
Unravelling the Antifibrinolytic Mechanism of Action of the 1,2,3-Triazole Derivatives

 ,
,  , and
, and
Abstract
1. Introduction
2. Results
2.1. Isolated Enzyme Assays
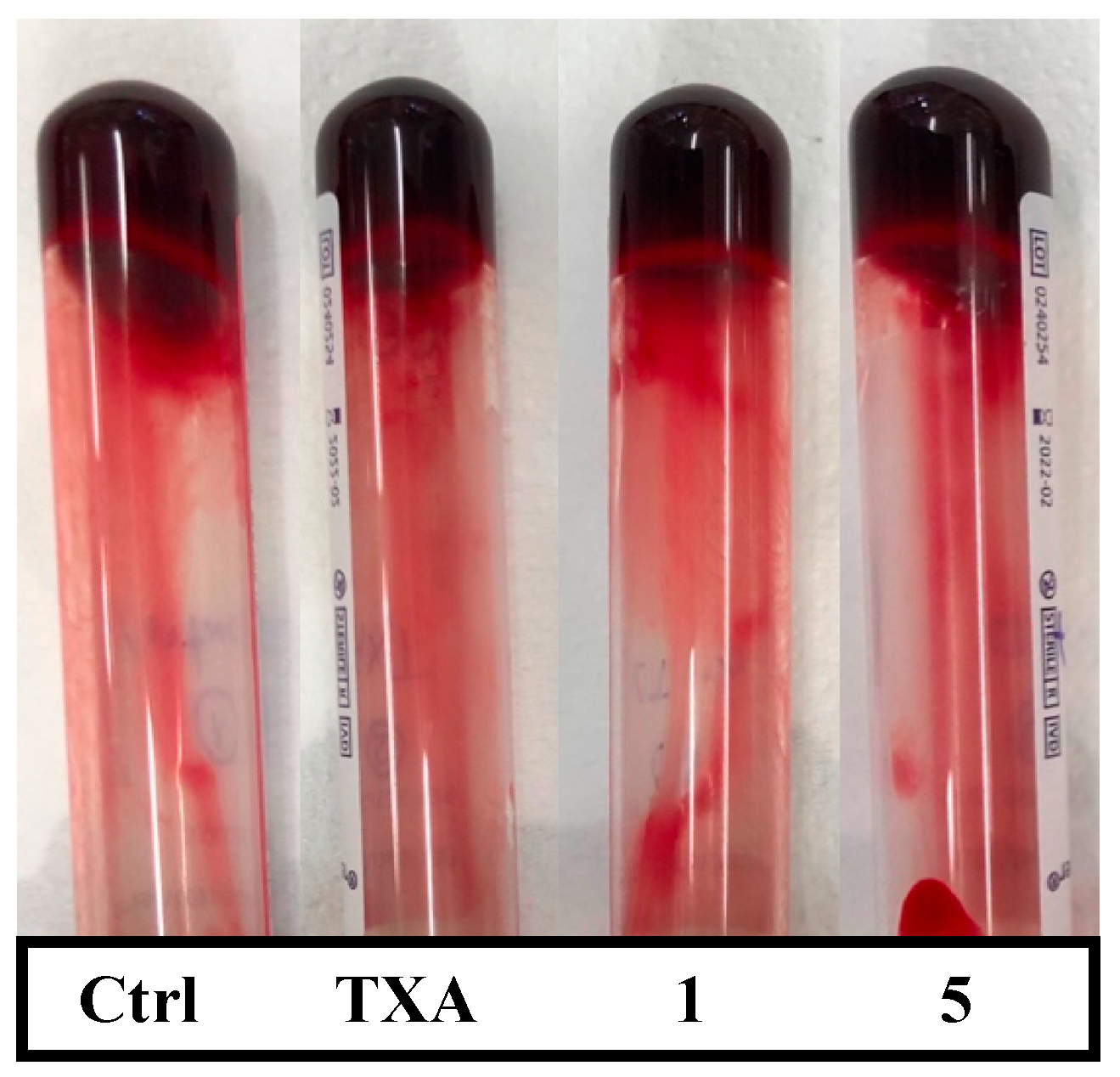
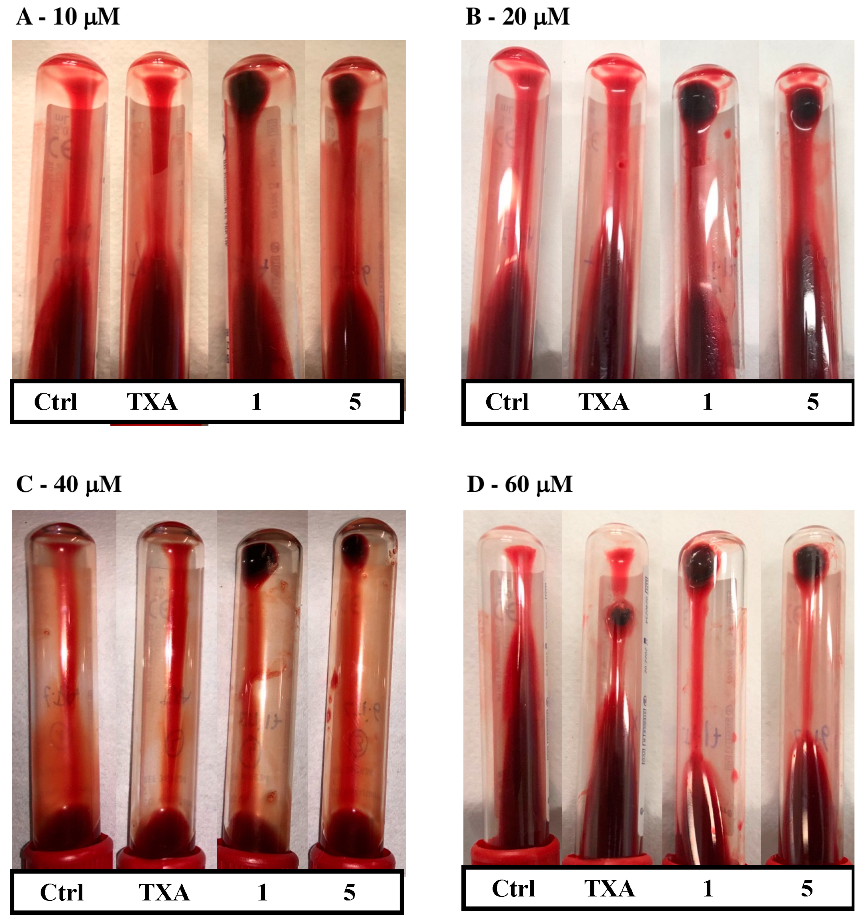
2.2. Ex Vivo Assays
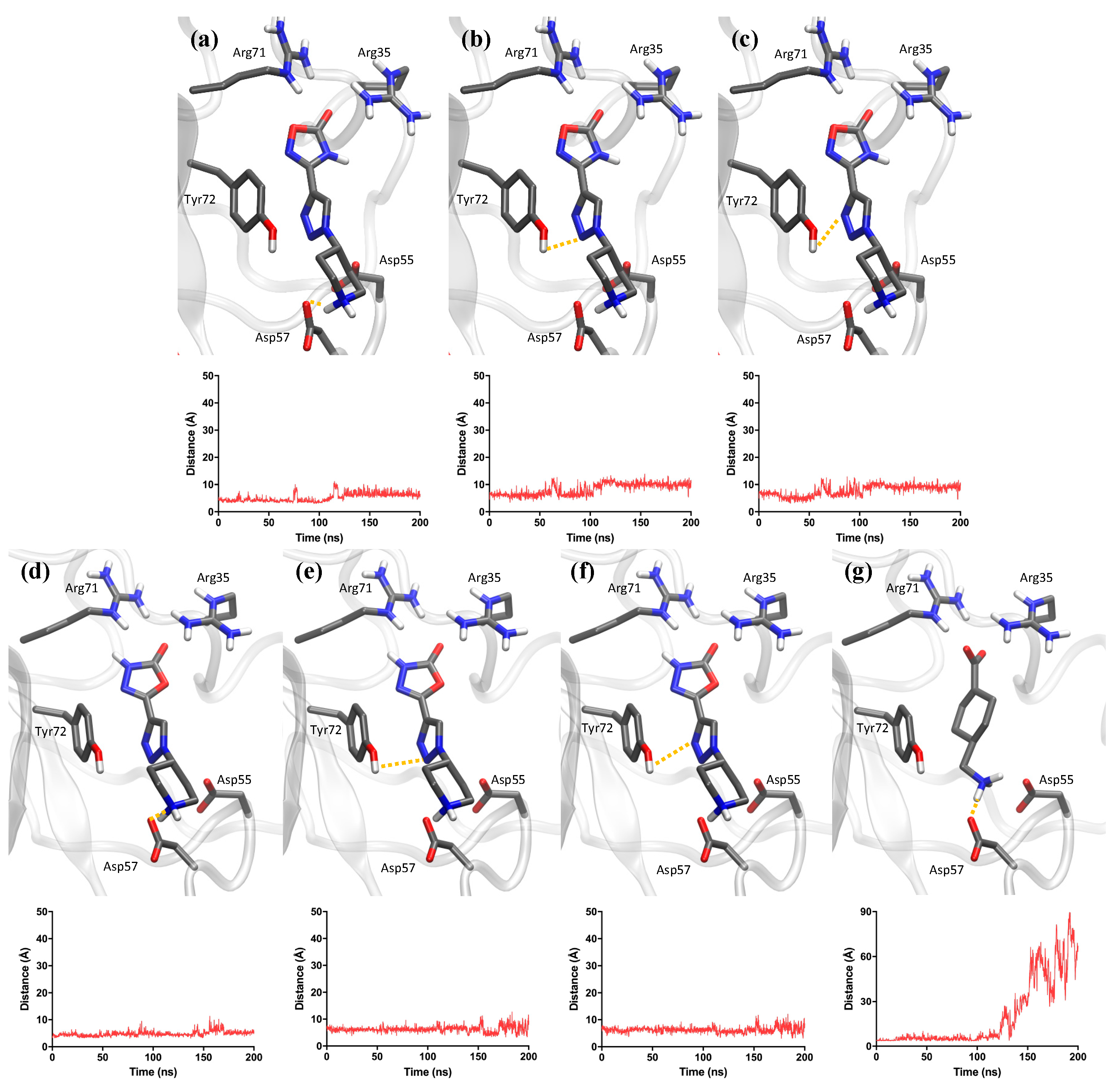
2.3. Computational Analysis
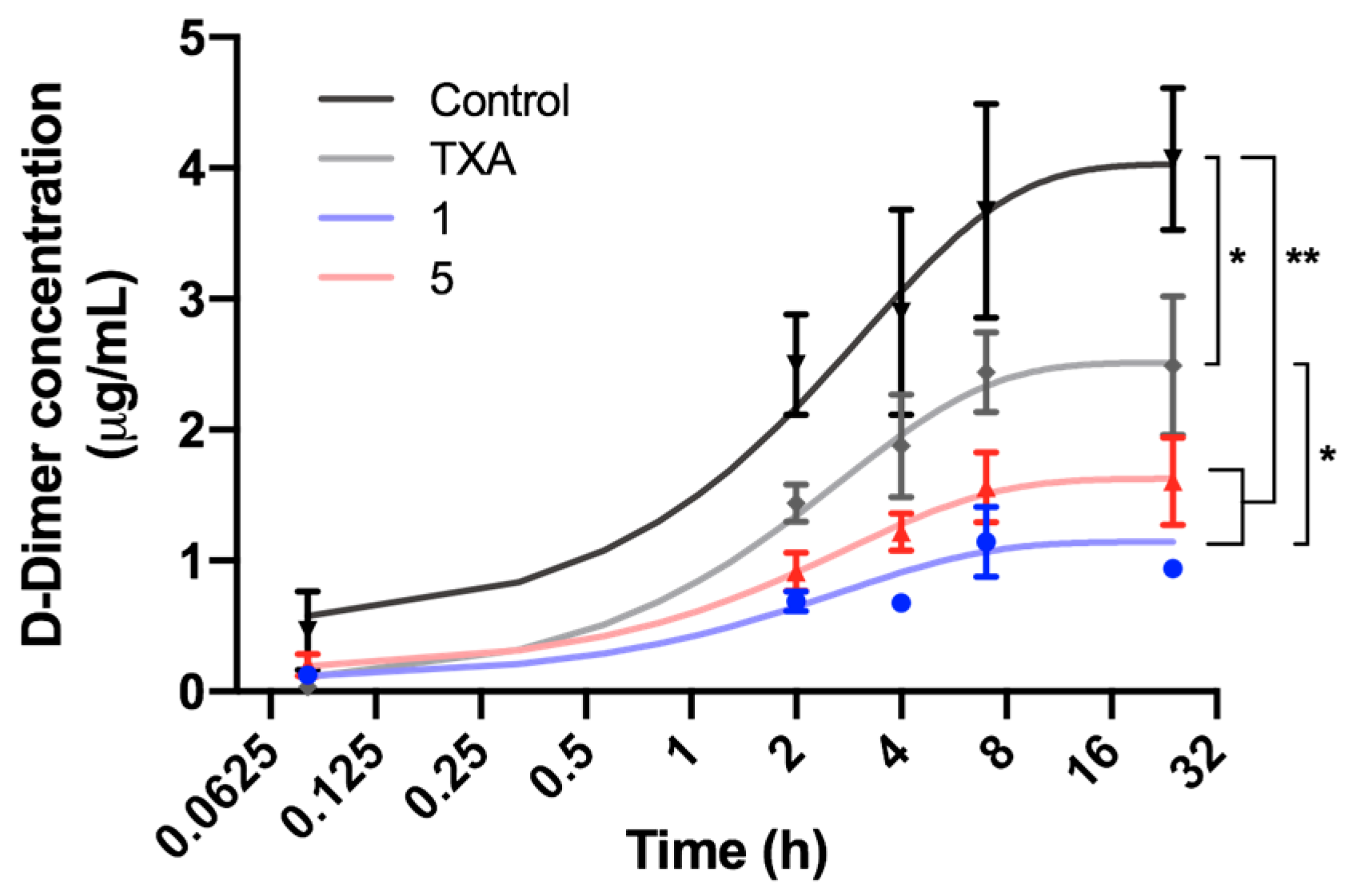
2.4. In Vivo Assays
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Isolated Enzyme Assays
4.2.1. Plasmin Activity Assay
4.2.2. tPA Activity Assay
4.2.3. Plasminogen Activation Assay
4.3. Ex Vivo Assays
4.3.1. Blood Extraction
4.3.2. Whole Blood Coagulation Assay
4.3.3. Whole Blood Dosage
4.3.4. Whole Blood Clot Lysis Assay
4.4. Molecular Dynamics Simulations
4.5. In Vivo Assays
4.5.1. Pharmacokinetic Study
4.5.2. Maximum Tolerated Dose Study
4.6. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lin, H.; Xu, L.; Yu, S.; Hong, W.; Huang, M.; Xu, P. Therapeutics targeting the fibrinolytic system. Exp. Mol. Med. 2020, 52, 367–379. [Google Scholar] [CrossRef] [PubMed]
- Medved, L.; Nieuwenhuizen, W. Molecular mechanisms of initiation of fibrinolysis by fibrin. Thromb. Haemost. 2003, 89, 409–419. [Google Scholar]
- Bhagavan, N.V.; Ha, C.-E. Biochemistry of Hemostasis. In Essentials of Medical Biochemistry, 2nd ed.; Academic Press: Cambridge, MA, USA; Elsevier Inc.: Amsterdam, The Netherlands, 2015; pp. 637–660. [Google Scholar]
- Norris, L.A. Blood coagulation. Best Pract. Res. Clin. Obstet. Gynaecol. 2003, 17, 369–383. [Google Scholar] [CrossRef] [PubMed]
