
Inter-Species Pharmacokinetic Modeling and Scaling for Drug Repurposing of Pyronaridine and Artesunate

Abstract
:
1. Introduction
2. Results
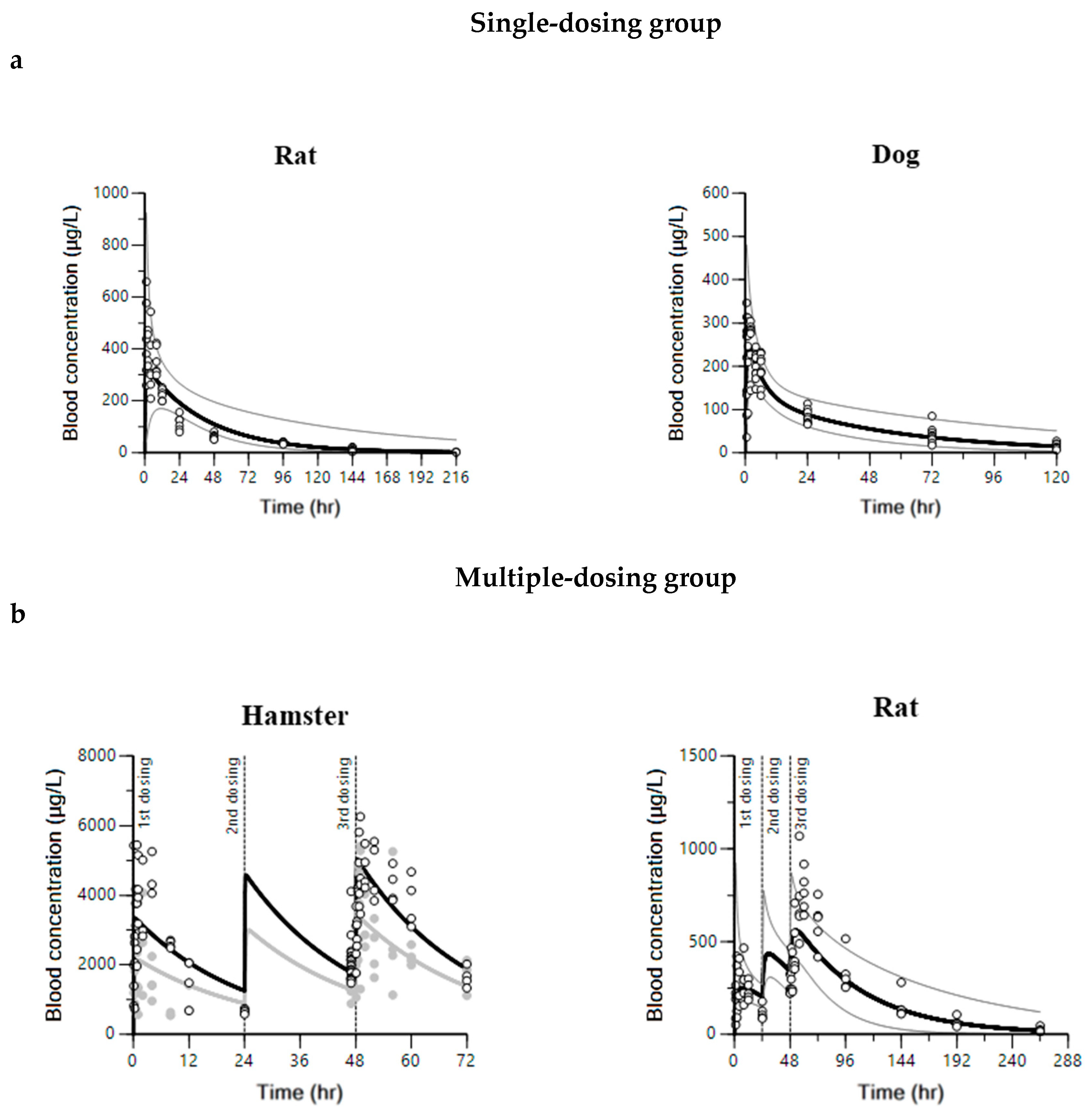
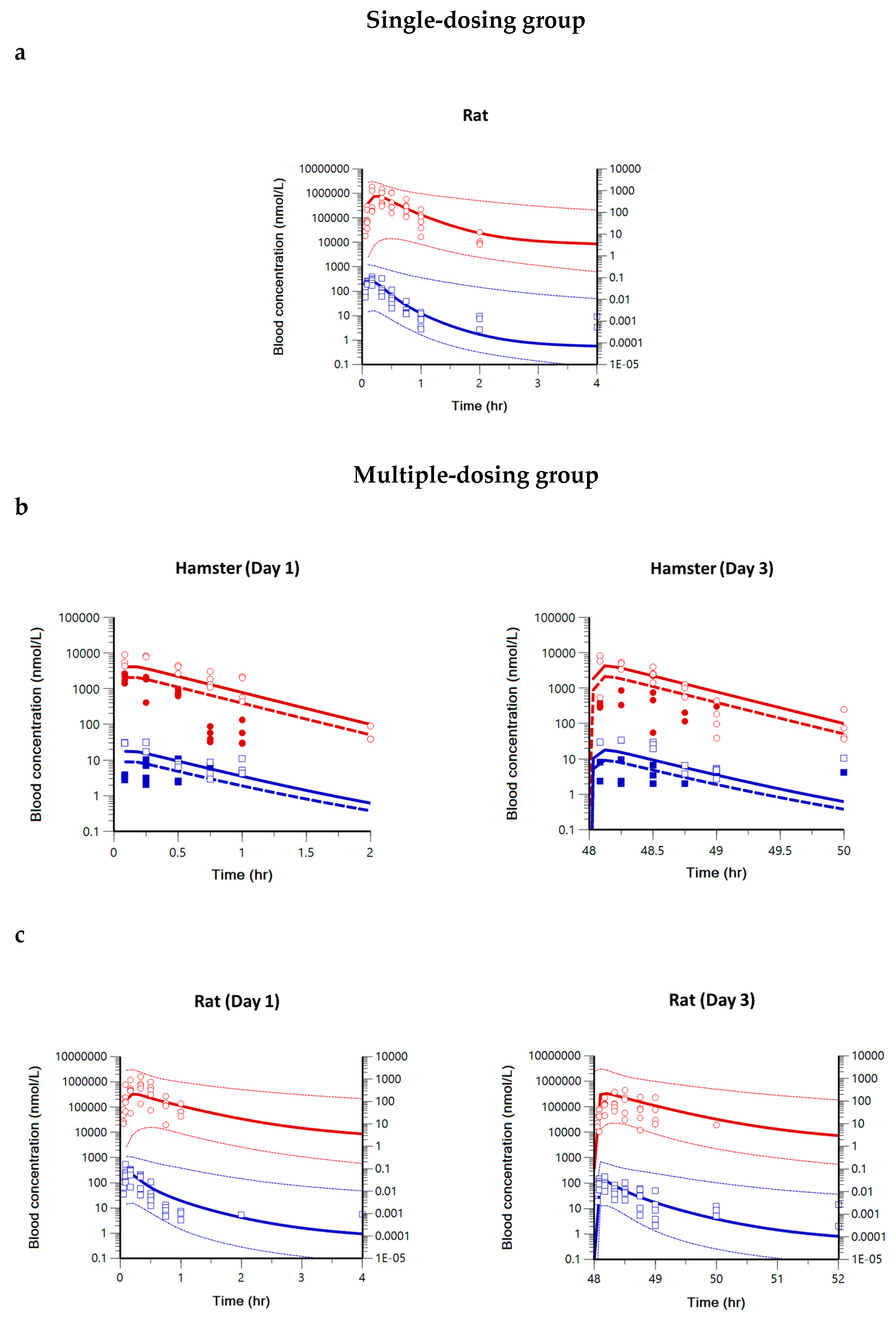
2.1. Pharmacokinetics of Pyronaridine, Artesunate, and Dihydroartemisinin in Hamsters, Rats, and Dogs
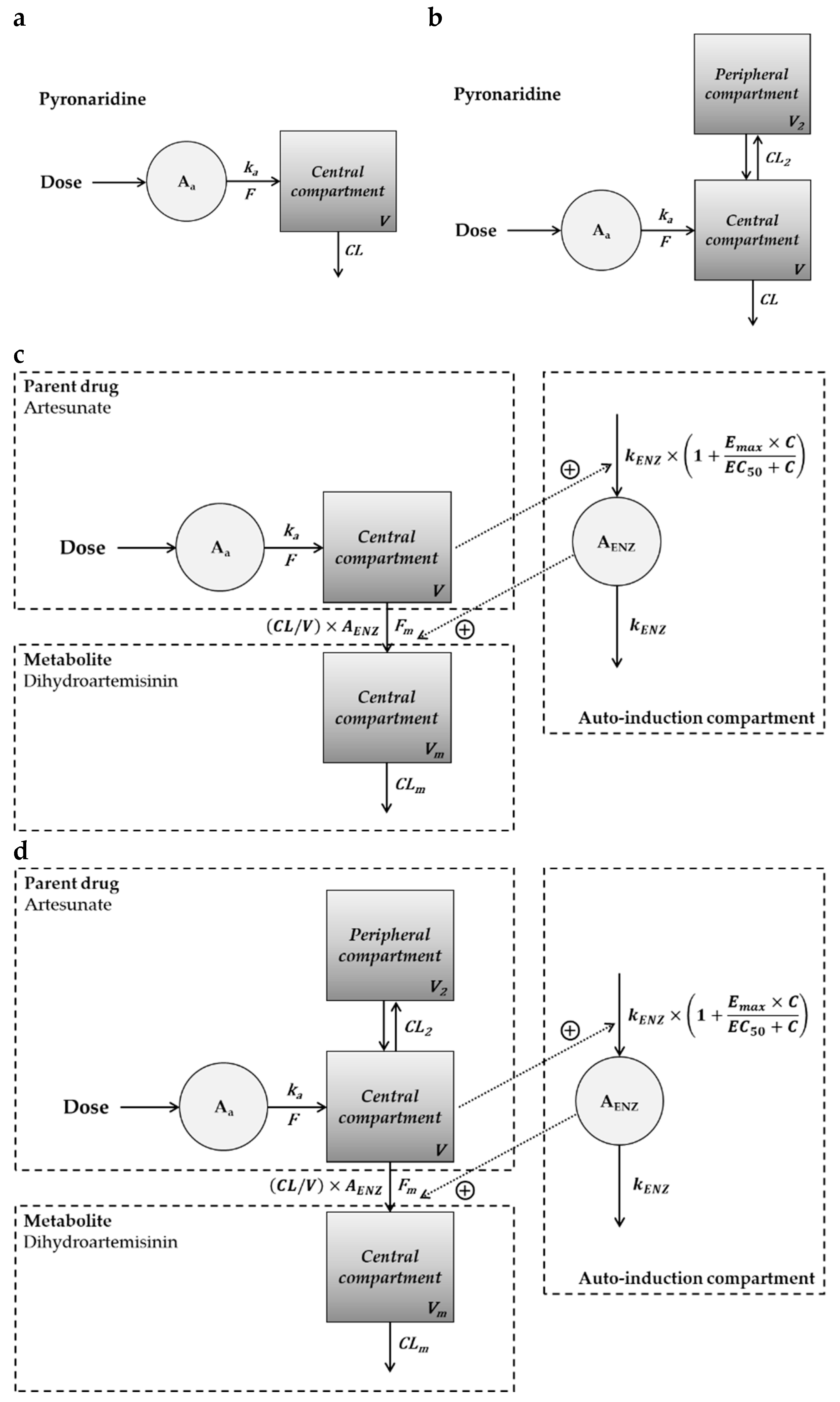
2.2. Compartment PK Modeling in Hamsters, Rats, and Dogs
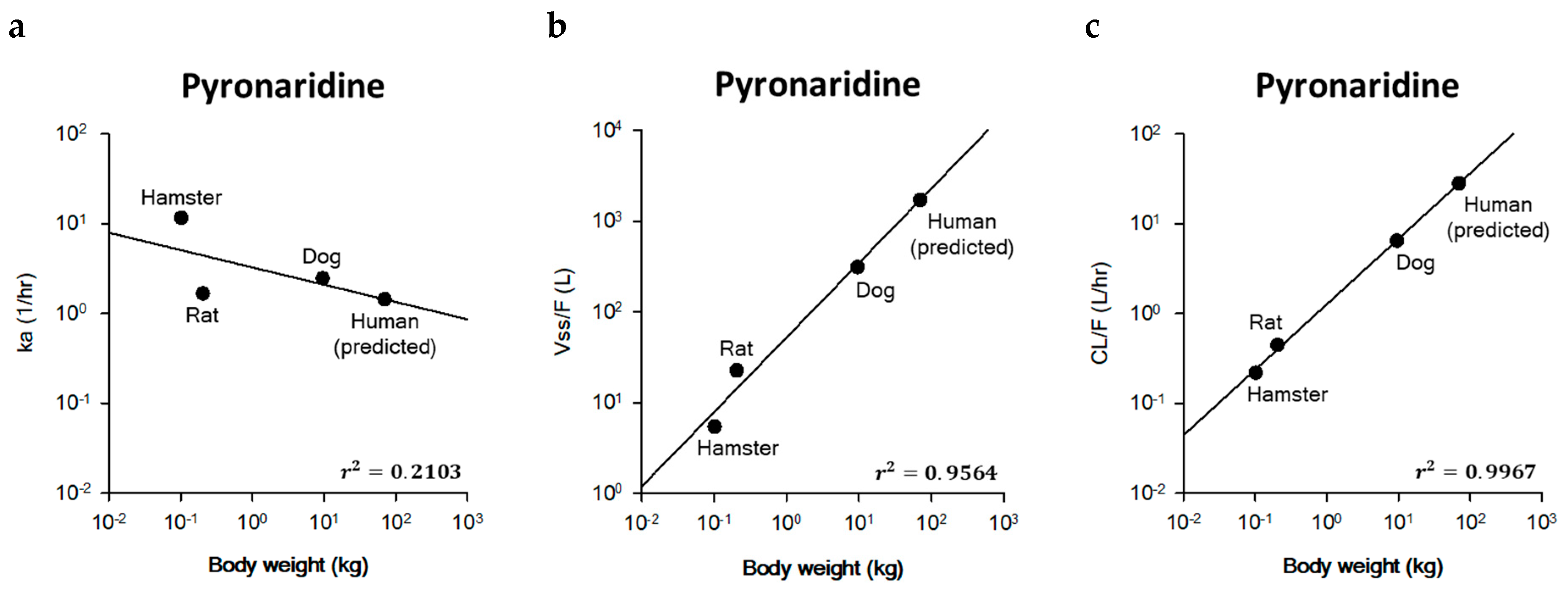
2.3. Prediction of Human PK Parameters Using Allometric Scaling
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. LC-MS/MS Conditions
4.3. Sample Preparation
4.4. Pharmacokinetic Study Design
4.5. Pharmacokinetic Evaluation
4.6. Pharmacokinetic Modeling
4.7. Extrapolation into Clinical PK Parameters by Allometric Scaling
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Croft, S.L.; Duparc, S.; Arbe-Barnes, S.J.; Craft, J.C.; Shin, C.S.; Fleckenstein, L.; Borghini-Fuhrer, I.; Rim, H.J. Review of Pyronaridine Anti-Malarial Properties and Product Characteristics. Malar. J. 2012, 11, 270. [Google Scholar] [CrossRef] [PubMed]
