcohols from electron-rich arenes have been introduced using aldehydes as a carbonyl-compound and catalysts such as zeolites, base and (Brönsted or Lewis) acid catalysts or organic amines by the degradation of Mannich bases. These types of products earned significance in pharmaceutical, supramolecular and petrol chemistry, as well as preparative organic chemistry, biosensor chemistry and organic photochemistry.
Regarding alkoxyalkylations, the Mannich or Betti reactions can be regarded as related. The latter reactions can occur via two main mechanisms: one starting with the formation of an iminium ion and its subsequent addition to the electron-rich aromatic ring, the other starting with the formation of a quinone methide and the subsequent aza-Michael addition of the amine to the methylidene group [1,2]. Taking the similarities of the hydroxy- or alkoxyalkylation of arenes into consideration, in scientific literature the latter mechanism, starting from quinone methides and the oxy-Michael addition of an alcohol, is currently the more frequently proposed mechanism [3,4]. However, considering the fact that alkoxyalkylation is reported as directly using acetals [5,6,7,8], the analogous compounds of imines, the hypothesis of their reaction with aromatic rings should not be neglected.
More interestingly, as reported, Mannich bases can undergo deaminative reactions yielding quinone methides [1,6,9,10,11,12,13,14,15], from which a broad molecular library can be synthesized of various biological activities. Alkoxyalkyl compounds are also prone to form quinone methides under certain conditions [16], thus opening the way towards aminoalkyl derivatives and a possible synthetic circle. (Figure 1)
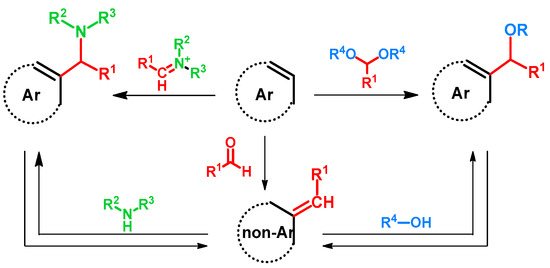
Figure 1.
Accepted or experimented reaction pathways of the alkoxymethylation of electron-rich arenes.
The aim of this review is to collect and summarize the aforementioned reactions, focusing on the synthesis of alkoxyalkylated derivatives. The literature processed is grouped with regard to the reaction pathways or substrates, such as the direct alkoxyalkylation via inorganic catalysis or through the Mannich and subsequent retro-Mannich reactions.
2. Hydroxy- and Alkoxyalkylation of Electron-Rich Arenes: Direct and Catalytic Methods
2.1. Hydroxy- and Alkoxymethylation of Monocylic Arenes
The hydroxymethylation of phenol was examined using formaldehyde. Due to the activation of hydroxyl function, two regioisomeric hydroxymethylated products (2 and 3) can be formed. Since both of the products are important chemicals in industry [17,18] or are important precursors toward the synthesis of bioactive compounds [19,20,21], the selectivity of the reaction is critical (Scheme 1). The effect of the divalent metal salts on the regioselectivity was examined by Komiyama and Morozumi [22]. It was found that without catalyst, or by using MgCl2, CuCl2 and FeCl2, the ratio 2aa/3aa was between 1 and 1.5. By applying divalent zinc salts such as Zn(H3CCOO)2, Zn(NO3)2 or ZnBr2, the selectivity could be shifted toward the formation of 2aa, and the ratio 2aa/3aa was around 10 [22]. The synthesis of 2aa was also published by Jeong and Hu[23]. For the synthesis, to provide the selectivity, Zn(NO3)2.6H2O was applied, and 2-hydroxymethylphenol (salicyl alcohol: 2aa) was further transformed to benzofuran derivatives [23].
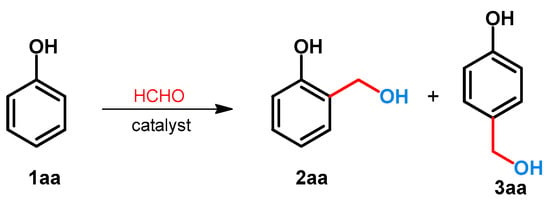
Scheme 1.
Regioselective catalytic hydroxymethylation of phenol with formaldehyde.
The prepared salicylic alcohol (2aa) was reported as a precursor to synthesizing boron-modified phenolic resin composites (BPR) [24,25]. It was proved that BPR has excellent heat resistance, ablative resistance, good mechanical and wear resistance and flame retardancy [25].
The C-ortho site-specific monohydroxymethylation of phenol 1aa was developed by Casiraghi et al. [26]. For the synthesis of salicyl alcohol 2aa, phenol 1aa was treated in xylene with an excess of paraformaldehyde in the presence of 1 mol equiv. of dimethoxyethane (DME) as an ether additive (Table 1, entry 1). The synthetic route was then extended for the synthesis of different salicyl alcohols summarized in Table 1. The ortho-selective monohydroxymethylation of a series of phenols was described by Wu et al. [27]. The reactions were performed in the presence of NaBO2 at 40 °C. The structures of the synthesized salicyl alcohols are listed in Table 1. It should be mentioned that by using NaBO2, the yields were found to be between 80 and 95% (Table 1). Mezzogori et al. reported that the H-mordenite zeolite catalyzed simultaneous synthesis of o- (2ax), m- and p-vanillols (Table 1, entry 30) [28].

Table 1.
Ortho-selective hydroxymethylation of substituted phenols using various catalysts.
Hydroxymethylation of phenol (1aa) and phenolic ketones (1bb-1be) was investigated by Goswami et al. [29]. The reactions were conducted at 60–70 °C. It was found that depending on the phenolic substrate:paraformaldehyde ratios, different products could be isolated. It was also assumed that for the formation of 1,3-dioxane derivatives 4, the necessary paraformaldehyde ratio depended on the phenol or phenolic ketones (1) in question. Starting from 1aa it was 1:3, from 1bb and 1bc 1:7, while from 1be the ratio was 1:12. It is interesting to note that starting from 1bd, even by using an extreme excess of paraformaldehyde (1:9), formation of 4bd was not observed (Scheme 2) [29].
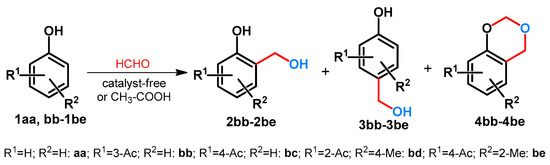
Scheme 2.
Divergent hydroxymethylation of phenolic compounds.
The previous syntheses, by using different additives or catalysts, attempted to avoid the dihydroxymethylation of phenols. In some cases, the extra hydroxymethyl function can be further used to build bioactive sidechains. For example, the hydroxymethyl group of compound 5 (Scheme 3) was further transformed to benzotriazole moiety [30], while 6 (Scheme 3) was used as an important crosslinking agent [31].
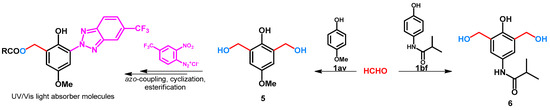
Scheme 3.
Ortho-ortho’ bis-hydroxymethylation of phenol derivatives.
Duan et al. reported the hydroxymethylation of platensimycin (7) as an excellent natural-product-derived lead molecule against various gram-positive pathogens. The synthesis was performed in MeOH, by using formaldehyde and inorganic additives KOH and CaCl2, leading to the formation of 8 in an excellent yield (Scheme 4) [32].
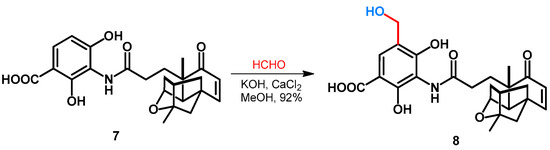
Scheme 4.
Hydroxymethylation of platensimycin.
The preparation of 3-hydroxymethyl-2,4,6-trimethylbenzoic acid (11) was reported by Stewart. The synthesis included a standard chloromethylation of mesitoic acid (9) with formaldehyde in the presence of hydrochloric acid, followed by the rapid hydrolysis of the chloromethyl group during the mildly basic aqueous wash stage incorporated in the work-up procedure (Scheme 5) [33].
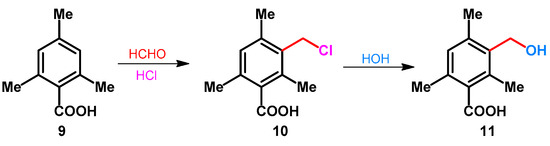
Scheme 5.
Chloromethylation and rapid hydrolysis of mesitoic acid.
The synthesis of 4-hydroxy-3-alkoxymethylbenzaldehydes (12a-d) was reported by Hirose et al. The role of hydrochloric acid is to form 3-chloromethyl intermediate that reacts immediately with the alcohols applied in the reaction (Scheme 6) [34]. Methoxymethylation of 2-hydroxy-5-methylbenzaldehyde was published by Jorgensen et al. (Scheme 6) [35]. Compound 13a was then transformed to new potential fibroblast growth factor (FGF) receptor 1 kinase inhibitors. Methoxymethylation of 2-hydroxy-5-methylacetophenone was performed in methanol by using paraformaldehyde in the presence of cc. hydrochloric acid (Scheme 6) [36]. Ortho-situated hydroxyl and acetyl functions enabled the building of substituted pyran moiety.
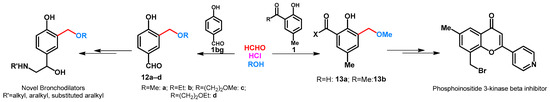
Scheme 6.
Alkoxymethylation of hydroxybenzaldehydes and phenolic ketones.
Ortho-selective ethoxymethylation was published by Bélanger et al. [37]. Phenol 1aa was reacted with formaldehyde in the presence of phenylboronic acid, leading to 1,3,2-benzodioxaborin 14 (Scheme 7). Treatment of 14 with ethanol in the presence of sulphuric acid catalyst gave the desired 2-ethoxymethylphenol 15. By treatment of 1,2-dihydroxybenzene (1bh) with dimethoxymethane in the presence of bistrifluoromethanesulfonimide (TFSI-H), a regioselective Friedel–Crafts alkylation took place and compound 16 was isolated in a yield of 60% (Scheme 7) [5].
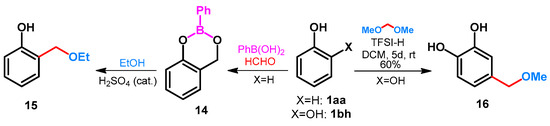
Scheme 7.
Alkoxymethylation of catechol and phenol with TFSI-H or phenylboronic acid.
The reaction among resorcinol derivatives and flavor-relevant saturated aldehydes was tested by Zomora et al. [38]. As a representative reaction, the synthesis of 4-(1-hydroxypentyl)-2-methyl-benzene-1,3-diol (17a) was carried out at 60 °C. It should be mentioned that thanks to the aliphatic alcohol function, the reaction provided different side products, e.g., methoxylated 17a (Table 2, entry 1), or by water elimination formation of double bond in the side chain. The asymmetric hydroxyalkylation of different phenols was developed by Casiraghi et al. [39]. The reactions were performed in toluene by using chloroacetaldehyde in the presence of chirally modified aluminum chloride derivatives. It was summarized that chiral Lewis acid promoter and the reactants fine-tuned both the regio- and enantioselectivity of the reaction (Table 2, entries 2–9). Ytterbium(III) trifluoromethanesulfonate (Yb(OTf)3)-catalyzed electrophilic aromatic substitution of substituted phenols with ethyl-glyoxylate was published by Wang and Zhang [40]. The reactions were performed at rt, and the lowest yield was observed when the starting phenol derivative was bearing tertiary nitrogen (Table 2, entry 13).
Table 2.
ortho-hydroxyalkylation of substituted phenols with different catalysts.
Methoxymethylation of electron-rich monocyclic arenes with formaldehyde-dimethylacetal by using β-Zeolite were performed by Müller et al. First, the method was developed starting from cumol (18a) [41], and was then extended to other arenes (18b–g) [42]. The structures of methoxymethylated derivatives are summarized in Table 3.
Table 3.