- Medcalf, R.L.; Keragala, C.B.; Draxler, D.F. Fibrinolysis and the Immune Response in Trauma. Semin. Thromb. Hemost. 2020, 46, 176–182. [Google Scholar] [CrossRef] [PubMed]
- Larsen, J.B.; Hvas, A.M. Fibrin Clot Formation and Lysis in Plasma. Methods Protoc. 2020, 3, 67. [Google Scholar] [CrossRef] [PubMed]
- Risman, R.A.; Paynter, B.; Percoco, V.; Shroff, M.; Bannish, B.E.; Tutwiler, V. Internal fibrinolysis of fibrin clots is driven by pore expansion. Sci. Rep. 2024, 14, 2623. [Google Scholar] [CrossRef] [PubMed]
- Urano, T.; Castellino, F.J.; Suzuki, Y. Regulation of plasminogen activation on cell surfaces and fibrin. J. Thromb. Haemost. 2018, 16, 1487–1497. [Google Scholar] [CrossRef] [PubMed]
- Castellino, F.J.; Ploplis, V.A. Structure and function of the plasminogen/plasmin system. Thromb. Haemost. 2005, 93, 647–654. [Google Scholar] [PubMed]
- Gale, A.J. Current Understanding of Hemostasis. Toxicol. Pathol. 2011, 39, 273–280. [Google Scholar] [CrossRef]
- Bannish, B.E.; Chernysh, I.N.; Keener, J.P.; Fogelson, A.L.; Weisel, J.W. Molecular and Physical Mechanisms of Fibrinolysis and Thrombolysis from Mathematical Modeling and Experiments. Sci. Rep. 2017, 7, 6914. [Google Scholar] [CrossRef]
- Xue, Y.; Bodin, C.; Olsson, K. Crystal structure of the native plasminogen reveals an activation-resistant compact conformation. J. Thromb. Haemost. 2012, 10, 1385–1396. [Google Scholar] [CrossRef] [PubMed]
- Law, R.H.P.; Caradoc-Davies, T.; Cowieson, N.; Horvath, A.J.; Quek, A.J.; Encarnacao, J.A.; Steer, D.; Cowan, A.; Zhang, Q.; Lu, B.G.C.; et al. The X-ray Crystal Structure of Full-Length Human Plasminogen. Cell Rep. 2012, 1, 185–190. [Google Scholar] [CrossRef] [PubMed]