- Kurth, F.; Pongratz, P.; Bélard, S.; Mordmüller, B.; Kremsner, P.G.; Ramharter, M. In Vitro Activity of Pyronaridine against Plasmodium Falciparum and Comparative Evaluation of Anti-Malarial Drug Susceptibility Assays. Malar. J. 2009, 8, 79. [Google Scholar] [CrossRef] [PubMed]
- Moore, L.R.; Fujioka, H.; Williams, P.S.; Chalmers, J.J.; Grimberg, B.; Zimmerman, P.A.; Zborowski, M. Hemoglobin Degradation in Malaria-infected Erythrocytes Determined from Live Cell Magnetophoresis. FASEB J. 2006, 20, 747–749. [Google Scholar] [CrossRef]
- Ma, W.; Balta, V.A.; Pan, W.; Rimer, J.D.; Sullivan, D.J.; Vekilov, P.G. Nonclassical Mechanisms to Irreversibly Suppress β-Hematin Crystal Growth. Commun. Biol. 2023, 6, 783. [Google Scholar] [CrossRef] [PubMed]
- EMA. Pyramax: Public Assessment Report (EMA/CHMP/61768/2012); European Medicines Agency: London, UK, 2021.
- Aweeka, F.T.; German, P.I. Clinical Pharmacology of Artemisinin-Based Combination Therapies. Clin. Pharmacokinet. 2008, 47, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Bukirwa, H.; Unnikrishnan, B.; Kramer, C.V.; Sinclair, D.; Nair, S.; Tharyan, P. Artesunate plus Pyronaridine for Treating Uncomplicated Plasmodium Falciparum Malaria. Cochrane Database Syst. Rev. 2014, 2014, CD006404. [Google Scholar] [CrossRef]