β-Zeolite-catalyzed para-methoxymethylation of electron-rich benzenes.
2.2. Hydroxy- and Alkoxymethylation of Phenol-Fused Carbocycles
Regarding alkoxyalkylations of naphthol and its derivatives, the first synthesis concerns such compounds as hydroxylated/alkoxylated bisarylmethylene (BAM) intermediates. In this regard, the reaction of 2-naphthol and benzyl alcohol occurring under high temperature and in the presence of a base—due to which the benzyl alcohol is in situ oxidized to benzaldehyde—yields compound 21. Subsequently, 21 has been observed to react with the still present benzyl alcohol, yielding compound 22, which through a base-catalyzed elimination loses benzaldehyde, leaving behind the final benzyl-substituted product 23 (Scheme 8) [43].
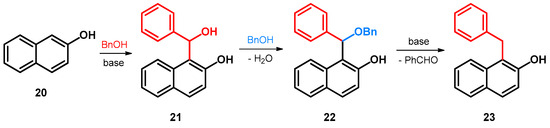
Scheme 8.
Synthesis of 1-benzylnaphthalen-2-ol (23) from 2-naphthol (20) and benzyl alcohol.
First synthesis of alkoxylated BAMs concerns the oxy-Michael-type addition reaction between aromatic aldehydes (25a–j), EtOH and 2-naphthol (24a) or 6-hydroxyquinoline (24b) assisted by 2,5-dihydroxy-1,4-benzoquinone (27) in the presence of HCl. The reaction afforded 1-[ethoxy(aryl)methyl]-2-naphthol (26a–g) and 5-[ethoxy(aryl)methyl]-6-hydroxyquinoline (26h–j) derivatives at room temperature (Scheme 9, route a, Table 4). The importance of 27 was outlined, as without it only xanthene byproducts could be achieved; however, though it was mentioned as catalyst, highest yields could be achieved at additive concentrations (Entries 6 and 7, Table 5) [44].
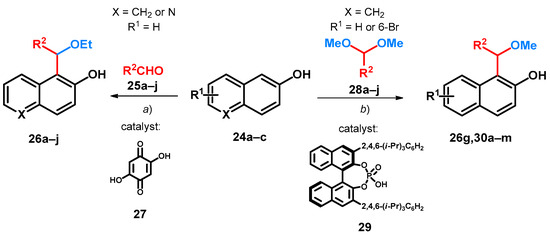
Scheme 9.
Substituted bisarylmethylene derivatives of 2-naphthol starting from 2-naphthol. (a) EtOH/HCl, 100 mol% 27, r.t., 24 h; (b) abs. C2H4Cl2, 20 mol% 29, 20 mol% AcOH, 35 °C.
Table 4.
Products and yields of reaction route a (check Scheme 9).
Acetals are widely used in carbon–carbon bond-forming reactions with a variety of nucleophiles for the synthesis of ethers [7,8]. Utilizing different chiral BINOL-based phosphoric acids as Brønsted acid catalysts with acetic acid as additive, the Friedel–Crafts-method synthesis of chiral alkoxylated BAMs from various acetals and naphthols (24a,b) was also described. After a thorough screening of solvent, catalyst and additive concentration, the chiral phosphoric acid (R)-TRIP (29) could efficiently catalyze the asymmetric Friedel–Crafts reaction of 24a,b with different aromatic acetals (28a–j), affording chiral ethers (26g,30a–m) of good enantioselectivity and yield (Scheme 9, Table 6) [6].
It has been previously demonstrated that organic ammonium tribromides (OATB) and in situ generated bromonium ion can be used for C–S bond cleavage in deprotection of dithioacetals [45] and in hydrolysis of 1-thioglycosides [46]. Starting out from the corresponding unsymmetrical sulfides (31a–g)—prepared according to literature methods—the synthesis of alkoxylated BAMs (32a–r) or hydroxylated 1-benzylnaphthalen-2-ol (33s–w) derivatives could be achieved by utilizing BDMS (bromodimethylsulfonium bromide) and either the corresponding alcohol or water (Table 7) [47].
Table 7.
Substituted bisarylmethylene derivatives of 2-naphthol starting out from thioethers. (a) 2 equiv. R3OH, 1 equiv. BDMS, DCM, r.t., 1–2 min. (b) 40 μL H2O, 1 equiv. BDMS, MeCN, r.t., 5 min.
Besides aromatic aldehydes, glyoxylic acid and its esters have also been investigated in the alkoxyalkylation of the electron-rich 1- and 2-napthol systems. Starting out from 1-naphthol (34a) and glyoxylic acid, utilizing tert-butoxy carbamate or acetamide as catalyst, the methyl 2-methoxyacetate derivative of 1-naphthol (35) could be achieved (Scheme 10). Though the syntheses yielded alkoxylated derivatives showing promising catalytic effects in this regard for tert-butoxy carbamate and acetamide, the aim of the described work was to synthesize amino acid derivatives through a modified Mannich reaction, thus highlighting the catalysts as unfavorable [48].
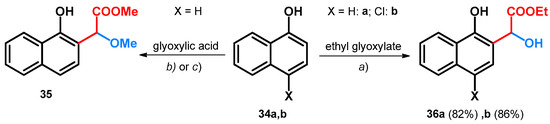
Scheme 10.
(a) NH2Boc, pTSA, MeOH, reflux, 98 h, 39%; (b) acetamide, pTSA, MeOH, reflux, 72 h, 34%; (c) 5 mol% Yb(OTf)3 CH2Cl2, r.t.
When starting out from ethyl glyoxylate and utilizing ytterbium(III) trifluoromethanesulfonate in DCM beside the previously described phenol derivatives (Scheme 10), 1-naphthol (34a) and its 4-chloro derivative (34b) gave hydroxyl derivatives (36a,b, Scheme 10) [40].
When starting out from 2-naphthol and glyoxylic acid and utilizing the same tert-butoxy carbamate or acetamide as catalyst, similar results could be achieved: the methyl 2-methoxyacetate derivative of 2-naphthol (37) could be achieved similar to 35 (Scheme 11) [48].
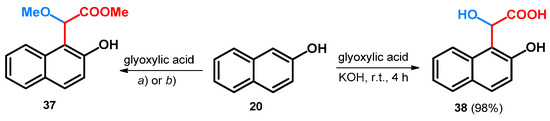
Scheme 11.
(a) NH2Boc, PTSA, MeOH, reflux, 93 h, 38%; (b) acetamide, PTSA, MeOH, reflux, 84 h, 32%;.
Without the use of such catalysts, applying only KOH, glyoxylic acid condenses with 20, yielding α-hydroxy-α-(2-hydroxy-1-naphthyl)acetic acid (38, Scheme 11). Unfortunately, the acquired enantiomers could not be resolved due to possible racemization under the resolving conditions [49].
The previously mentioned glyoxylic acid esters have also been utilized on phenols fused with heterocycles. However, in the case of compound 39—despite the EDGs on the aromatic ring—titanium catalyst was needed most probably due to the strong inactivating effect of the triflate protecting group on the pyrrolidine moieties’ nitrogen. The reaction yielded the ethyl ester derivative 40a in high yield (89%); however, as far as we know there was no published yield for the relatively more interesting (–)–menthol ester (40b, Scheme 12) [50].
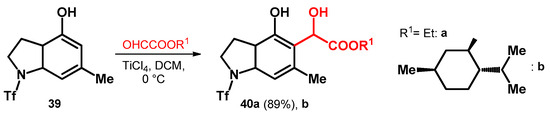
Scheme 12.
Alkoxyalkylations of pyrrolidine-fused phenol system.
Regarding phenol-fused ring-systems, another interesting reaction concerns the transformation of chrysin, a naturally occurring flavonoid. The reaction conditions specified in the patent were not discoverable, but similar conditions are also discussed later, in the case of indole derivatives in Section 2.3.1.: the flavonoid (41) was reacted with a formaldehyde source in the presence of an alkali hydroxide that gave rise to the formation of the methoxymethylated derivative 42 (Scheme 13) [51].
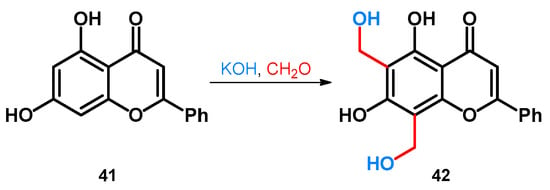
Scheme 13.
Synthesis of chrysin-based hydroxymethylene derivative 42.
2.3. Hydroxy- and Alkoxymethylation of N-heterocycles
2.3.1. Hydroxy- and Alkoxymethylation of Indoles
During the development of the total synthesis of penitrems—a family of tremorgenic mycotoxins isolated from Penicillium crustosum—researchers were looking for indole nitrogen protecting groups that would not reduce the reactivity of the indole substructure in a modified Mannich reaction. When trying to build in a methoxylated benzyl protecting group, starting out from the acetal of the corresponding aldehydes under acidic conditions with a free alcoholic group—conditions like what later was exploited as described in Section 2.2. Scheme 9—yielded alkoxyalkylated BAMs 44a,b (Scheme 14) [52].
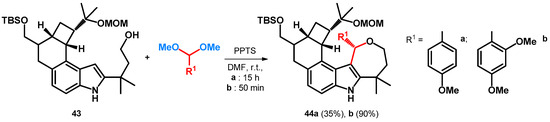
Scheme 14.
Unsuccessful protection of indole nitrogen yielding BAMs. PPTS = pyridinium p-toluenesulfonate, MOM = methoxymethyl ether.
Later, MPM (p-methoxybenzyl) was found to be a sufficient protecting group and the N-protected aldehyde 45 was prepared to take part in the aforementioned modified Mannich reaction, yielding the alkoxyalkylated indole-fused oxocine 47 as the final product via intermediate Mannich-base 46 and a tandem Mannich cyclization/gramine fragmentation sequence (Scheme 15). Subsequently, compound 47 was further transformed to give the A–F rings of penitrems with appropriate functionalization [52].
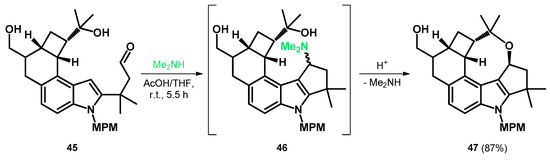
Scheme 15.
Tandem Mannich cyclization/gramine fragmentation sequence yielding 47.
The same research group later described the complete total synthesis of the penitrem ring-systems utilizing a different reaction pathway. This reaction also required the building of the oxocine ring condensed—among other rings—on the indole substructure that required no Mannich-type intermediate. Alcohols 48 and 49a,b were synthesized and were tandem-cyclized into the oxocine-condensed ring-system (50) using scandium triflate (Scheme 16) [53].
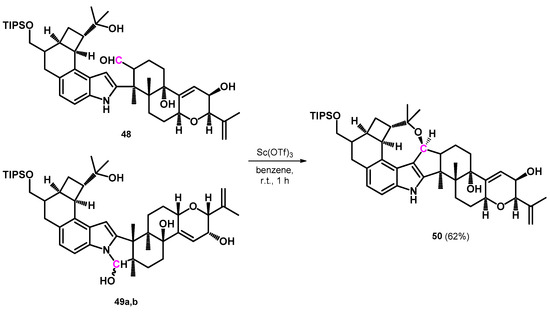
Scheme 16.
Oxidation of three different intermediates (48,49a,b) into 50 by scandium triflate.
Starting out from less functionalized indole derivatives, the first publication describes the synthesis of a novel Nav1.7 sodium channel inhibitor discovery. Compound 52 was synthesized uniquely among the other derivatives described using a gold salt as oxidizer starting out from 51 (Scheme 17). Compound 52 was later transformed; however, the final product had no inhibitory effect [54].
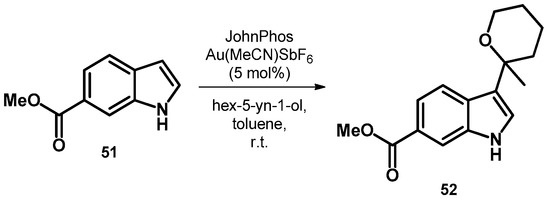
Scheme 17.