- Saes, J.L.; Schols, S.E.M.; van Heerde, W.L.; Nijziel, M.R. Hemorrhagic disorders of fibrinolysis: A clinical review. J. Thromb. Haemost. 2018, 16, 1498–1509. [Google Scholar] [CrossRef] [PubMed]
- Boström, J.; Grant, J.A.; Fjellström, O.; Thelin, A.; Gustafsson, D. Potent fibrinolysis inhibitor discovered by shape and electrostatic complementarity to the drug tranexamic acid. J. Med. Chem. 2013, 56, 3273–3280. [Google Scholar] [CrossRef] [PubMed]
- Pabinger, I.; Fries, D.; Schöchl, H.; Streif, W.; Toller, W. Tranexamic acid for treatment and prophylaxis of bleeding and hyperfibrinolysis. Wien. Klin. Wochenschr. 2017, 129, 303–316. [Google Scholar] [CrossRef]
- Chapin, J.; Hajjar, K.A. Fibrinolysis and the control of blood coagulation. Blood Rev. 2015, 29, 17–24. [Google Scholar] [CrossRef]
- Solomon, C.; Collis, R.E.; Collins, P.W. Haemostatic monitoring during postpartum haemorrhage and implications for management. Br. J. Anaesth. 2012, 109, 851–863. [Google Scholar] [CrossRef]
- Rossaint, R.; Bouillon, B.; Cerny, V.; Coats, T.J.; Duranteau, J.; Fernández-Mondéjar, E.; Filipescu, D.; Hunt, B.J.; Komadina, R.; Nardi, G.; et al. The European guideline on management of major bleeding and coagulopathy following trauma: Fourth edition. Crit. Care 2016, 20, 100. [Google Scholar] [PubMed]
- Prudovsky, I.; Kacer, D.; Zucco, V.V.; Palmeri, M.; Falank, C.; Kramer, R.; Carter, D.; Rappold, J. Tranexamic acid: Beyond antifibrinolysis. Transfusion 2022, 62 (Suppl. S1), S301–S312. [Google Scholar] [CrossRef]