- Thomas, C.M.; Stauffer, W.M.; Alpern, J.D. Food and Drug Administration Approval of Artesunate for Severe Malaria: Enough to Achieve Best Practice? Clin. Infect. Dis. 2023, 76, e864–e866. [Google Scholar] [CrossRef] [PubMed]
- Barradell, L.B.; Fitton, A. Artesunate: A Review of Its Pharmacology and Therapeutic Efficacy in the Treatment of Malaria. Drugs 1995, 50, 714–741. [Google Scholar] [CrossRef] [PubMed]
- Vivas, L.; Rattray, L.; Stewart, L.; Bongard, E.; Robinson, B.L.; Peters, W.; Croft, S.L. Anti-Malarial Efficacy of Pyronaridine and Artesunate in Combination in Vitro and in Vivo. Acta Trop. 2008, 105, 222–228. [Google Scholar] [CrossRef]
- Newton, P.N.; Van Vugt, M.; Teja-Isavadharm, P.; Siriyanonda, D.; Rasameesoroj, M.; Teerapong, P.; Ruangveerayuth, R.; Slight, T.; Nosten, F.; Suputtamongkol, Y.; et al. Comparison of Oral Artesunate and Dihydroartemisinin Antimalarial Bioavailabilities in Acute Falciparum Malaria. Antimicrob. Agents Chemother. 2002, 46, 1125–1127. [Google Scholar] [CrossRef]
- EMA. Artesunate Amivas: EPAR—Medicine Overview (EMA/531550/2021); European Medicines Agency: London, UK, 2022.