Synthesis of tetrahydro-2H-pyran-2-yl-indole derivative 52.
Utilizing similar techniques, two research papers describe the oxy-Michael-type modification of indole derivatives (51,53a–p) using aromatic aldehydes and methanol in a similar way as discussed previously in Section 2.2, Scheme 9; however, in these cases the acidic catalysts were exchanged to NaOH still yielding BAMs 54aa–xa (Table 8) [4,55]. It would be interesting to carry out the previously discussed alterations on 2-naphthols with similar conditions.
Table 8.
Alkoxyalkylation of indoles (a) 2 equiv. NaOH, 120 °C, 2–24 h; (b) 1.5 equiv. NaOH, 20 °C, 24 h.
Standing out from the transformations discussed previously, a site-selective 1,1-difunctionalization of unactivated alkenes was described that was enabled by cationic palladium catalysis. The scope and limitations of the synthesis were investigated in the case of alkenes comprising alcohols and carboxylic acids (55a–k) utilizing palladium(II) acetate and silver hexafluoroantimonate(V) as catalysts yielding 57a–k (Table 9). It is worth mentioning that for the reaction to take place, directing group AQ was necessary and crucial [56].
Table 9.
Substituents and yields of final products.
Another reaction standing out from the ones discussed previously concerns the synthesis of spiro-pyridoindolone derivatives (59). However, one similarity ties it to the transformation discussed in Scheme 17, as the reaction concerns an alkyne moiety reacting with an alcohol—intramolecularly in 58—in the presence of gold salt catalyst (Table 10) [57].
Table 10.
Gold-catalyzed intramolecular alkynol cyclization/hydroindolylation.
2.3.2. Hydroxy- and Alkoxymethylation of Uracil
Uracil (60) bearing an enamino ketone character was successfully utilized in a hydroxyalkylation reaction using electron-deficient aromatic aldehydes in water. Instead of the more commonly used halogeno derivatives of aromatic aldehydes, the scope and limitations were investigated for p-nitrobenzaldehyde with heteroaromatic aldehyde derivatives, showing moderate yields (Table 11) [58].
Table 11.
Synthesis and achieved yields of 5-(arylhydroxymethyl)uracil derivatives.
Building upon these transformations, Hlavác et al. described the synthesis of 61a in very similar reaction conditions. However, instead of the hydroxy derivative, first they obtained the chloro derivative (62) that had to be further hydrolyzed without the presence of hydrochloric acid. The scope and limitations was also investigated by supplementing water with different aliphatic alcohols, all yielding the alkoxymethylated BAMs incorporating the uracil moiety (63a–o, Scheme 18) [59].
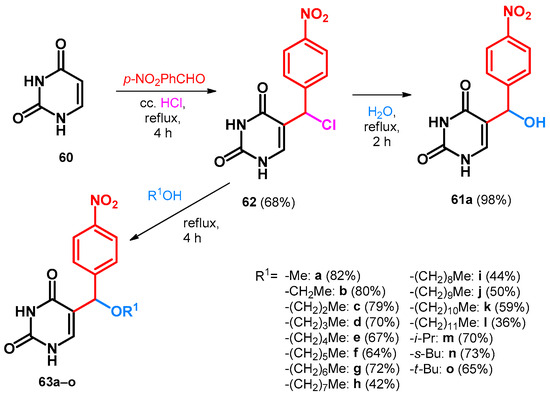
Scheme 18.
Synthesis of alkoxymethylated BAMs incorporating uracil moiety.
Besides aromatic aldehydes, paraformaldehyde was also investigated in the case of 6-methyluracil. The reaction was carried out using KOH as additive, and in both cases the final product was a hydroxymethylated derivative (65). However, it is interesting to mention the differences between the two publications: one describes no solvent and a much longer reaction (route a)) [60], while the other describes the solvent and, despite only a 5 °C difference, half the reaction time and much better yields (route b)) (Scheme 19) [61]. Both reactions share similarities with the incomplete transformation described in Scheme 13 [51].
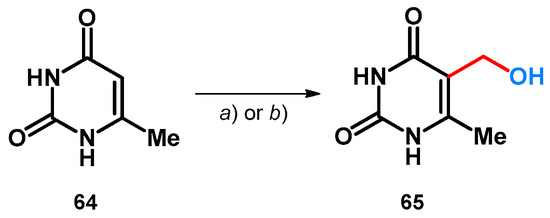
Scheme 19.
(a) (CH2O)n, KOH, 50 °C, 73 h (76%) [60]; (b) (CH2O)n, KOH, H2O, 0 °C then 55 °C for 36 h (98%).
3. Hydroxy- and Alkoxyalkylation of Electron-Rich Arenes via Aminoalkyl Intermediates
3.1. Transformations of Phenolic Mannich Bases
Transformations of benzylamines with various nucleophiles have been widely investigated applying conditions on these substances, which favor the retro-Mannich degradation yield ortho-quinone-methides (oQM). As these compounds are prone to rearomatization, nucleophilic attack on the methine group leads to novel molecules bearing benzylalcohol or benzyl-alkyl ether fragments. It was published that by the methylation of compounds 66a,b, unstable quaternary ammonium species formed (67a,b) which are easily transformed to benzyl-methyl ethers 68a,b under treatment with NaOMe in MeOH [62] (Scheme 20).
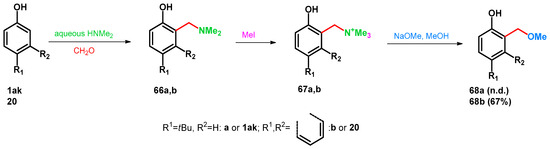
Scheme 20.
Mannich-subsequent retro-Mannich reaction of phenols towards benzyl-methyl ethers.
Based on the findings of Gardner et al., Freccero and his research group in the year 2000 published various transformations of o-hydroxybenzyl-trimethylammonium iodide (69), testing multiple N- (and S-) nucleophiles and conditions. UV- and heat-driven eliminative deamination and subsequent nucleophilic addition methods were tested in pH-buffered aqueous solutions [63] (Table 12).
Table 12.
Transformations of 69 with amino acids and glutathione as N-, O- or S-nucleophiles.
It was proved that in aqueous conditions, not only is the used N-nucleophile capable of the addition towards compounds 70a–e, but the solvent too can act as a nucleophile yielding salicyl alcohol 2aa. It was found that via thermodeamination, pH = 6–7 favored the formation of compound 2aa, while higher pH favored 70a–e. Using photodeaminative methods (UV 254 nm), the ratio of 2aa rose higher than formerly in most cases (Table 12). Interestingly, when tyrosine was used, a measurable rate of O-alkylation was observed at pH = 10–12 besides the N-alkylation.
In 1971 H. J. Roth and K. Michel found that the photodeamination-driven transformation can be applied on tertiary amine 72 in 2-propanol-HCl medium, yielding products 1ab, 73–75 (Scheme 21) [64]. The radical deamination favored the formation of 73.
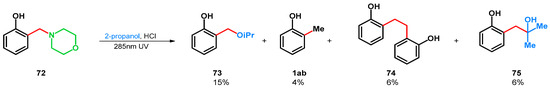
Scheme 21.
UV-driven retro-Mannich reactions of o-morpholinomethyl phenol Mannich base.
Saito et al. in 1997 published the [65] highly efficient oQM formation of dimethylaminomethyl phenols and subsequent transformations toward different acetals. They investigated the oQM formation of o-hydroxybenzyl alcohols; however, it was found that Mannich bases are much more prone to transformation.
Carrying on the study, Freccero et al. [66] transformed Mannich bases of BINOLs towards L-proline-ester based diastereomers with excellent diastereomeric excess (>99%), which were subsequently transformed to chiral hydroxymethyl derivatives using water/acetonitrile solvent mixture, reaching enantiomeric excess higher than 99%, although they reported the reversibility of the ligand exchange step (77a–d and 78a,b, Scheme 22).
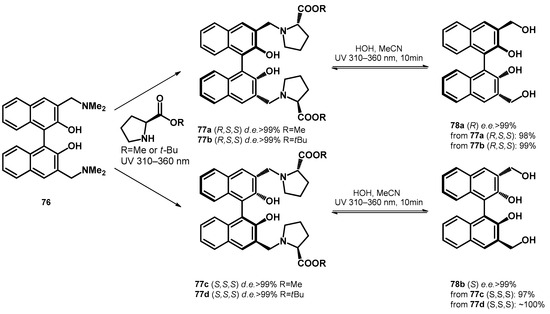
Scheme 22.
Enantio- and diastereoselective photochemical synthesis of bis-hydroxymethyl BINOLs.
In the same year, they reported that using thermal incubation or flash photolysis tests, electron-donating substituents facilitate the formation of oQMs and suppress the rearomative transformation stabilizing the oQM. It can be concluded that electron-withdrawing groups suppress the deamination and facilitate the rearomatization. During flash photolysis test, quaternary ammonium compounds had significantly higher quantum yields than the corresponding tertiary amines (Table 13) [67].
Table 13.
Thermal- and photochemical degradation of Mannich bases 79a–k.
Basarić et al. in 2015 also supported this while testing the photodeamination of p-cresol-derived tertiary amines and their hydrochlorides via laser flash photolysis at 266 nm. It was found that the quantum yield can be elevated by protonation also. (Scheme 23) [68].
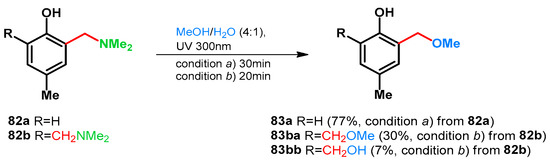
Scheme 23.
Acid driven photodeamination of tertiary Mannich-base 82a,b.
Takaki et al., while synthetizing chromene derivatives starting from Mannich bases or α-substituted salicyl alcohols, tested the equilibrium of compounds 84, 80c and 2aa. (Scheme 24)
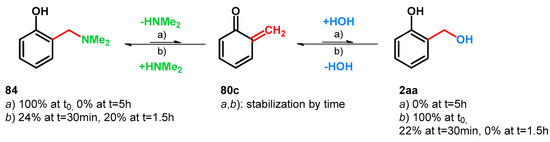
Scheme 24.
Equilibrium between Mannich-base 84 and salicyl alcohol (2aa). Conditions: UV 280 nm, D2O/CD3CN, Ph2CH2 (an internal standard); (a) 84 100% at t0, HNMe2 (40 wt.% in H2O, ~1 equiv.); (b) 2aa 100% at t0, H2O (~1 equiv.).
They also found that in contrast to using quaternary benzylammonium salts, treating secondary Mannich-base 84 with water, UV (>280 nm) in D2O/CD3CN, no trace of hydrated oQM 2aa was observable (via 1H-NMR analysis) (condition a); however, starting from 2aa and reacting it with HNMe2 under the same reaction conditions, full conversion was achievable towards 80c and 84 (condition b) [16]. These findings prove the importance of the formation of quaternary ammonium salts of elevated leaving properties regarding retro-Mannich transformations. However, elevation of the leaving property of a tertiary amine can be applied through protonation [64,68].
Later, Freccero’s research group broadened the investigations in 2016. Various Mannich bases were synthesized from phenols, bearing 4- or 5-arylethynyl substituents for the investigation of the substituent effect on the photogeneration of oQM. An aspect of the oQM detection was the study of reactivity with water or mercaptoethanol. Overall findings were that the good-leaving quaternary ammonium moiety is needed for a good quantum-yield photolysis, the 5-arylethynyl derivatives showed lower reactivity and that the electron donating substituents fasten the formation of oQMs. Three compounds (85a–c) were transformed to hydroxymethyl derivatives as test reactions with water-trapping of 86a–c (Scheme 25) [69].
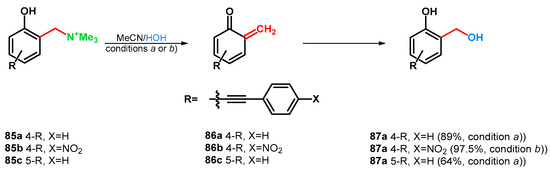
Scheme 25.