- Lam, T.; Medcalf, R.L.; Cloud, G.C.; Myles, P.S.; Keragala, C.B. Tranexamic acid for haemostasis and beyond: Does dose matter? Thromb. J. 2023, 21, 94. [Google Scholar] [CrossRef]
- Gall, L.S.; Brohi, K.; Davenport, R.A. Diagnosis and Treatment of Hyperfibrinolysis in Trauma (A European Perspective). Semin. Thromb. Hemost. 2017, 43, 224–234. [Google Scholar] [CrossRef] [PubMed]
- Mahdy, A.M.; Webster, N.R. Perioperative systemic haemostatic agents. Br. J. Anaesth. 2004, 93, 842–858. [Google Scholar] [CrossRef] [PubMed]
- Roberts, I. Tranexamic acid in trauma: How should we use it? J. Thromb. Haemost. 2015, 13, S195–S199. [Google Scholar] [CrossRef] [PubMed]
- Mannucci, P.M. Hemostatic drugs. N. Engl. J. Med. 1998, 339, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Levy, J.H.; Koster, A.; Quinones, Q.J.; Milling, T.J.; Key, N.S. Antifibrinolytic therapy and perioperative considerations. Anesthesiology 2018, 128, 657–670. [Google Scholar] [CrossRef]
- McCormack, P.L. Tranexamic acid: A review of its use in the treatment of hyperfibrinolysis. Drugs 2012, 72, 585–617. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Mazzitelli, B.A.; Quek, A.J.; Veldman, M.J.; Conroy, P.J.; Caradoc-Davies, T.T.; Ooms, L.M.; Tuck, K.L.; Schoenecker, J.G.; Whisstock, J.C.; et al. Tranexamic acid is an active site inhibitor of urokinase plasminogen activator. Blood Adv. 2019, 3, 729–733. [Google Scholar] [CrossRef] [PubMed]
- Goobie, S.M. Tranexamic acid: Still far to go. Br. J. Anaesth. 2017, 118, 293–295. [Google Scholar] [CrossRef]