- Yu, R.; Jin, G.; Fujimoto, M. Dihydroartemisinin: A Potential Drug for the Treatment of Malignancies and Inflammatory Diseases. Front. Oncol. 2021, 11, 722331. [Google Scholar] [CrossRef] [PubMed]
- Ruwizhi, N.; Maseko, R.B.; Aderibigbe, B.A. Recent Advances in the Therapeutic Efficacy of Artesunate. Pharmaceutics 2022, 14, 504. [Google Scholar] [CrossRef]
- Moss, S.; Mańko, E.; Krishna, S.; Campino, S.; Clark, T.G.; Last, A. How Has Mass Drug Administration with Dihydroartemisinin-Piperaquine Impacted Molecular Markers of Drug Resistance? A Systematic Review. Malar. J. 2022, 21, 186. [Google Scholar] [CrossRef] [PubMed]
- Otta, D.A.; De Araújo, F.F.; De Rezende, V.B.; Souza-Fagundes, E.M.; Elói-Santos, S.M.; Costa-Silva, M.F.; Santos, R.A.; Costa, H.A.; Siqueira-Neto, J.L.; Martins-Filho, O.A.; et al. Identification of Anti-Trypanosoma Cruzi Lead Compounds with Putative Immunomodulatory Activity. Antimicrob. Agents Chemother. 2018, 62, e01834-17. [Google Scholar] [CrossRef] [PubMed]
- Qi, J.; Yang, C.Z.; Wang, C.Y.; Wang, S.B.; Yang, M.; Wang, J.H. Function and Mechanism of Pyronaridine: A New Inhibitor of P-Glycoprotein-Mediated Multidrug Resistance. Acta Pharmacol. Sin. 2002, 23, 544–550. [Google Scholar]
- Villanueva, P.J.; Gutierrez, D.A.; Contreras, L.; Parra, K.; Segura-Cabrera, A.; Varela-Ramirez, A.; Aguilera, R.J. The Antimalarial Drug Pyronaridine Inhibits Topoisomerase II in Breast Cancer Cells and Hinders Tumor Progression In Vivo. Clin. Cancer Drugs 2021, 8, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, W.; Yao, J.; Wang, T.; Li, S.; Qi, W.; Han, S.; Ren, Y.; Dang, Z.; Han, X.; et al. Old Drug Repurposing for Neglected Disease: Pyronaridine as a Promising Candidate for the Treatment of Echinococcus Granulosus Infections. eBioMedicine 2020, 54, 102711. [Google Scholar] [CrossRef] [PubMed]
- Lane, T.R.; Massey, C.; Comer, J.E.; Anantpadma, M.; Freundlich, J.S.; Davey, R.A.; Madrid, P.B.; Ekins, S. Repurposing the Antimalarial Pyronaridine Tetraphosphate to Protect against Ebola Virus Infection. PLoS Negl. Trop. Dis. 2019, 13, e0007890. [Google Scholar] [CrossRef] [PubMed]
- Jackson, N.; Alhussan, A.; Bromma, K.; Jay, D.; Donnelly, J.C.; West, F.G.; Lavasanifar, A.; Weinfeld, M.; Beckham, W.; Chithrani, D.B. Repurposing Antimalarial Pyronaridine as a DNA Repair Inhibitor to Exploit the Full Potential of Gold-Nanoparticle-Mediated Radiation Response. Pharmaceutics 2022, 14, 2795. [Google Scholar] [CrossRef]
- Zuo, S.; Li, Q.; Liu, X.; Feng, H.; Chen, Y. The Potential Therapeutic Effects of Artesunate on Stroke and Other Central Nervous System Diseases. Biomed. Res. Int. 2016, 2016, 1489050. [Google Scholar] [CrossRef]
- Ho, W.E.; Peh, H.Y.; Chan, T.K.; Wong, W.S.F. Artemisinins: Pharmacological Actions beyond Anti-Malarial. Pharmacol. Ther. 2014, 142, 126–139. [Google Scholar] [CrossRef] [PubMed]
- Raffetin, A.; Bruneel, F.; Roussel, C.; Thellier, M.; Buffet, P.; Caumes, E.; Jauréguiberry, S. Use of Artesunate in Non-Malarial Indications. Med. Mal. Infect. 2018, 48, 238–249. [Google Scholar] [CrossRef]
- Puhl, A.C.; Fritch, E.J.; Lane, T.R.; Tse, L.V.; Yount, B.L.; Sacramento, C.Q.; Fintelman-Rodrigues, N.; Tavella, T.A.; Maranhão Costa, F.T.; Weston, S.; et al. Repurposing the Ebola and Marburg Virus Inhibitors Tilorone, Quinacrine, and Pyronaridine: In Vitro Activity against SARS-CoV-2 and Potential Mechanisms. ACS Omega 2021, 6, 7454–7468. [Google Scholar] [CrossRef]
- Jeong, D.E.; Song, H.J.; Lim, S.; Lee, S.J.; Lim, J.E.; Nam, D.H.; Joo, K.M.; Jeong, B.C.; Jeon, S.S.; Choi, H.Y.; et al. Repurposing the Anti-Malarial Drug Artesunate as a Novel Therapeutic Agent for Metastatic Renal Cell Carcinoma Due to Its Attenuation of Tumor Growth, Metastasis, and Angiogenesis. Oncotarget 2015, 6, 33046–33064. [Google Scholar] [CrossRef]
- Vandewynckel, Y.P.; Laukens, D.; Geerts, A.; Vanhove, C.; Descamps, B.; Colle, I.; Devisscher, L.; Bogaerts, E.; Paridaens, A.; Verhelst, X.; et al. Therapeutic Effects of Artesunate in Hepatocellular Carcinoma: Repurposing an Ancient Antimalarial Agent. Eur. J. Gastroenterol. Hepatol. 2014, 26, 861–870. [Google Scholar] [CrossRef] [PubMed]
- Bhise, N.; Agarwal, M.; Thakur, N.; Akshay, P.S.; Cherian, S.; Lole, K. Repurposing of Artesunate, an Antimalarial Drug, as a Potential Inhibitor of Hepatitis E Virus. Arch. Virol. 2023, 168, 147. [Google Scholar] [CrossRef]
- Huang, S.; Galaj, E.; Wang, J.; Guo, Y.; Wang, S.; Shi, M.; Yin, X.; Liu, K.; Luo, Y.; Meng, L.; et al. Repurposing Antimalarial Artesunate for the Prophylactic Treatment of Depression: Evidence from Preclinical Research. Brain Behav. 2023, 13, e2833. [Google Scholar] [CrossRef]
- Ramirez, I.; Marchion, D.; Xiong, Y.; Abbasi, F.; Boac, B.; McClung, C.; Al Sawah, E.; Judson, P.; Apte, S.M.; Lancaster, J.M. Repurposing Artesunate, an Anti-Malarial, for Ovarian Cancer Treatment: Mechanistic Insights from Cell Line Models. Gynecol. Oncol. 2015, 137, 207. [Google Scholar] [CrossRef]
- Lu, X.; Efferth, T. Repurposing of Artemisinin-Type Drugs for the Treatment of Acute Leukemia. Semin. Cancer Biol. 2021, 68, 291–312. [Google Scholar] [CrossRef]