Study on substituent effects in correspondence with oQM generation condition (a) UV 310nm, 5 min; condition (b) UV 360nm, 450 min.
Basarić et al. in 2020 synthesized fluorescent BODIPY-Mannich dyes which, via oQM state, can react with the thiol-groups of BSA (bovine serum albumin). The dyes had strong fluorescence that, in the used concentrations, did not affect cells. Based on the previous studies, hydrochlorides of Mannich bases (88a,b) were successfully probed for the transformation with methanol, and the photomethanolysis occurred via irradiation (Table 14) [70].
Table 14.
Photomethanolysis probe on BODIPY-Mannich dyes.
As already stated, polarized N-centers favor the retro-Mannich transformation towards oQMs. Non-UV-driven base and acid catalysis have also been reported to promote such transformations towards O-alkylated products. Regarding the synthesis of various chromanecarboxylic acids, the alkoxylmethylation step was reported via the usage of acetic acid additive [71]. However, it was found that a Mannich reaction of disubstituted phenols in alcohols gave alkoxymethylated products while synthesizing antioxidant molecules [72] (Scheme 26).
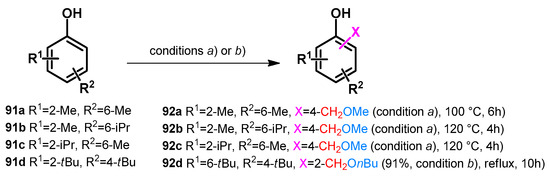
Scheme 26.
One-pot alkoxymethylation of phenols via subsequent Mannich–retro-Mannich reactions: condition (a) HNEt2 (0.14 equiv.), paraformaldehyde (0.87 equiv.), MeOH; condition (b) HN(nBu)2 (0.10 equiv.), paraformaldehyde (1.7 equiv.), HOAc (0.5 equiv.), nBuOH.
For the alkoxymethylation of substituted (alkyl, halogeno or 2H-benzo[d][1,2,3]triazol-2-yl) phenols, numerous examples were reported via the treatment of Mannich bases with acetic anhydride and subsequent alkylation with different alcohols. Treating Mannich bases with acetic anhydride yielded O-acetyl-O-acetyloxymethyl phenols, which under acidic [73] or basic [74] conditions gave the O-alkyl products (Table 15). It was found by Crisp and Turner that under mild basic conditions in water, hydroxymethyl derivatives were also able to form, and by the multistep Mannich and subsequent retro-Mannich route, in contrast to alkali-mediated hydroxymethylation, higher yields can be achieved (Table 15) [75]. Interestingly, synthesizing benzotriazole derivatives with 2-hydroxyethoxymethyl moieties for step iii, 5 equivalents of paraformaldehyde were added. The reason behind this might be that via an in situ acetal formation with ethylene glycol, the formation of bis(benzyloxy)ethylene side-products can be subdued [74].
Table 15.
Mannich and subsequent retro-Mannich reaction of substituted phenols via acetyloxylation and hydro- or alcoholysis.
The method itself can be applied to yield tetrakisalkoxymethylated bisphenols as resin raw materials, which have high storage stability and excellent solvent solubility, heat resistance and optical properties [76]. However, it should be noted that from 97a–c to 99a–c, multigram scale reactions (0.3 mol of 97a yielding 0.275 mol of 99a) were reported, although from 99a–c to 100a–c, the data represent a small-scale (0.8 mmol of 99a) synthesis (Scheme 27).
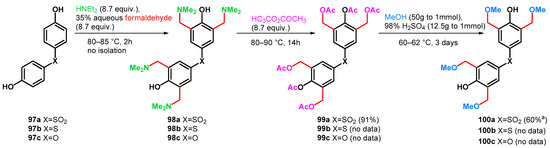
Scheme 27.
Methoxymethylation of bisphenols 97a–c via Mannich–retro-Mannich reactions.
Alkoxymethylation methods are studied in correspondence with resorcinarene chemistry. Resorcinarenes, which are versatile building blocks for supramolecular chemistry, have been modified yielding alkoxymethylated products. In 2004, Rissanen, Shivanyuk et al. conducted the synthesis of tetrakis(alkoxymethyl)resorcinarenes. Testing reaction conditions, it was found that no conversion was observable without using tromethamine, thus hypothesizing an in situ Mannich reaction by which products transform towards the desired alkoxymethylated products (Table 16) [77]. In 2006, Urbaniak et al. carried on the study using iminodiacetic acid as an N-nucleophile. Iminodiacetic acid acts as both N-nucleophile and an in situ protonating agent, being an optimal additive for the transformation of electron-rich arenes towards their alkoxymethyl derivatives. It was found that a stoichiometric amount of iminodiacetic acid is not necessary, as a 9.4 mol% catalytic amount was sufficient. They proposed a reaction mechanism which involves autoprotonation of the Mannich-type intermediate [78]. Later, selective alkoxymethylation was carried out using iminodiacetic acid catalyst. They reported selectivity driven by the alteration of the reaction time, as shorter reaction times yielded the less-substituted resorcinarenes (Table 16) [79].
Table 16.
Alkoxymethylation of resorcinarenes using tromethamine (TRIS) or iminodiacetic acid catalyst.
3.2. Transformations of Semi-Synthetic Phenols and N-heterocycles
3.2.1. Transformations of Phenol-Fused Molecules
Numerous Mannich–retro-Mannich-driven hydroxy- and alkoxymethylation reactions have been reported in correspondence with hydroxyfunctionalized heterocycles. In 1995, Hara et al. reported the unexpected methoxymethylation side-reaction of tetrahydroisoquinolines (103a–c, 105a,b). The reaction is driven by an intramolecular retro-Mannich-type reaction in which o- or pQM molecules form with simultaneous ring opening and turnover (Scheme 28) [80].
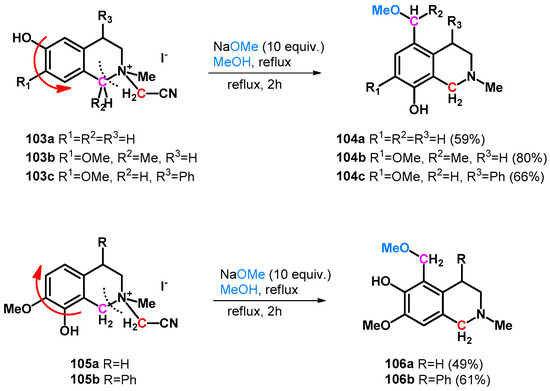
Scheme 28.
Intramolecular retro-Mannich reaction and ring turnover of tetrahydroisoquinolines 103a–c and 105a,b yielding methoxymethyl derivatives.
Zhang et al. preformed the alcoholysis of topotecan, the highly water-soluble analogue of 10-hydroxycamptothecine, synthesized via the Mannich reaction [81,82]. The already presented equilibrium was also found between topotecan (107a) and compounds 107b–e, which was explained with the hydrogen bonding ability between the dimethylamino and the phenolic hydroxyl group, thus positively polarizing the nitrogen atom, facilitating the N–C bond cleavage. Compound 106e showed lower IC50 values than the parent molecule on HepG2 and C26 cell lines, presenting the importance of the N–O swap in pharmaceutical chemistry and drug research (Scheme 29) [83]. Carrying on the study, the same research group broadened the scope of the reaction using thiols and enol-ethers [84].
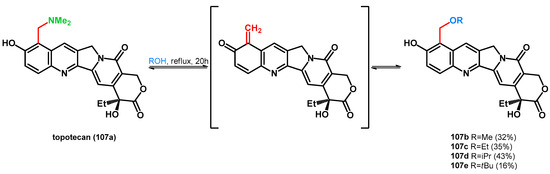
Scheme 29.
retro-Mannich driven N–O swap of topotecan (107a), a widely known Mannich base.
Homer and Sperry in 2018 targeted the total synthesis of Tricholoma alkaloids [85]. The synthesis started from the Mannich reaction of 5-hydroxy-2-methylindole (108) [86] yielding 109, which was subsequently transformed in one-batch towards 110 with moderate yields (Scheme 30). It is interesting to mention that 6.0 equivalent MeI was added in two portions. It functioned as an N-methylating agent [63], facilitating the retro-Mannich reaction and also methylating the phenolic hydroxyl group. That compound 110 was isolated with moderate yields might have been caused by the undesired reaction with sodium methoxide and methyl iodide trapping the reagents. The possible product-bearing unmethylated phenolic hydroxyl group was not isolated, as the desired product directly formed in the reaction chamber.
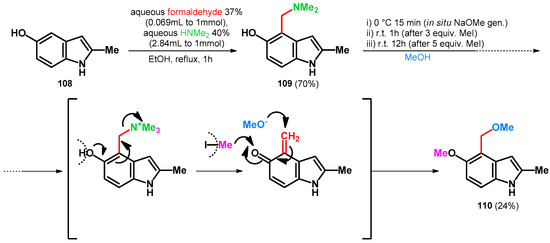
Scheme 30.
Total synthesis of tricholoma alkaloid 110 utilizing N–O swap.
In 2006, Bew et al. reported the synthesis of methylene-bridged (S)-tyrosine-phenol dimers. While conducting the syntheses, two methods were described, one exploiting the potential of retro-Mannich reactions, the other utilizing inorganic additives while reducing reaction steps. Utilizing pathway A, slightly higher yields could be achieved than utilizing B, despite the greater number of steps and longer reaction time (Scheme 31) [87].
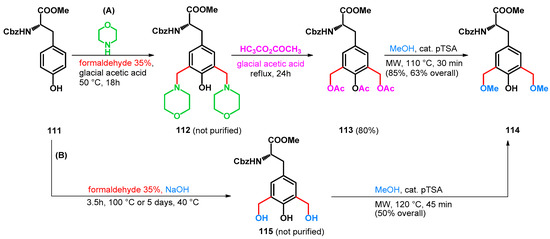
Scheme 31.
Synthesis of bis-methoxymethyl compound 114, a building block for methylene-bridged tyrosine derivatives.
It is worth mentioning, however, that the usage of amino-acid catalysis (i.e., iminodiacetic acid) has not been tested during the study. In 2006, Basarić et al. reported the photochemical transformations of Mannich bases of dipeptides towards methoxymethylated blocks, presenting photoswitchable units for peptide modification and fine-tuning [88]. Bis(dimethylaminomethyl)-derivative 116d was investigated in the reaction towards 117da,db. At 30 min reaction time, the ratio was 1:1, running the reaction for a longer time, 68% conversion towards 117db was achieved (Scheme 32).
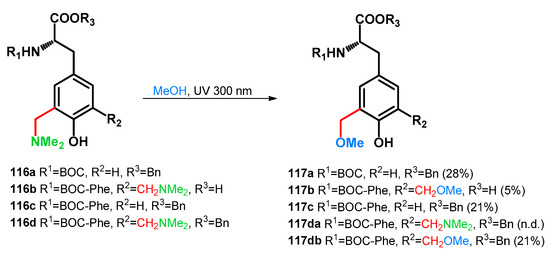
Scheme 32.
Photochemical synthesis of methoxymethyl tyrosine derivatives and dipeptides.
In patented literature, two research groups have successfully synthesized 2-methoxymethyl estrone or 2-methoxymethyl-17α-estradiol derivatives via different reaction routes starting from Mannich bases 118a–c [89,90]. It was found that the usage of dimethyl sulfate as an initiator of the retro-Mannich subreaction (condition a), being a more aggressive alkylating agent than methyl iodide, also alkylated the 3-OH group (119b). While using MeI (condition b)), as formerly presented, no O-alkylation was observed under the investigated conditions. While the latter findings may be advantageous, designing a one-pot method may cause undesired side-reactions, and this should be taken into consideration when conducting alkylative retro-Mannich transformations (Scheme 33).
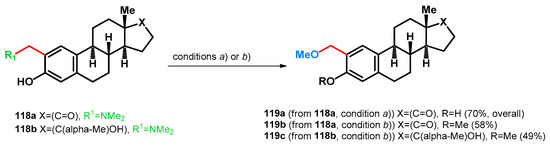
Scheme 33.