- Montroy, J.; Hutton, B.; Moodley, P.; Fergusson, N.A.; Cheng, W.; Tinmouth, A.; Lavallée, L.T.; Fergusson, D.A.; Breau, R.H. The efficacy and safety of topical tranexamic acid: A systematic review and meta-analysis. Transfus. Med. Rev. 2018, 32, 165–178. [Google Scholar] [CrossRef]
- Akosman, I.; Lovecchio, F.; Fourman, M.; Sarmiento, M.; Lyons, K.; Memtsoudis, S.; Kim, H.J. Is High-Dose Tranexamic Safe in Spine Surgery? A Systematic Review and Meta-Analysis. Glob. Spine J. 2023, 13, 2085–2095. [Google Scholar] [CrossRef]
- Sundström, A.; Seaman, H.; Kieler, H.; Alfredsson, L. The risk of venous thromboembolism associated with the use of tranexamic acid and other drugs used to treat menorrhagia: A case-control study using the General Practice Research Database. BJOG Int. J. Obstet. Gynaecol. 2009, 116, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Myers, S.P.; Kutcher, M.E.; Rosengart, M.R.; Sperry, J.L.; Peitzman, A.B.; Brown, J.B.; Neal, M.D. Tranexamic acid administration is associated with an increased risk of posttraumatic venous thromboembolism. J. Trauma Acute Care Surg. 2019, 86, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Perel, P.; Ker, K.; Morales Uribe, C.H.; Roberts, I. Tranexamic acid for reducing mortality in emergency and urgent surgery. Cochrane Database Syst. Rev. 2013, 2013, CD010245. [Google Scholar] [CrossRef] [PubMed]
- Ng, W.; Jerath, A.; Wasowicz, M. Tranexamic acid: A clinical review. Anaesthesiol. Intensiv. Ther. 2015, 47, 339–350. [Google Scholar] [CrossRef]