- Lane, T.R.; Dyall, J.; Mercer, L.; Goodin, C.; Foil, D.H.; Zhou, H.; Postnikova, E.; Liang, J.Y.; Holbrook, M.R.; Madrid, P.B.; et al. Repurposing Pyramax® for the Treatment of Ebola Virus Disease: Additivity of the Lysosomotropic Pyronaridine and Non-Lysosomotropic Artesunate. bioRxiv 2020, 13, 061333. [Google Scholar]
- Lane, T.R.; Dyall, J.; Mercer, L.; Goodin, C.; Foil, D.H.; Zhou, H.; Postnikova, E.; Liang, J.Y.; Holbrook, M.R.; Madrid, P.B.; et al. Repurposing Pyramax®, Quinacrine and Tilorone as Treatments for Ebola Virus Disease. Antivir. Res. 2020, 182, 104908. [Google Scholar] [CrossRef] [PubMed]
- Bae, J.-Y.; Lee, G.E.; Park, H.; Cho, J.; Kim, Y.-E.; Lee, J.-Y.; Ju, C.; Kim, W.-K.; Kim, J.I.; Park, M.-S. Pyronaridine and Artesunate Are Potential Antiviral Drugs against COVID-19 and Influenza. bioRxiv 2020. [Google Scholar] [CrossRef]
- Mithun, R.; Shubham, J.K.; Anil, G.J. Drug Repurposing—Hypothesis, Molecular Aspects and Therapeutic Applications; IntechOpen: London, UK, 2020. [Google Scholar]
- Kulkarni, V.S.; Alagarsamy, V.; Solomon, V.R.; Jose, P.A.; Murugesan, S. Drug Repurposing: An Effective Tool in Modern Drug Discovery. Russ. J. Bioorg. Chem. 2023, 49, 157–166. [Google Scholar] [CrossRef]
- Kang, D.W.; Kim, K.M.; Kim, J.H.; Cho, H.Y. Application of Minimal Physiologically-Based Pharmacokinetic Model to Simulate Lung and Trachea Exposure of Pyronaridine and Artesunate in Hamsters. Pharmaceutics 2023, 15, 838. [Google Scholar] [CrossRef] [PubMed]
- Morris, C.A.; Lopez-Lazaro, L.; Jung, D.; Methaneethorn, J.; Duparc, S.; Borghini-Fuhrer, I.; Pokorny, R.; Shin, C.S.; Fleckenstein, L. Drug-Drug Interaction Analysis of Pyronaridine/Artesunate and Ritonavir in Healthy Volunteers. Am. J. Trop. Med. Hyg. 2012, 86, 489–495. [Google Scholar] [CrossRef] [PubMed]
- Lindegardh, N.; Hanpithakpong, W.; Kamanikom, B.; Pattayaso, J.; Singhasivanon, P.; White, N.J.; Day, N.P.J. Quantification of Dihydroartemisinin, Artesunate and Artemisinin in Human Blood: Overcoming the Technical Challenge of Protecting the Peroxide Bridge. Bioanalysis 2011, 3, 1613–1624. [Google Scholar] [CrossRef] [PubMed]
- Stein, A.M.; Peletier, L.A. Predicting the Onset of Nonlinear Pharmacokinetics. CPT Pharmacomet. Syst. Pharmacol. 2018, 7, 670–677. [Google Scholar] [CrossRef]
- Cho, H.; Lee, E.K. Pkconverter: R Package to Convert the Pharmacokinetic Parameters. Transl. Clin. Pharmacol. 2019, 27, 73–79. [Google Scholar] [CrossRef]
- Bennett, K.; Si, Y.; Steinbach, T.; Zhang, J.; Li, Q. Pharmacokinetic and Pharmacodynamic Evaluation of Intramuscular Artesunate in Healthy Beagle Dogs. Am. J. Trop. Med. Hyg. 2008, 79, 36–41. [Google Scholar] [CrossRef]
- Kenyon, E.M. Interspecies Extrapolation. Methods Mol. Biol. 2012, 959, 501–520. [Google Scholar]
- Feng, Z.; Jiang, N.X.; Wang, C.Y.; Zhang, W. Pharmacokinetics of Pyronaridine, an Antimalarial in Rabbits. Acta Pharm. Sin. 1986, 21, 801–805. [Google Scholar]
- Wages, S.A.; Patchen, L.C.; Churchill, F.C. Analysis of Blood and Urine Samples from Macaca Mulata for Pyronaridine by High-Performance Liquid Chromatography with Electrochemical Detection. J. Chromatogr. B Biomed. Sci. Appl. 1990, 527, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.C.; Fleckenstein, L. Improved Assay Method for the Determination of Pyronaridine in Plasma and Whole Blood by High-Performance Liquid Chromatography for Application to Clinical Pharmacokinetic Studies. J. Chromatogr. B Biomed. Sci. Appl. 2001, 752, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Ni, Y.C.; Zhan, C.Q.; HA, S.H.; Shao, B.R. The Embryotoxicity of a New Antimalarial Pyronaridine in Rats. Acta Pharm. Sin. 1982, 17, 401–406. [Google Scholar]
- Zou, H.X.; Zhang, Y.F.; Zhong, D.F.; Jiang, Y.; Liu, F.; Zhao, Q.Y.; Zuo, Z.; Zhang, Y.F.; Yan, X.Y. Effect of Autoinduction and Food on the Pharmacokinetics of Furmonertinib and Its Active Metabolite Characterized by a Population Pharmacokinetic Model. Acta Pharmacol. Sin. 2022, 43, 1865–1874. [Google Scholar] [CrossRef] [PubMed]
- Cosson, V.; Jorga, K.; Fuseau, E. Modeling of Metabolite Pharmacokinetics in a Large Pharmacokinetic Data Set: An Application. In Pharmacometrics: The Science of Quantitative Pharmacology; Wiley: Hoboken, NJ, USA, 2006. [Google Scholar]
- Keiser, J.; Gruyer, M.S.; Perrottet, N.; Zanolari, B.; Mercier, T.; Decosterd, L. Pharmacokinetic Parameters of Artesunate and Dihydroartemisinin in Rats Infected with Fasciola Hepatica. J. Antimicrob. Chemother. 2009, 63, 543–549. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Xie, L.H.; Si, Y.; Wong, E.; Upadhyay, R.; Yanez, D.; Weina, P.J. Toxicokinetics and Hydrolysis of Artelinate and Artesunate in Malaria-Infected Rats. Int. J. Toxicol. 2005, 24, 241–250. [Google Scholar] [CrossRef] [PubMed]
- Zhu, F.; Du, F.; Li, X.; Xing, J. An Investigation of the Auto-Induction of and Gender-Related Variability in the Pharmacokinetics of Dihydroartemisinin in the Rat. Malar. J. 2012, 11, 379. [Google Scholar] [CrossRef] [PubMed]