N–O swap of estrogen-based Mannich bases 118a,b (a) (i) MeI (26 equiv.), in Et2O 20 h, r.t.; isolated (ii) KOH (excess), MeOH, 3h, reflux [90] (b) MeOH, 17% aqueous KOH (in 2 steps, 12 + 2 weight equiv.), Me2SO4 (9–10 molar equiv.), 60 °C, 5 h [89].
In 2016, transformations of apigenin (120a), luteolin (120b) and quercetin (120c) via Mannich reaction were reported [91]. It was found that under the investigated conditions (a–c), running the reaction in MeOH, simultaneous methoxymethylation (at position C–6) and aminomethylation (at position C–8) took place yielding products 121a–c. Although it has not been investigated, it is noteworthy that a C–6,8 bis-(4-methylpiperazino)methyl derivative is hypothetically possible to form in the reaction mixture. The possible molecules, however, might be prone to transformation towards 121a–c based on the polarizing properties of the proximal two hydroxyl groups [83] or acidity of the parent molecules themselves [78]; however, the yields have not been reported (Scheme 34).
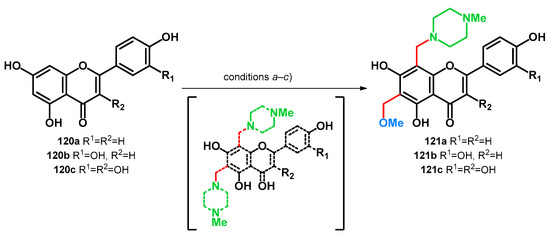
Scheme 34.
Transformation of flavonoids 120a–c yielding methoxymethyl–morpholinomethyl derivatives: condition (a) MeOH, 2.5 equiv. HCHO, 0.8 equiv. N-Me-piperazine, 63 °C, 1 h; condition (b) MeOH, 1.5 equiv. HCHO, 1.5 equiv. N-Me-piperazine, 20 °C, 12 h; condition (c) MeOH, 2.0 equiv. HCHO, 1.2 equiv. N-Me-piperazine, 46 °C, 6 h.
In 2023, Bereczki et al. synthesized Mannich derivatives of cannabidiol (122) in order to enhance their penetrability and/or water solubility [92]. Mannich reaction was an optimal tool; however, conducting the reaction using n-buthylamine in ethanol, formation of the ethoxymethylated side-product 124, in addition to 123, was observed (condition b). The conditions were investigated and optimized, using different solvents (MeOH, 1,4-dioxane) and reaction times. While an O-nucleophile alkoxymethylation did not take place in methanol (condition a), neither did it using ethanol at lower temperatures (condition c) or 1,4-dioxane (condition d). The formation of 124 was investigated, and it was found that without an amine component (condition f), the desired product did not form. Under condition e [38], 124 was isolable with moderate yields (Table 17).
Table 17.
Mannich-type modifications of (–)–cannabidiol under different conditions.
The information that an equilibrium was found [16] between alkoxymethyl and aminomethyl phenolic compounds, and that product 124 did not form at lower temperatures, suggests that the mentioned equilibrium can be shifted towards the alkoxymethyl compounds by raising temperature. It is notable that higher temperatures might be able to facilitate proton exchange and thus the polarization of the proximal Mannich-N-atom.
Modified Mannich reaction of 8-hydroxyquinoline, a widely cited bioactive agent [93,94,95,96,97] (125), was reported using diethylamine hydrochloride and paraformaldehyde; however, the isolated products were not identified as Mannich bases. The reaction was carried out in ethanol that most probably served as an O-nucleophile on the intermediate Mannich base and exchanged the diethylamino function giving 5,7-bis(ethoxymethyl)quinolin-8-ol (126a) as a major product. A dimer (126b) was also isolated, formation of which is hypothesized via a para-quinone-methide intermediate (Scheme 35) [98].
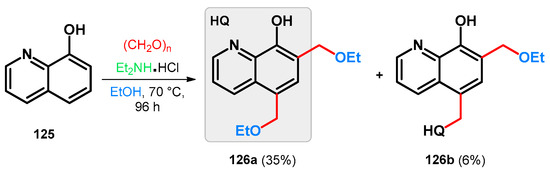
Scheme 35.
Synthesis of 8-hydroxyquinoline-derived ethoxymethylene derivatives 126a and b.
Taking the aforementioned information into consideration, a hypothetical reason behind the formation of 126a can be the in situ protonating ability of the diethylamine hydrochloride, thus constituting the driving force of the retro-Mannich substep. This is supported by the fact that by utilizing methylene bromide as a methylene source with diethylamine base, the previously targeted diethylaminomethyl-derived 8-hydroxyquinolines were able to be successfully isolated.
3.2.2. Transformations of N-heterocycles
In 2016, Youssif et al. synthesized antiproliferative 3-alkoxymethyl or 3-phenyl indole-2-carboxamides, which showed good activity on MCF7 and HCT116 cell lines (Scheme 36) [99]. In 2022, they carried on the study, synthesizing 2,3-dihydropyrazino [1,2-a]indole-1,4-dione derivatives, which showed activity on BRAFV600E and EGFR [100]. It is worth mentioning that in correspondence with both articles, retro-Mannich and subsequent alkoxylation reactions were utilized in order to yield the desired products.
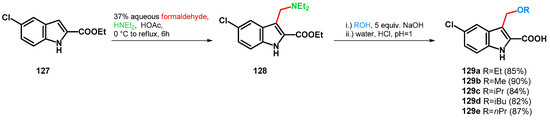
Scheme 36.
Design of 3-alkoxymethyl indole-2-carboxylic acid antiproliferative agents.
Transformation of 128 towards 129a–e was conducted in the corresponding alcohols and NaOH with excellent yields. Retro-Mannich reaction was reported using both acid and base catalysis in correspondence with the synthesis of indole-based triarylmethanes. The base catalysis is promoted by the deprotonation of the indole and its subsequent deamination at position C–3 [101]. Note, however, that in the case of 129a–e, theoretically the proximal carboxylic function could act as an autoprotonating agent if the reaction mixture reaches neutral pH and ester function is hydrolized while the aminomethyl moiety is still intact. Also, the retro-Mannich reaction can be conducted under acidic conditions present during work-up.
The C–3-alkoxymethyl transformations of kynurenic acid ethyl ester (130) were reported by Szatmári et al. [102]. Kynurenic acid being an endogenous neuroprotective agent [103,104,105,106,107,108,109,110,111] of prosperous research, its skeleton has been fine-tuned via the Mannich reaction [112,113,114,115,116,117]. It was found that utilizing secondary amino acid nucleophiles and paraformaldehyde in ethanol, the substrate did not undergo aminomethylation. The isolated product was compound 131a. Based on previous research, an expansive study was conducted focusing on the optimization of the reaction conditions in order to yield different C–3-alkoxyalkylated kynurenic acid derivatives. Additives triethylamine, NaOEt, pTSA and HOAc did not facilitate the formation of 131a. Most probably in the case of the electron-deficient arenes (such as 130), the alkoxyalkylation reaction could be conducted via the Mannich–retro-Mannich route with better conversions.
As a next step for secondary amines, their corresponding acetate salts or amino acid derivatives along with various ampholytic additives were tested in the reaction in order to investigate the simultaneous amino- and alkoxymethylation (Table 18).
Table 18.
C–3 alkoxymethylation of kynurenic acid ethyl ester (130) via organic additives.
It was reported that the low basicity of the nucleophile facilitates the reaction, but it was proposed that using morpholine-based additives, the O-atom of the nucleophile might be able to coordinate the solvent, thus elevating the conversion towards 131a,b. The optimal equivalence was found to be 0.5, able to subdue side-reactions.
The scope of the reaction was investigated using different aldehydes, although no conversion was observed. However, using different alcohols, isopropyl derivative 131b was isolated. The transesterification and C–3-isopropoxymethylation were found to run simultaneously. Similarly to what was previously reported [92], the desired product did not form using methanol, neither did it using tertiary butanol, which was proposed as being an excessively bulky O-nucleophile.
However, the mechanism of the Mannich–retro-Mannich-driven alkoxymethylation has been previously proposed [78]. The authors in that case reported indirect proof of the mechanism via the esterification of compound 132c with ethanol, which instantaneously transformed towards 131a under the investigated conditions.
4. Conclusions and Outlook
The main footsteps of the former years’ research concerning hydroxy- and alkoxyalkylation of electron-rich arenes have been reviewed. First, the focus was pointed towards the direct alkoxy- or hydroxyalkylation of phenols, phenolic ketones and phenol-fused carbo- and heterocycles regarding their hydroxy-functionalized benzene ring, utilizing various catalysts such as sodium metaborate, zeolites as well as homogenous acid and base catalysts. Regioselective hydroxyalkylation has been also successfully conducted. Reviewing the corresponding syntheses, various alkylene sources have been used, such as aldehydes, acetals or, in some cases, dehydrated analogues of oxo-compounds, and alkynes functioned as appropriate substrates.
In the second part of this review, alkoxy- and hydroxyalkylation via retro-Mannich reaction was in focus. The widely used Mannich-reaction, being a versatile tool for C–C bonding, yielded benzylic-type amines using various bioactive compounds (indoles, amino acids and small peptides, cannabinoids, flavonoids, anti-proliferative alkaloids or small heterocycles) as substrates. The Mannich bases already bearing the aminoalkyl moieties were reported to transform towards quinone methides, which compounds subsequently underwent reactions with O-nucleophiles. For the quinone methide generation, several methods were grouped, such as photodeamination, N-overalkylation, using acid catalysis and N–O swap via acetyloxylation and subsequent hydro- or alcoholysis. Heterocyclic compounds bearing pyrrol-type nitrogen atoms were reported to undergo such reactions via base catalysis. One-pot methods were also presented in this review, examining the potential of amino-acid catalysis or the driving force of intramolecular proton transfer.
Research focusing on novel C–C bonding formation techniques using water or various alcohols as nucleophiles has already been widely published; however, the recent progress of the field proves its impact.
Author Contributions
Conceptualization, I.S.; investigation, I.S., B.L. and P.S.; writing—original draft preparation, I.S., B.L. and P.S.; writing—review and editing, I.S., B.L. and P.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not Applicable.
Data Availability Statement
Not Applicable.