- Xing, J.; Bai, K.H.; Liu, T.; Wang, R.L.; Zhang, L.F.; Zhang, S.Q. The Multiple-Dosing Pharmacokinetics of Artemether, Artesunate, and Their Metabolite Dihydroartemisinin in Rats. Xenobiotica 2011, 41, 252–258. [Google Scholar] [CrossRef]
- Smith, D.A.; Beaumont, K.; Maurer, T.S.; Di, L. Volume of Distribution in Drug Design. J. Med. Chem. 2015, 58, 5691–5698. [Google Scholar] [CrossRef]
- Murad, N.; Pasikanti, K.K.; Madej, B.D.; Minnich, A.; McComas, J.M.; Crouch, S.; Polli, J.W.; Weber, A.D. Predicting Volume of Distribution in Humans: Performance of in Silico Methods for a Large Set of Structurally Diverse Clinical Compounds. Drug Metab. Dispos. 2021, 49, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, H.; Yoshida, K.; Nakada, T.; Kojima, K.; Ogasawara, A.; Nakamaru, Y.; Yamazaki, H. Prediction of Human Distribution Volumes of Compounds in Various Elimination Phases Using Physiologically Based Pharmacokinetic Modeling and Experimental Pharmacokinetics in Animals. Drug Metab. Dispos. 2019, 47, 114–123. [Google Scholar] [CrossRef] [PubMed]
- del Amo, E.M.; Ghemtio, L.; Xhaard, H.; Yliperttula, M.; Urtti, A.; Kidron, H. Applying Linear and Non-Linear Methods for Parallel Prediction of Volume of Distribution and Fraction of Unbound Drug. PLoS ONE 2013, 8, e74758. [Google Scholar] [CrossRef] [PubMed]
- Chai, L.; Wang, R.; Wang, Y.; Guo, W.; Li, N.; Zuo, H.; Wang, Y.; Duan, D.; Ren, G.; Zheng, B.; et al. Auto-Induction of Intestinal First-Pass Effect Related Time-Dependent Pharmacokinetics of Artemisinin Rather than Dihydroartemisinin. J. Pharm. Sci. 2021, 110, 458–466. [Google Scholar] [CrossRef] [PubMed]
- Smythe, W.; Khandelwal, A.; Merle, C.; Rustomjee, R.; Gninafon, M.; Lo, M.B.; Sow, O.B.; Olliaro, P.L.; Lienhardt, C.; Horton, J.; et al. A Semimechanistic Pharmacokinetic-Enzyme Turnover Model for Rifampin Autoinduction in Adult Tuberculosis Patients. Antimicrob. Agents Chemother. 2012, 56, 2091–2098. [Google Scholar] [CrossRef] [PubMed]
- Svensson, R.J.; Aarnoutse, R.E.; Diacon, A.H.; Dawson, R.; Gillespie, S.H.; Boeree, M.J.; Simonsson, U.S.H. A Population Pharmacokinetic Model Incorporating Saturable Pharmacokinetics and Autoinduction for High Rifampicin Doses. Clin. Pharmacol. Ther. 2018, 103, 674–683. [Google Scholar] [CrossRef] [PubMed]
- Shaw, J.P.; Jaw-Tsai, S. An Interspecies Extrapolation of the Pharmacokinetics of Telavancin, a Rapidly Bactericidal, Concentration-Dependent Antibiotic. Xenobiotica 2011, 41, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Boxenbaum, H. Interspecies Scaling, Allometry, Physiological Time, and the Ground Plan of Pharmacokinetics. J. Pharmacokinet. Biopharm. 1982, 10, 201–227. [Google Scholar] [CrossRef]
- Mahmood, I.; Balian, J.D. Interspecies Scaling: Predicting Clearance of Drugs in Humans. Three Different Approaches. Xenobiotica 1996, 26, 887–895. [Google Scholar] [CrossRef]
- Huang, Q.; Riviere, J.E. The Application of Allometric Scaling Principles to Predict Pharmacokinetic Parameters across Species. Expert Opin. Drug Metab. Toxicol. 2014, 10, 1241–1253. [Google Scholar] [CrossRef]
- Mahmood, I. Application of Allometric Principles for the Prediction of Pharmacokinetics in Human and Veterinary Drug Development. Adv. Drug Deliv. Rev. 2007, 59, 1177–1192. [Google Scholar] [CrossRef] [PubMed]
- Jolling, K.; Perez Ruixo, J.J.; Hemeryck, A.; Vermeulen, A.; Greway, T. Mixed-Effects Modelling of the Interspecies Pharmacokinetic Scaling of Pegylated Human Erythropoietin. Eur. J. Pharm. Sci. 2005, 24, 465–475. [Google Scholar] [CrossRef] [PubMed]
- Chu, W.Y.; Dorlo, T.P.C. Pyronaridine: A Review of Its Clinical Pharmacology in the Treatment of Malaria. J. Antimicrob. Chemother. 2023, 78, 2406–2418. [Google Scholar] [CrossRef] [PubMed]
- Ayyoub, A.; Methaneethorn, J.; Ramharter, M.; Djimde, A.A.; Tekete, M.; Duparc, S.; Borghini-Fuhrer, I.; Shin, J.S.; Fleckenstein, L. Population Pharmacokinetics of Pyronaridine in Pediatric Malaria Patients. Antimicrob. Agents Chemother. 2016, 60, 1450–1458. [Google Scholar] [CrossRef]
- Miller, R.S.; Li, Q.; Cantilena, L.R.; Leary, K.J.; Saviolakis, G.A.; Melendez, V.; Smith, B.; Weina, P.J. Pharmacokinetic Profiles of Artesunate Following Multiple Intravenous Doses of 2, 4, and 8mg/Kg in Healthy Volunteers: Phase 1b Study. Malar. J. 2012, 11, 255. [Google Scholar] [CrossRef] [PubMed]
- Tarning, J.; Rijken, M.J.; McGready, R.; Phyo, A.P.; Hanpithakpong, W.; Day, N.P.J.; White, N.J.; Nosten, F.; Lindegardh, N. Population Pharmacokinetics of Dihydroartemisinin and Piperaquine in Pregnant and Nonpregnant Women with Uncomplicated Malaria. Antimicrob. Agents Chemother. 2012, 56, 1997–2007. [Google Scholar] [CrossRef] [PubMed]
- Birgersson, S.; Valea, I.; Tinto, H.; Traore-Coulibaly, M.; Toe, L.C.; Hoglund, R.M.; Van Geertruyden, J.P.; Ward, S.A.; D’Alessandro, U.; Abelö, A.; et al. Population Pharmacokinetics of Artesunate and Dihydroartemisinin in Pregnant and Non-Pregnant Women with Uncomplicated Plasmodium Falciparum Malaria in Burkina Faso: An Open Label Trial. Wellcome Open Res. 2020, 4, 45. [Google Scholar] [CrossRef] [PubMed]