Acknowledgments
The authors thank the Hungarian Research Foundation (OTKA No. K-138871), the Ministry of Human Capacities, Hungary grant, TKP-2021-EGA-32. P. S. was supported by the ÚNKP-23-3-SZTE-185 New National Excellence Program of the Ministry for Innovation and Technology from the source of the National Research, Development and Innovation Fund.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
| BAM | bisarylmethylene |
| BINOL | 1,1′-bi-2-naphthol |
| BDMS | bromodimethylsulfonium bromide |
| BODIPY | dipyrrometheneboron difluoride |
| BPR | boron-modified phenolic resin |
| BSA | bovine serum albumin |
| DCM | dichloromethane |
| EDG | electron donating group |
| MOM | methoxymethyl |
| MPM | p-methoxybenzyl |
| PPTS | pyridinium p-toluenesulfonate |
| pTSA | p-toluenesulfonic acid |
| QM | quinone methide |
| (R)-TRIP | (R)-3,3′-bis(2,4,6-triisopropylphenyl)-1,1′-binaphthyl-2,2′-diyl hydrogenphosphate |
| TBSO | tert-butyl-dimethylsilyloxy |
| TRIS | tris(hydroxymethyl)aminomethane, tromethamine |
References
- Barta, P.; Fülöp, F.; Szatmári, I. Mannich Base-Connected Syntheses Mediated by Ortho-Quinone Methides. Beilstein J. Org. Chem. 2018, 14, 560–575. [Google Scholar] [CrossRef] [PubMed]
- Cardellicchio, C.; Capozzi, M.A.M.; Naso, F. The Betti Base: The Awakening of a Sleeping Beauty. Tetrahedron Asymmetry 2010, 21, 507–517. [Google Scholar] [CrossRef]
- Li, T.; Cao, M.; Liang, J.; Xie, X.; Du, G. Mechanism of Base-Catalyzed Resorcinol-Formaldehyde and Phenol-Resorcinol-Formaldehyde Condensation Reactions: A Theoretical Study. Polymers 2017, 9, 426. [Google Scholar] [CrossRef] [PubMed]
- Pi, C.; Yin, X.; Cui, X.; Ma, Y.; Wu, Y. Directed C3-Alkoxymethylation of Indole via Three-Component Cascade Reaction. Org. Lett. 2019, 21, 2081–2084. [Google Scholar] [CrossRef] [PubMed]
- Cossy, J.; Lutz, F.; Alauze, V.; Meyer, C. Carbon-Carbon Bond Forming Reactions by Using Bistrifluoromethanesulfonimide. Synlett 2002, 2002, 0045–0048. [Google Scholar] [CrossRef]
- Qin, L.; Wang, P.; Zhang, Y.; Ren, Z.; Zhang, X.; Da, C.-S. Direct Asymmetric Friedel–Crafts Reaction of Naphthols with Acetals Catalyzed by Chiral Brønsted Acids. Synlett 2015, 27, 571–574. [Google Scholar] [CrossRef][Green Version]
- Zerth, H.M.; Leonard, N.M.; Mohan, R.S. Synthesis of Homoallyl Ethers via Allylation of Acetals in Ionic Liquids Catalyzed by Trimethylsilyl Trifluoromethanesulfonate. Org. Lett. 2003, 5, 55–57. [Google Scholar] [CrossRef] [PubMed]
- Maity, P.; Srinivas, H.D.; Watson, M.P. Copper-Catalyzed Enantioselective Additions to Oxocarbenium Ions: Alkynylation of Isochroman Acetals. J. Am. Chem. Soc. 2011, 133, 17142–17145. [Google Scholar] [CrossRef]
- Szatmári, I.; Fülöp, F. Simple Access to Pentacyclic Oxazinoisoquinolines via an Unexpected Transformation of Aminomethylnaphthols. Tetrahedron Lett. 2011, 52, 4440–4442. [Google Scholar] [CrossRef]
- Csütörtöki, R.; Szatmári, I.; Koch, A.; Heydenreich, M.; Kleinpeter, E.; Fülöp, F. Synthesis and Conformational Analysis of New Naphth[1,2-e][1,3]Oxazino[3,4-c]Quinazoline Derivatives. Tetrahedron 2011, 67, 8564–8571. [Google Scholar] [CrossRef]
- Csütörtöki, R.; Szatmári, I.; Koch, A.; Heydenreich, M.; Kleinpeter, E.; Fülöp, F. Syntheses and Conformational Analyses of New Naphth[1,2-e][1,3]Oxazino[3,2-c]Quinazolin-13-Ones. Tetrahedron 2012, 68, 4600–4608. [Google Scholar] [CrossRef]
- Szatmári, I.; Barta, P.; Csámpai, A.; Fülöp, F. Synthesis and Detailed Conformational Analysis of New Naphthoxazino[2,3-a]Benz[c]Azepine and Naphthoxazino[2,3-a]Thieno[3,2-c]Pyridine Derivatives. Tetrahedron 2017, 73, 4790–4804. [Google Scholar] [CrossRef]
- Szatmári, I.; Barta, P.; Tóth, G.; Balázs, A.; Halász, J.; Fülöp, F. Synthesis and Conformational Behaviour of Enantiomeric Naphthoxazinoquinoxalinone Derivatives. Eur. J. Org. Chem. 2017, 2017, 5537–5545. [Google Scholar] [CrossRef]
- Szatmári, I.; Belasri, K.; Heydenreich, M.; Koch, A.; Kleinpeter, E.; Fülöp, F. Ortho-Quinone Methide Driven Synthesis of New O, N- or N,N-Heterocycles. ChemistryOpen 2019, 8, 961–971. [Google Scholar] [CrossRef] [PubMed]
- Hegedűs, D.; Szemerédi, N.; Spengler, G.; Szatmári, I. Application of Partially Aromatic Ortho-Quionone-Methides for the Synthesis of Novel Naphthoxazines with Improved Antibacterial Activity. Eur. J. Med. Chem. 2022, 237, 114391. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, M.; Sakamoto, M.; Komeyama, K.; Yoshida, H.; Takaki, K. Convenient Synthesis of 2-Amino-4 H -chromenes from Photochemically Generated o-Quinone Methides and Malononitrile. J. Heterocycl. Chem. 2015, 52, 59–66. [Google Scholar] [CrossRef]
- Makarov, A.S.; Kekhvaeva, A.E.; Chalikidi, P.N.; Abaev, V.T.; Trushkov, I.V.; Uchuskin, M.G. A Simple Synthesis of Densely Substituted Benzofurans by Domino Reaction of 2-Hydroxybenzyl Alcohols with 2-Substituted Furans. Synthesis 2019, 51, 3747–3757. [Google Scholar] [CrossRef]
- Merkushev, A.A.; Strel’nikov, V.N.; Uchuskin, M.G.; Trushkov, I.V. A Simple Synthesis of Benzofurans by Acid-Catalyzed Domino Reaction of Salicyl Alcohols with N-Tosylfurfurylamine. Tetrahedron 2017, 73, 6523–6529. [Google Scholar] [CrossRef]
- Choi, J.; Yeo, S.; Kim, M.; Lee, H.; Kim, S. P-Hydroxybenzyl Alcohol Inhibits Four Obesity-related Enzymes in Vitro. J. Biochem. Mol. Toxicol. 2018, 32, e22223. [Google Scholar] [CrossRef]
- Swami Vetha, B.S.; Adam, A.G.; Aileru, A. Redox Responsive Copolyoxalate Smart Polymers for Inflammation and Other Aging-Associated Diseases. Int. J. Mol. Sci. 2021, 22, 5607. [Google Scholar] [CrossRef]
- Wendt, B.; Riem-Ha, H.; Hesse, M. Synthesis of Two Metabolites of the Antiarrythmicum Amiodarone. Helv. Chim. Acta 2002, 85, 2990–3001. [Google Scholar] [CrossRef]
- Morozumi, T.; Komiyama, M. Quantitative Analysis on Ortho-Directing Activities of Divalent Metal Salts in Hydroxymethylation of Phenol by Formaldehyde. J. Mol. Catal. 1991, 69, 339–346. [Google Scholar] [CrossRef]
- Hu, K.; Jeong, J.-H. A Convergent Synthetic Study of Biologically Active Benzofuran Derivatives. Arch. Pharm. Res. 2006, 29, 476–478. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Huang, Z.; Liu, Y.; Li, Y. Novel Cardanol-Containing Boron-Modified Phenolic Resin Composites: Non-Isothermal Curing Kinetics, Thermal Properties, and Ablation Mechanism. High Perform. Polym. 2017, 29, 279–288. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, X.; Wang, R.; Zhang, Y.; Wu, J.; Zhou, Z.; Yin, P. Research Progress in Boron-Modified Phenolic Resin and Its Composites. Polymers 2023, 15, 3543. [Google Scholar] [CrossRef] [PubMed]
- Casiraghi, G.; Casnati, G.; Puglia, G.; Sartori, G. Selective Reactions between Phenols and Formaldehyde. A Superior Synthesis of Salicyl Alcohols. Synthesis 1980, 1980, 124–125. [Google Scholar] [CrossRef]
- Li, H.-J.; Wu, Y.-Y.; Wu, Q.-X.; Wang, R.; Dai, C.-Y.; Shen, Z.-L.; Xie, C.-L.; Wu, Y.-C. Water-Promoted Ortho-Selective Monohydroxymethylation of Phenols in the NaBO2 System. Org. Biomol. Chem. 2014, 12, 3100–3107. [Google Scholar] [CrossRef] [PubMed]
- Cavani, F.; Pozzo, L.D.; Maselli, L.; Mezzogori, R. Hydroxymethylation of 2-Methoxyphenol Catalyzed by H-Mordenite: Analysis of the Reaction Scheme. Stud. Surf. Sci. Catal. 2002, 142, 565–572, ISBN 978-0-444-51174-4. [Google Scholar]
- Goswami, J.; Borthakur, N.; Goswami, A. A Water Based Method for Hydroxymethylation of Phenols and Phenolic Ketones. J. Chem. Res. 2003, 2003, 200–203. [Google Scholar] [CrossRef]
- Laredo, W.R. UV/Visible Light Absorbers for Ophthalmic Lens Materials. U.S. Patent 2010113641A1, 6 May 2010. [Google Scholar]
- Adegawa, Y. Chemical Amplification Type Negative-Working Resist Composition for Electron Beams or X-rays. Patent EP1109066A1, 20 June 2001. [Google Scholar]
- Tian, K.; Deng, Y.; Qiu, L.; Zhu, X.; Shen, B.; Duan, Y.; Huang, Y. Semisynthesis and Biological Evaluation of Platensimycin Analogues with Varying Aminobenzoic Acids. ChemistrySelect 2018, 3, 12625–12629. [Google Scholar] [CrossRef]
- Stewart, F.H.C. 3-Chloromethyl-2,4,6-trimethylbenzoic acid. Org. Prep. Proced. Int. 1981, 13, 116–118. [Google Scholar] [CrossRef]
- Sohda, S.; Fujimoto, M.; Tamegai, T.; Hirose, N. New Bronchodilators. Synthesis and Bronchodilating Activity of Some 3-(Alkoxymethy1)-a-(N-Substituted Aminomethy1)-4-Hydroxybenzyl Alcohols. J. Med. Chem. 1979, 3, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Ravindranathan, K.P.; Mandiyan, V.; Ekkati, A.R.; Bae, J.H.; Schlessinger, J.; Jorgensen, W.L. Discovery of Novel Fibroblast Growth Factor Receptor 1 Kinase Inhibitors by Structure-Based Virtual Screening. J. Med. Chem. 2010, 53, 1662–1672. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.P.; Robertson, A.D.; Kenche, V.; Thompson, P.; Prabaharan, H.; Anderson, K.; Abbott, B.; Goncalves, I.; Nesbitt, W.; Shoenwaelder, S.; et al. Inhibition of Phosphoinositide 3-Kinase Beta. Patent WO2004016607A1, 26 February 2004. [Google Scholar]
- Lau, C.K.; Williams, H.W.R.; Tardiff, S.; Dufresne, C.; Scheigetz, J.; Bélanger, P.C. Ortho-Specific Alkylation of Phenols via 1,3,2-Benzodioxaborins. Can. J. Chem. 1989, 67, 1384–1387. [Google Scholar] [CrossRef]
- Hidalgo, F.J.; Aguilar, I.; Zamora, R. Model Studies on the Effect of Aldehyde Structure on Their Selective Trapping by Phenolic Compounds. J. Agric. Food Chem. 2017, 65, 4736–4743. [Google Scholar] [CrossRef] [PubMed]