- Simpson, J.A.; Agbenyega, T.; Barnes, K.I.; Di Perri, G.; Folb, P.; Gomes, M.; Krishna, S.; Krudsood, S.; Looareesuwan, S.; Mansor, S.; et al. Population Pharmacokinetics of Artesunate and Dihydroartemisinin Following Intra-Rectal Dosing of Artesunate in Malaria Patients. PLoS Med. 2006, 3, e444. [Google Scholar] [CrossRef]
- Tan, B.; Naik, H.; Jang, I.J.; Yu, K.S.; Kirsch, L.E.; Shin, C.S.; Craft, J.C.; Fleckenstein, L. Population Pharmacokinetics of Artesunate and Dihydroartemisinin Following Single-and Multiple-Dosing of Oral Artesunate in Healthy Subjects. Malar. J. 2009, 8, 304. [Google Scholar] [CrossRef]
- Newton, P.N.; Barnes, K.I.; Smith, P.J.; Evans, A.C.; Chierakul, W.; Ruangveerayuth, R.; White, N.J. The Pharmacokinetics of Intravenous Artesunate in Adults with Severe Falciparum Malaria. Eur. J. Clin. Pharmacol. 2006, 62, 1003–1009. [Google Scholar] [CrossRef]
- Nealon, C.; Dzeing, A.; Müller-Römer, U.; Planche, T.; Sinou, V.; Kombila, M.; Kremsner, P.G.; Parzy, D.; Krishna, S. Intramuscular Bioavailability and Clinical Efficacy of Artesunate in Gabonese Children with Severe Malaria. Antimicrob. Agents Chemother. 2002, 46, 3933–3939. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Cantilena, L.R.; Leary, K.J.; Saviolakis, G.A.; Miller, R.S.; Melendez, V.; Weina, P.J. Pharmacokinetic Profiles of Artesunate after Single Intravenous Doses at 0.5,1,2,4, and 8 Mg/Kg in Healthy Volunteers: A Phase I Study. Am. J. Trop. Med. Hyg. 2009, 81, 615–621. [Google Scholar] [CrossRef] [PubMed]
- Morris, C.A.; Duparc, S.; Borghini-Fuhrer, I.; Jung, D.; Shin, C.S.; Fleckenstein, L. Review of the Clinical Pharmacokinetics of Artesunate and Its Active Metabolite Dihydroartemisinin Following Intravenous, Intramuscular, Oral or Rectal Administration. Malar. J. 2011, 10, 263. [Google Scholar] [CrossRef] [PubMed]
- Kouakou, Y.I.; Tod, M.; Leboucher, G.; Lavoignat, A.; Bonnot, G.; Bienvenu, A.L.; Picot, S. Systematic Review of Artesunate Pharmacokinetics: Implication for Treatment of Resistant Malaria. Int. J. Infect. Dis. 2019, 89, 30–44. [Google Scholar] [CrossRef]
- Batty, K.T.; Thu, L.T.A.; Davis, T.M.E.; Ilett, K.F.; Mai, T.X.; Hung, N.C.; Tien, N.P.; Powell, S.M.; Van Thien, H.; Binh, T.Q.; et al. A Pharmacokinetic and Pharmacodynamic Study of Intravenous vs Oral Artesunate in Uncomplicated Falciparum Malaria. Br. J. Clin. Pharmacol. 1998, 45, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Davis, T.M.E. The Pharmacokinetic Properties of Intramuscular Artesunate and Rectal Dihydroartemisinin in Uncomplicated Falciparum Malaria. Br. J. Clin. Pharmacol. 2002, 53, 23–30. [Google Scholar] [CrossRef]
- Davis, T.M.E.; Phuong, H.L.; Ilett, K.F.; Hung, N.C.; Batty, K.T.; Phuong, V.D.B.; Powell, S.M.; Thien, H.V.; Binh, T.Q. Pharmacokinetics and Pharmacodynamics of Intravenous Artesunate in Severe Falciparum Malaria. Antimicrob. Agents Chemother. 2001, 45, 181–186. [Google Scholar] [CrossRef]
- Krishna, S.; Planche, T.; Agbenyega, T.; Woodrow, C.; Agranoff, D.; Bedu-Addo, G.; Owusu-Ofori, A.K.; Appiah, J.A.; Ramanathan, S.; Mansor, S.M.; et al. Bioavailability and Preliminary Clinical Efficacy of Intrarectal Artesunate in Ghanaian Children with Moderate Malaria. Antimicrob. Agents Chemother. 2001, 45, 509–516. [Google Scholar] [CrossRef] [PubMed]
- Hien, T.T.; Davis, T.M.E.; Chuong, L.V.; Ilett, K.F.; Sinh, D.X.T.; Phu, N.H.; Agus, C.; Chiswell, G.M.; White, N.J.; Farrar, J. Comparative Pharmacokinetics of Intramuscular Artesunate and Artemether in Patients with Severe Falciparum Malaria. Antimicrob. Agents Chemother. 2004, 48, 4234–4239. [Google Scholar] [CrossRef]
- Holt, K.; Nagar, S.; Korzekwa, K. Methods to Predict Volume of Distribution. Curr. Pharmacol. Rep. 2019, 5, 391–399. [Google Scholar] [CrossRef]
- Gillette, J.R. Factors Affecting Drug Metabolism. Ann. N. Y. Acad. Sci. 1971, 179, 43–66. [Google Scholar] [CrossRef] [PubMed]
- Sawada, Y.; Hanano, M.; Sugiyama, Y.; Harashima, H.; Iga, T. Prediction of the Volumes of Distribution of Basic Drugs in Humans Based on Data from Animals. J. Pharmacokinet. Biopharm. 1984, 12, 587–596. [Google Scholar] [CrossRef]
- Li, Q.; Xie, L.; Zhang, J.; Weina, P.J. The Distribution Pattern of Intravenous [14C] Artesunate in Rat Tissues by Quantitative Whole-Body Autoradiography and Tissue Dissection Techniques. J. Pharm. Biomed. Anal. 2008, 48, 876–884. [Google Scholar] [CrossRef]
- Li, Q.; Xie, L.H.; Haeberle, A.; Zhang, J.; Weina, P. The Evaluation of Radiolabeled Artesunate on Tissue Distribution in Rats and Protein Binding in Humans. Am. J. Trop. Med. Hyg. 2006, 75, 817–826. [Google Scholar] [CrossRef]
- Xie, L.H.; Li, Q.; Zhang, J.; Weina, P.J. Pharmacokinetics, Tissue Distribution and Mass Balance of Radiolabeled Dihydroartemisinin in Male Rats. Malar. J. 2009, 8, 112. [Google Scholar] [CrossRef] [PubMed]
- Doke, S.K.; Dhawale, S.C. Alternatives to Animal Testing: A Review. Saudi Pharm. J. 2015, 23, 223–229. [Google Scholar] [CrossRef]
- FDA. Facts about FDA and Animal Welfare, Testing & Research; FDA: Silver Spring, MA, USA, 2024.
- Mahmood, I. Interspecies Scaling: Predicting Volumes, Mean Residence Time and Elimination Half-Life. Some Suggestions. J. Pharm. Pharmacol. 1998, 50, 493–499. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Unit | Estimate | CV% | IIV * (%RSE) |
|---|---|---|---|---|
| Pyronaridine | ||||
| Hamster | ||||
| ka | 1/h | 11.62 | 30.21 | - |
| V/F | L/kg | 53.32 | 6.58 | - |
| CL/F | L/h/kg | 2.15 | 4.36 | - |
| ε | - | 0.36 | 7.95 | - |
| Rat | ||||
| ka | 1/h | 1.67 | 8.84 | 0.02 (13.44) |
| V/F | L/kg | 110.00 | 4.96 | 1.69 (16.25) |
| CL/F | L/h/kg | 2.18 | 6.15 | 0.04 (28.17) |
| ε | - | 0.34 | 4.13 | - |
| Dog | ||||
| ka | 1/h | 2.46 | 22.43 | 0.46 (60.48) |
| V/F | L/kg | 18.98 | 10.20 | 0.03 (142.17) |
| V2/F | L/kg | 13.70 | 17.82 | - |
| CL/F | L/h/kg | 0.68 | 8.54 | 0.05 (48.35) |
| CL2/F | L/h/kg | 1.30 | 28.88 | - |
| ε | - | 0.20 | 10.15 | - |
| Artesunate and dihydroartemisinin | ||||
| Hamster | ||||
| ka | 1/h | 2.08 | 15.02 | - |
| V/F | L/kg | 1571.71 | 18.95 | - |
| CL/F | L/h/kg | 3555.94 | 9.60 | - |
| Vm/(F∙Fm) | L/kg | 0.02 | 11.70 | - |
| CLm/(F∙Fm) | L/h/kg | 117.68 | 9.66 | - |
| kenz | 1/h | 47.40 | 13.19 | - |
| Emax | - | 7.06 | 10.70 | - |
| EC50 | nmol/L | 0.42 | 13.37 | - |
| ε (artesunate) | - | 4.38 | 14.21 | - |
| ε (dihydroartemisinin) | - | 1033.76 | 12.12 | - |
| Rat | ||||
| ka | 1/h | 1.54 | 24.85 | |
| V/F | L/kg | 33.03 | 14.81 | |
| V2/F | L/kg | 481.05 | 21.47 | |
| CL/F | L/h/kg | 13.33 | 14.61 | |
| CL2/F | L/h/kg | 82.69 | 13.45 | 0.18 (0.06) |
| Vm/(F∙Fm) | L/kg | 0.16 | 25.23 | |
| CLm/(F∙Fm) | L/h/kg | 237.25 | 34.09 | 1.13 (0.29) |
| kenz | 1/h | 1.04 | 28.48 | 1.32 (0.41) |
| Emax | - | 76.89 | 19.64 | |
| EC50 | nmol/L | 0.49 | 42.60 | |
| ε (artesunate) | - | 35.19 | 18.81 | - |
| ε (dihydroartemisinin) | - | 194.67 | 25.47 | - |
| Species | Group | N | Dosage Regimen (Pyronaridine/Artesunate) | Blood Sampling Time (Hour) | Ref. |
|---|---|---|---|---|---|
| Hamster | H1 | 60 | 180/60 mg/kg daily (for 3 days) | 0, 0.08, 0.25, 0.5, 0.75, 1, 2, 4, 8, 12, 24, 47, 48.08, 48.25, 48.5, 48.75, 49, 50, 52, 56, 60, 72 | [37] |
| Hamster | H2 | 48 | 360/120 mg/kg daily (for 3 days) | 0, 0.08, 0.25, 0.5, 0.75, 1, 2, 4, 8, 12, 24, 47, 48.08, 48.25, 48.5, 48.75, 49, 50, 52, 56, 60, 72 | [37] |
| Rat | R1 | 3 | 60/20 mg/kg once | 0, (0.05, 0.08, 0.16, 0.33, 0.5, 0.75), 1, 2, 4, 8, 12, 24, 48, 96, 144 | - |
| 3 | 60/20 mg/kg once | 0, (0.05, 0.08, 0.16, 0.33, 0.5, 0.75), 1, 2, 4, 8, 12, 24, 48, 96, 144, 216 | - | ||
| Rat | R2 | 5 | 60/20 mg/kg daily (for 3 days) | 0, (0.05, 0.08, 0.16, 0.33, 0.5, 0.75), 1, 2, 4, 8, 12, 24, 48, (48.05, 48.17, 48.33, 48.5, 48.75), 49, 50, 52, 56, 60, 72, 96, 144, 192, 264 | - |
| Dog | D1 | 9 | 90/- mg/head once | 0, 0.5, 1, 2, 4, 6, 24, 72, 120 | - |
| One-compartment model of pyronaridine in hamsters and rats |
| Two-compartment model of pyronaridine in dogs |
| The parent-metabolite model with auto-induction for artesunate and dihydroartemisinin in hamsters |
| The parent-metabolite model with auto-induction for artesunate and dihydroartemisinin in rats |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kang, D.W.; Kim, J.H.; Kim, K.M.; Cho, S.-j.; Choi, G.-W.; Cho, H.-Y. Inter-Species Pharmacokinetic Modeling and Scaling for Drug Repurposing of Pyronaridine and Artesunate. Int. J. Mol. Sci. 2024, 25, 6998. https://doi.org/10.3390/ijms25136998
Kang DW, Kim JH, Kim KM, Cho S-j, Choi G-W, Cho H-Y. Inter-Species Pharmacokinetic Modeling and Scaling for Drug Repurposing of Pyronaridine and Artesunate. International Journal of Molecular Sciences. 2024; 25(13):6998. https://doi.org/10.3390/ijms25136998
Chicago/Turabian StyleKang, Dong Wook, Ju Hee Kim, Kyung Min Kim, Seok-jin Cho, Go-Wun Choi, and Hea-Young Cho. 2024. "Inter-Species Pharmacokinetic Modeling and Scaling for Drug Repurposing of Pyronaridine and Artesunate" International Journal of Molecular Sciences 25, no. 13: 6998. https://doi.org/10.3390/ijms25136998