- Bigi, F.; Cadraghi, G.; Casnati, G.; Sartori, G.; Fava, G.G.; Belicchi, M.F. Asymmetric Electrophilic Substitution on Phenols. 1. Enantioselective Ortho-Hydroxyalkylation Mediated by Chiral Alkoxyaluminum Chlorides. J. Org. Chem. 1985, 50, 5018–5022. [Google Scholar] [CrossRef]
- Zhang, W.; Wang, P.G. Ytterbium(III) Trifluoromethanesulfonate Catalyzed Electrophilic Aromatic Substitution with Glyoxalate and Lipase-Mediated Product Resolution: A Convenient Route to Optically Active Aromatic r-Hydroxy Esters. J. Org. Chem. 2000, 65, 4732–4735. [Google Scholar] [CrossRef] [PubMed]
- Griesbach, U.; Mueller, U.; Puetter, H.; Quaiser, C.; Winsel, H. Electrochemical Method for Producing Benzaldehyde Dimethyl Acetylene. Patent WO2009059944A1, 14 May 2009. [Google Scholar]
- Griesbach, U.; Mueller, U.; Ebel, K.; Botzem, J.; Yilmaz, B.; Stock, C. Method for the Regioselective Alkoxyalkylation of Substituted Benzenes. Patent WO2009059941A1, 14 May 2009. [Google Scholar]
- Kito, T.; Yoshinaga, K.; Ohkami, S.; Ikeda, K.; Yamaye, M. Precursors in the Alkylation of 2-Naphthol with Benzyl Alcohol in the Presence of a Base. J. Org. Chem. 1985, 50, 4628–4630. [Google Scholar] [CrossRef]
- Keshiour, S.; Shaabani, A.; Pedarpour, M.; Sarvary, A. An oxy-Michael addition: 2,5-dihydroxy1,4-benzoquinone-assisted synthesis of 1-[ethoxy(phenyl)methyl]-2-naphthol and 5-[ethoxy(phenyl)methyl]-6-hydroxyquinoline derivatives. Res. Chem. Intermed. 2012, 40, 149–156. [Google Scholar] [CrossRef]
- Mondal, E.; Bose, G.; Khan, A.T. An Expedient and Efficient Method for the Cleavage of Dithioacetals to the Corresponding Carbonyl Compounds Using Organic Ammonium Tribromide (OATB). Synlett 2001, 6, 785–786. [Google Scholar] [CrossRef]
- Mondal, E.; Sahu, P.R.; Khan, A.T. A Useful and Catalytic Method for Protection of Carbonyl Compounds into the Corresponding 1,3-Oxathiolanes and Deprotection to the Parent Carbonyl Compounds. Synlett 2002, 3, 463–467. [Google Scholar] [CrossRef]
- Islam, K.; Basha, R.S.; Dar, A.A.; Das, D.K.; Khan, A.T. A Direct Approach for the Expedient Synthesis of Unsymmetrical Ethers by Employing Bromodimethylsulfonium Bromide (BDMS) Mediated C-S Bond Cleavage of Naphthalene-2-Ol Sulfides. RSC Adv. 2015, 5, 79759–79764. [Google Scholar] [CrossRef]
- Csütörtöki, R.; Szatmári, I.; Mándi, A.; Kurtán, T.; Fülöp, F. Synthesis of Hydroxynaphthyl-Substituted α-Amino Acid Derivatives via a Modified Mannich Reaction. Synlett 2011, 2011, 1940–1946. [Google Scholar] [CrossRef]
- Yamaye, M.; Okano, H.; Motoyanagi, Y.; Cho (Toh), N.; Yoshinaga, T.; Tsuru, T.; Mukae, K. Novel α-Hydroxy- α-(2-Hydroxy-1-Naphthyl)Acetic Acid and Its Derivatives. Preparation and Optical Resolution. Synthesis 2004, 3, 341–344. [Google Scholar] [CrossRef]
- Velthuisen, E.J.; Weatherhead, J.G. Indoline Derivatives. Patent WO2018020357A1, 1 February 2018. [Google Scholar]
- Li, F.; Zhang, Z.; Chen, L.; Wu, Z.; Wang, Z. 6,8-Dimethylol Chrysin and 6,8-Dimethylol Ether Chrysin, Method for Preparing Same and Pharmaceutical Use. Patent CN101307043A, 19 November 2008. [Google Scholar]
- Smith, A.B.; Haseltine, J.N.; Visncik, M. Tremorgenic Indole Alkaloid Studies 6. Preparation of an Advanced Intermediate for the Synthesis of Penitrem D. Synthesis of an Indole-Oxocane. Tetrahedron 1989, 45, 2431–2449. [Google Scholar] [CrossRef]
- Smith, A.B.; Kanoh, N.; Ishiyama, H.; Hartz, R.A. Total Synthesis of (−)-Penitrem D. J. Am. Chem. Soc. 2000, 122, 11254–11255. [Google Scholar] [CrossRef]
- Wu, Y.-J.; Venables, B.; Guernon, J.; Chen, J.; Sit, S.-Y.; Rajamani, R.; Knox, R.J.; Matchett, M.; Pieschl, R.L.; Herrington, J.; et al. Discovery of New Indole-Based Acylsulfonamide Nav1.7 Inhibitors. Bioorg. Med. Chem. Lett. 2019, 29, 659–663. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Zha, D.; Cui, P.; Zhang, H.; Li, C.; Shi, J.; Han, B. Friedel–Crafts Reaction of Indoles for (3-Indolyl)Methyl Ethers under Basic Condition: Application in Unsymmetrical Bis(Indolyl)Methanes. Results Chem. 2021, 3, 100247. [Google Scholar] [CrossRef]
- Jeon, J.; Ryu, H.; Lee, C.; Cho, D.; Baik, M.-H.; Hong, S. Site-Selective 1,1-Difunctionalization of Unactivated Alkenes Enabled by Cationic Palladium Catalysis. J. Am. Chem. Soc. 2019, 141, 10045–10059. [Google Scholar] [CrossRef]
- Shinde, M.H.; Ramana, C.V. Facile Synthesis of the Spiro-Pyridoindolone Scaffold via a Gold-Catalysed Intramolecular Alkynol Cyclisation/Hydroindolylation. Org. Biomol. Chem. 2022, 20, 2086–2095. [Google Scholar] [CrossRef]
- Lam, B.L.; Pridgen, L.N. An Acid-Catalyzed Hydroxyalkylation of Uracil: A Facile Synthesis of 5-(Arylhydroxymethyl)Uracils. J. Org. Chem. 1986, 51, 2592–2594. [Google Scholar] [CrossRef]
- Spáčilová, L.; Džubák, P.; Hajdúch, M.; Křupková, S.; Hradil, P.; Hlaváč, J. Synthesis and Cytotoxic Activity of Various 5-[Alkoxy-(4-Nitro-Phenyl)-Methyl]-Uracils in Their Racemic Form. Bioorg. Med. Chem. Lett. 2007, 17, 6647–6650. [Google Scholar] [CrossRef] [PubMed]
- Batinac, S.; Sermek, D.M.; Cetina, M.; Pavelić, K.; Mintas, M.; Raić-Malić, S. Synthesis of the novel bicyclicoxepino-pyrimidine and fluorinated pyrrolidinopyrimidines. Heterocycles 2004, 63, 2523–2536. [Google Scholar]
- Alexander, M.D.; Chuaqui, C.; Malona, J.; McDonald, J.J.; Ni, Y.; Niu, D.; Petter, R.C.; Singh, J. MK2 Inhibitors and Uses Thereof. U.S. Patent 20160075720A1, 17 March 2016. [Google Scholar]
- Gardner, P.D.; Rafsanjani, H.S.; Rand, L. Reaction of Phenolic Mannich Base Methiodides and Oxides with Various Nucleophiles. J. Am. Chem. Soc. 1959, 81, 3364–3367. [Google Scholar] [CrossRef]
- Modica, E.; Zanaletti, R.; Freccero, M.; Mella, M. Alkylation of Amino Acids and Glutathione in Water by o -Quinone Methide. Reactivity and Selectivity. J. Org. Chem. 2001, 66, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Roth, H.J.; Michel, K. Bestrahlung von 2-Morpholinomethyl-phenol in Isopropanol 5. Mitt.: Zur Photochemie der 2-Aminomethyl-phenole. Arch. Pharm. 1971, 304, 493–498. [Google Scholar] [CrossRef] [PubMed]
- Nakatani, K.; Higashida, N. Highly Efficient Photochemical Generation of O-Quinone Methide from Mannich Bases of Phenol Derivatives. Tetrahedron Lett. 1997, 38, 5005–5008. [Google Scholar] [CrossRef]
- Colloredo-Mels, S.; Doria, F.; Verga, D.; Freccero, M. Photogenerated Quinone Methides as Useful Intermediates in the Synthesis of Chiral BINOL Ligands. J. Org. Chem. 2006, 71, 3889–3895. [Google Scholar] [CrossRef] [PubMed]
- Weinert, E.E.; Dondi, R.; Colloredo-Melz, S.; Frankenfield, K.N.; Mitchell, C.H.; Freccero, M.; Rokita, S.E. Substituents on Quinone Methides Strongly Modulate Formation and Stability of Their Nucleophilic Adducts. J. Am. Chem. Soc. 2006, 128, 11940–11947. [Google Scholar] [CrossRef]
- Škalamera, Đ.; Bohne, C.; Landgraf, S.; Basarić, N. Photodeamination Reaction Mechanism in Aminomethyl p -Cresol Derivatives: Different Reactivity of Amines and Ammonium Salts. J. Org. Chem. 2015, 80, 10817–10828. [Google Scholar] [CrossRef]
- Doria, F.; Lena, A.; Bargiggia, R.; Freccero, M. Conjugation, Substituent, and Solvent Effects on the Photogeneration of Quinone Methides. J. Org. Chem. 2016, 81, 3665–3673. [Google Scholar] [CrossRef] [PubMed]
- Zlatić, K.; Antol, I.; Uzelac, L.; Mikecin Dražić, A.-M.; Kralj, M.; Bohne, C.; Basarić, N. Labeling of Proteins by BODIPY-Quinone Methides Utilizing Anti-Kasha Photochemistry. ACS Appl. Mater. Interfaces 2020, 12, 347–351. [Google Scholar] [CrossRef] [PubMed]
- Hayashibara, T.; Sato, J.; Torihara, M. Process for Producing Chroman-Carboxylic Acid. Patent EP1184378A1, 6 March 2002. [Google Scholar]
- Wang, Y.; Ren, H.; He, S.; Yan, Y.; Wang, D.; Zhao, Z.; Wu, S.; Niu, N.; Zhang, M.; Zhang, R.; et al. Macromolecular Hindered Phenol Antioxidant and Preparation Method and Application Thereof. Patent CN113512232A, 19 October 2021. [Google Scholar]
- Kunita, K.; Aoshima, K.; Nakamura, I.; Nakamura, T. Negative Working Image Recording Material. Patent EP0874282A1, 28 October 1998. [Google Scholar]
- Okumuto, H.; Yamazaki, M.; Niina, N. Benzotriazole Derivative. Patent JP2015151369A, 24 August 2015. [Google Scholar]
- Crisp, G.T.; Turner, P.D. Preparation of Functionalised Aryl Alkynes as Precursors to Extended Cyclophanes. Tetrahedron 2000, 56, 407–415. [Google Scholar] [CrossRef]
- Nasu, H. Novel Alkoxymethyl-Substituted Bisphenol Compound. Patent JP2018115120A, 26 July 2018. [Google Scholar]
- Nummelin, S.; Falabu, D.; Shivanyuk, A.; Rissanen, K. Alkoxy-, Acyloxy-, and Bromomethylation of Resorcinarenes. Org. Lett. 2004, 6, 2869–2872. [Google Scholar] [CrossRef] [PubMed]
- Urbaniak, M.; Iwanek, W. Synthesis of Alkoxymethyl Derivatives of Resorcinarene via the Mannich Reaction Catalysed with Iminodiacetic Acid. Tetrahedron 2006, 62, 1508–1511. [Google Scholar] [CrossRef]
- Urbaniak, M.; Pedrycz, A.; Gawdzik, B.; Wzorek, A. Preparation of Partially Functionalised Resorcinarene Derivatives. Supramol. Chem. 2013, 25, 777–781. [Google Scholar] [CrossRef]
- Hara, H.; Kaneko, K.; Endoh, M.; Uchida, H.; Hoshino, O. A Novel Ring Cleavage and Recyclization of N-Cyanomethyl-1,2,3,4-tetrahydroisoquinolinium Methioidides: A Biomimetic Synthesis of Libetamine. Tetrahedron 1995, 51, 10189–10204. [Google Scholar] [CrossRef]
- Boehm, J.C.; Johnson, R.K.; Hecht, S.M.; Kingsbury, W.D.; Holden, K.G. Water Soluble Camptothecin Analogs. Patent EP0321122A2, 21 June 1989. [Google Scholar]
- Bracher, F.; Tremmel, T. From Lead to Drug Utilizing a Mannich Reaction: The Topotecan Story. Arch. Pharm. 2017, 349, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, G.; Dong, M.; Zhang, Q. The Solvolysis of Topotecan in Alcohols and Acetic Anhydride. Bioorg. Med. Chem. Lett. 2011, 21, 2324–2326. [Google Scholar] [CrossRef]
- Tan, H.; Wang, G.; Li, J.; Meng, G.; Liu, Z.; Dong, M.; Li, Y.; Ju, D.; Zhang, Q. Synthesis of Novel 10-Hydroxycamptothecin Derivatives Utilizing Topotecan Hydrochloride as Ortho-Quinonemethide Precursor. Bioorg. Med. Chem. 2015, 23, 118–125. [Google Scholar] [CrossRef]
- Homer, J.A.; Sperry, J. Synthesis of Three Tricholoma-Derived Indoles via an Ortho-Quinone Methide. Arkivoc 2018, 2018, 6–12. [Google Scholar] [CrossRef]
- Monti, S.A.; Johnson, W.O. Position Selective Mannich Reactions of Some 5- and 6-Hydroxyindoles. Tetrahedron 1970, 26, 3685–3694. [Google Scholar] [CrossRef]
- Bew, S.P.; Hughes, D.L.; Sharma, S.V. Acid-Catalyzed Synthesis of Methylene-Bridged (S)-Tyrosine−Phenol Dimers. J. Org. Chem. 2006, 71, 7881–7884. [Google Scholar] [CrossRef] [PubMed]
- Husak, A.; Noichl, B.P.; Šumanovac Ramljak, T.; Sohora, M.; Škalamera, Đ.; Budiša, N.; Basarić, N. Photochemical Formation of Quinone Methides from Peptides Containing Modified Tyrosine. Org. Biomol. Chem. 2016, 14, 10894–10905. [Google Scholar] [CrossRef] [PubMed]
- Tanabe, M.; Peters, R.H.; Chao, W.-R.; Shigeno, K. Estrone Sulfamate Inhibitors of Estrone Sulfatase, and Associated Pharmaceutical Compositions and Methods of Use. Patent WO1999033858A2, 8 July 1999. [Google Scholar]
- Kaneko, H.; Hashimoto, M.; Kawase, K. 2-Methoxymethyl-17alpha-Substituted Estradiol-3-Methyl Ethers and Their Preparation. U.S. Patent 3132162A, 5 May 1964. [Google Scholar]
- Meng, Z.; Jia, J.; Wang, Z.; Qiao, L.; Song, J.; Yao, B.; Liang, J.; Chang, Q.; Guo, Z.; Li, H.X.; et al. Flavone Derivatives, Preparation Method and Application Thereof. Patent CN108017608B, 17 January 2023. [Google Scholar]
- Lőrincz, E.B.; Tóth, G.; Spolárics, J.; Herczeg, M.; Hodek, J.; Zupkó, I.; Minorics, R.; Ádám, D.; Oláh, A.; Zouboulis, C.C.; et al. Mannich-Type Modifications of (−)-Cannabidiol and (−)-Cannabigerol Leading to New, Bioactive Derivatives. Sci. Rep. 2023, 13, 19618–19637. [Google Scholar] [CrossRef] [PubMed]
- Csuvik, O.; Szatmári, I. Synthesis of Bioactive Aminomethylated 8-Hydroxyquinolines via the Modified Mannich Reaction. Int. J. Mol. Sci. 2023, 24, 7915. [Google Scholar] [CrossRef] [PubMed]
- Mészáros, J.P.; Poljarević, J.M.; Szatmári, I.; Csuvik, O.; Fülöp, F.; Szoboszlai, N.; Spengler, G.; Enyedy, É.A. An 8-Hydroxyquinoline–Proline Hybrid with Multidrug Resistance Reversal Activity and the Solution Chemistry of Its Half-Sandwich Organometallic Ru and Rh Complexes. Dalton Trans. 2020, 49, 7977–7992. [Google Scholar] [CrossRef]
- Pape, V.F.S.; Palkó, R.; Tóth, S.; Szabó, M.J.; Sessler, J.; Dormán, G.; Enyedy, É.A.; Soós, T.; Szatmári, I.; Szakács, G. Structure–Activity Relationships of 8-Hydroxyquinoline-Derived Mannich Bases with Tertiary Amines Targeting Multidrug-Resistant Cancer. J. Med. Chem. 2022, 65, 7729–7745. [Google Scholar] [CrossRef]
- Chan, S.H.; Chui, C.H.; Chan, S.W.; Kok, S.H.L.; Chan, D.; Tsoi, M.Y.T.; Leung, P.H.M.; Lam, A.K.Y.; Chan, A.S.C.; Lam, K.H.; et al. Synthesis of 8-Hydroxyquinoline Derivatives as Novel Antitumor Agents. ACS Med. Chem. Lett. 2013, 4, 170–174. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Xu, H.; Chen, W.; Zhan, P.; Liu, X. 8-Hydroxyquinoline: A Privileged Structure with a Broad-Ranging Pharmacological Potential. Med. Chem. Commun. 2015, 6, 61–74. [Google Scholar] [CrossRef]
- Hon, Y.; Chou, Y.; Wu, I. Dibromomethane as One-Carbon Source in Organic Synthesis: The Mannich Base Formation from the Reaction of Phenolic Compounds with a Preheated Mixture of Dibromomethane and Diethylamine. Synth. Commun. 2004, 34, 2253–2267. [Google Scholar] [CrossRef]
- Abdelrahman, M.H.; Aboraia, A.S.; Youssif, B.G.M.; Elsadek, B.E.M. Design, Synthesis and Pharmacophoric Model Building of New 3-alkoxymethyl/3-phenyl Indole-2-carboxamides with Potential Antiproliferative Activity. Chem. Biol. Drug Des. 2017, 90, 64–82. [Google Scholar] [CrossRef]
- Gomaa, H.A.M.; Shaker, M.E.; Alzarea, S.I.; Hendawy, O.M.; Mohamed, F.A.M.; Gouda, A.M.; Ali, A.T.; Morcoss, M.M.; Abdelrahman, M.H.; Trembleau, L.; et al. Optimization and SAR Investigation of Novel 2,3-Dihydropyrazino[1,2-a]Indole-1,4-Dione Derivatives as EGFR and BRAFV600E Dual Inhibitors with Potent Antiproliferative and Antioxidant Activities. Bioorg. Chem. 2022, 120, 105616. [Google Scholar] [CrossRef]
- Deb, M.; Das, C.; Deka, B.; Saikia, P.; Baruah, P. Hydrogen-Bond-Catalyzed Arylation of 3-(Aminoalkyl)Indoles via C–N Bond Cleavage with Thiourea under Microwave Irradiation: An Approach to 3-(α,α-Diarylmethyl)Indoles. Synlett 2016, 27, 2788–2794. [Google Scholar] [CrossRef]
- Simon, P.; Lőrinczi, B.; Szatmári, I. C–3 Alkoxymethylation of 4-Oxo-1,4-Dihydroquinoline 2-Carboxylic Acid Esters via Organic Additives. Heliyon 2024, 10, e32188. [Google Scholar] [CrossRef]
- Zádori, D.; Veres, G.; Szalárdy, L.; Klivényi, P.; Toldi, J.; Vécsei, L. Glutamatergic Dysfunctioning in Alzheimer’s Disease and Related Therapeutic Targets. J. Alzheimers Dis. 2014, 42, 177–187. [Google Scholar] [CrossRef]
- Zádori, D.; Nyiri, G.; Szőnyi, A.; Szatmári, I.; Fülöp, F.; Toldi, J.; Freund, T.F.; Vécsei, L.; Klivényi, P. Neuroprotective Effects of a Novel Kynurenic Acid Analogue in a Transgenic Mouse Model of Huntington’s Disease. J. Neural Transm. 2011, 118, 865–875. [Google Scholar] [CrossRef]
- Rózsa, É.; Robotka, H.; Vécsei, L.; Toldi, J. The Janus-Face Kynurenic Acid. J. Neural Transm. 2008, 115, 1087–1091. [Google Scholar] [CrossRef]
- Sas, K.; Robotka, H.; Toldi, J.; Vécsei, L. Mitochondria, Metabolic Disturbances, Oxidative Stress and the Kynurenine System, with Focus on Neurodegenerative Disorders. J. Neurol. Sci. 2007, 257, 221–239. [Google Scholar] [CrossRef]
- Gigler, G.; Szénási, G.; Simó, A.; Lévay, G.; Hársing, L.G.; Sas, K.; Vécsei, L.; Toldi, J. Neuroprotective Effect of L-Kynurenine Sulfate Administered before Focal Cerebral Ischemia in Mice and Global Cerebral Ischemia in Gerbils. Eur. J. Pharm. 2007, 564, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Kiss, C.; Shepard, P.D.; Bari, F.; Schwarcz, R. Cortical Spreading Depression Augments Kynurenate Levels and Reduces Malonate Toxicity in the Rat Cortex. Brain Res. 2004, 1002, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Wielosz, M.; Turski, W.A.; Urbañska, E.M. Endogenous level of kynurenic acid and activities of kynurenine aminotransferases following transient global ischemia in the gerbil hippocampus. Pol. J. Pharmacol. 2003, 55, 443–447. [Google Scholar]
- Nilsson, L.K.; Linderholm, K.R.; Engberg, G.; Paulson, L.; Blennow, K.; Lindström, L.H.; Nordin, C.; Karanti, A.; Persson, P.; Erhardt, S. Elevated Levels of Kynurenic Acid in the Cerebrospinal Fluid of Male Patients with Schizophrenia. Schizophr. Res. 2005, 80, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Fejes, A.; Párdutz, A.; Toldi, J.; Vécsei, L. Kynurenine Metabolites and Migraine: Experimental Studies and Therapeutic Perspectives. Curr. Neuropharmacol. 2011, 9, 376–387. [Google Scholar] [CrossRef] [PubMed]
- Nagy, K.; Plangár, I.; Tuka, B.; Gellért, L.; Varga, D.; Demeter, I.; Farkas, T.; Kis, Z.; Marosi, M.; Zádori, D.; et al. Synthesis and Biological Effects of Some Kynurenic Acid Analogs. Bioorg. Med. Chem. 2011, 19, 7590–7596. [Google Scholar] [CrossRef] [PubMed]
- Tiszlavicz, Z.; Németh, B.; Fülöp, F.; Vécsei, L.; Tápai, K.; Ocsovszky, I.; Mándi, Y. Different Inhibitory Effects of Kynurenic Acid and a Novel Kynurenic Acid Analogue on Tumour Necrosis Factor-α (TNF-α) Production by Mononuclear Cells, HMGB1 Production by Monocytes and HNP1-3 Secretion by Neutrophils. Naunyn-Schmiedebergs Arch. Pharmacol. 2011, 383, 447–455. [Google Scholar] [CrossRef] [PubMed]
- Gellért, L.; Fuzik, J.; Göblös, A.; Sárközi, K.; Marosi, M.; Kis, Z.; Farkas, T.; Szatmári, I.; Fülöp, F.; Vécsei, L.; et al. Neuroprotection with a New Kynurenic Acid Analog in the Four-Vessel Occlusion Model of Ischemia. Eur. J. Pharm. 2011, 667, 182–187. [Google Scholar] [CrossRef] [PubMed]
- Marosi, M.; Nagy, D.; Farkas, T.; Kis, Z.; Rózsa, É.; Robotka, H.; Fülöp, F.; Vécsei, L.; Toldi, J. A Novel Kynurenic Acid Analogue: A Comparison with Kynurenic Acid. An in Vitro Electrophysiological Study. J. Neural Transm. 2010, 117, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Vámos, E.; Párdutz, Á.; Varga, H.; Bohár, Z.; Tajti, J.; Fülöp, F.; Toldi, J.; Vécsei, L. L-Kynurenine Combined with Probenecid and the Novel Synthetic Kynurenic Acid Derivative Attenuate Nitroglycerin-Induced nNOS in the Rat Caudal Trigeminal Nucleus. Neuropharmacology 2009, 57, 425–429. [Google Scholar] [CrossRef] [PubMed]
- Lőrinczi, B.; Csámpai, A.; Fülöp, F.; Szatmári, I. Synthesis of New C-3 Substituted Kynurenic Acid Derivatives. Molecules 2020, 25, 937. [Google Scholar] [CrossRef]


















































































Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).