- Lecker, I.; Wang, D.S.; Whissell, P.D.; Avramescu, S.; Mazer, C.D.; Orser, B.A. Tranexamic acid-associated seizures: Causes and treatment. Ann. Neurol. 2016, 79, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Lecker, I.; Wang, D.S.; Romaschin, A.D.; Peterson, M.; Mazer, C.D.; Orser, B.A. Tranexamic acid concentrations associated with human seizures inhibit glycine receptors. J. Clin. Investig. 2012, 122, 4654–4666. [Google Scholar] [CrossRef]
- Lin, Z.; Xiaoyi, Z. Tranexamic acid-associated seizures: A meta-analysis. Seizure 2016, 36, 70–73. [Google Scholar] [CrossRef]
- Sharma, V.; Katznelson, R.; Jerath, A.; Garrido-Olivares, L.; Carroll, J.; Rao, V.; Wasowicz, M.; Djaiani, G. The association between tranexamic acid and convulsive seizures after cardiac surgery: A multivariate analysis in 11 529 patients. Anaesthesia 2014, 69, 124–130. [Google Scholar] [CrossRef]
- Koster, A.; Börgermann, J.; Zittermann, A.; Lueth, J.U.; Gillis-Januszewski, T.; Schirmer, U. Moderate dosage of tranexamic acid during cardiac surgery with cardiopulmonary bypass and convulsive seizures: Incidence and clinical outcome. Br. J. Anaesth. 2013, 110, 34–40. [Google Scholar] [CrossRef]
- Keyl, C.; Uhl, R.; Beyersdorf, F.; Stampf, S.; Lehane, C.; Wiesenack, C.; Trenk, D. High-dose tranexamic acid is related to increased risk of generalized seizures after aortic valve replacement. Eur. J. Cardio-thoracic Surg. 2011, 39, 114–121. [Google Scholar] [CrossRef]
- Jerath, A.; Yang, A.J.; Pang, K.S.; Looby, N.; Reyes-Garces, N.; Vasiljevic, T.; Bojko, B.; Pawliszyn, J.; Wijeysundera, D.; Beattie, W.S. Tranexamic acid dosing for cardiac surgical patients with chronic renal dysfunction: A new dosing regimen. Anesth. Analg. 2018, 127, 1323–1332. [Google Scholar] [CrossRef] [PubMed]
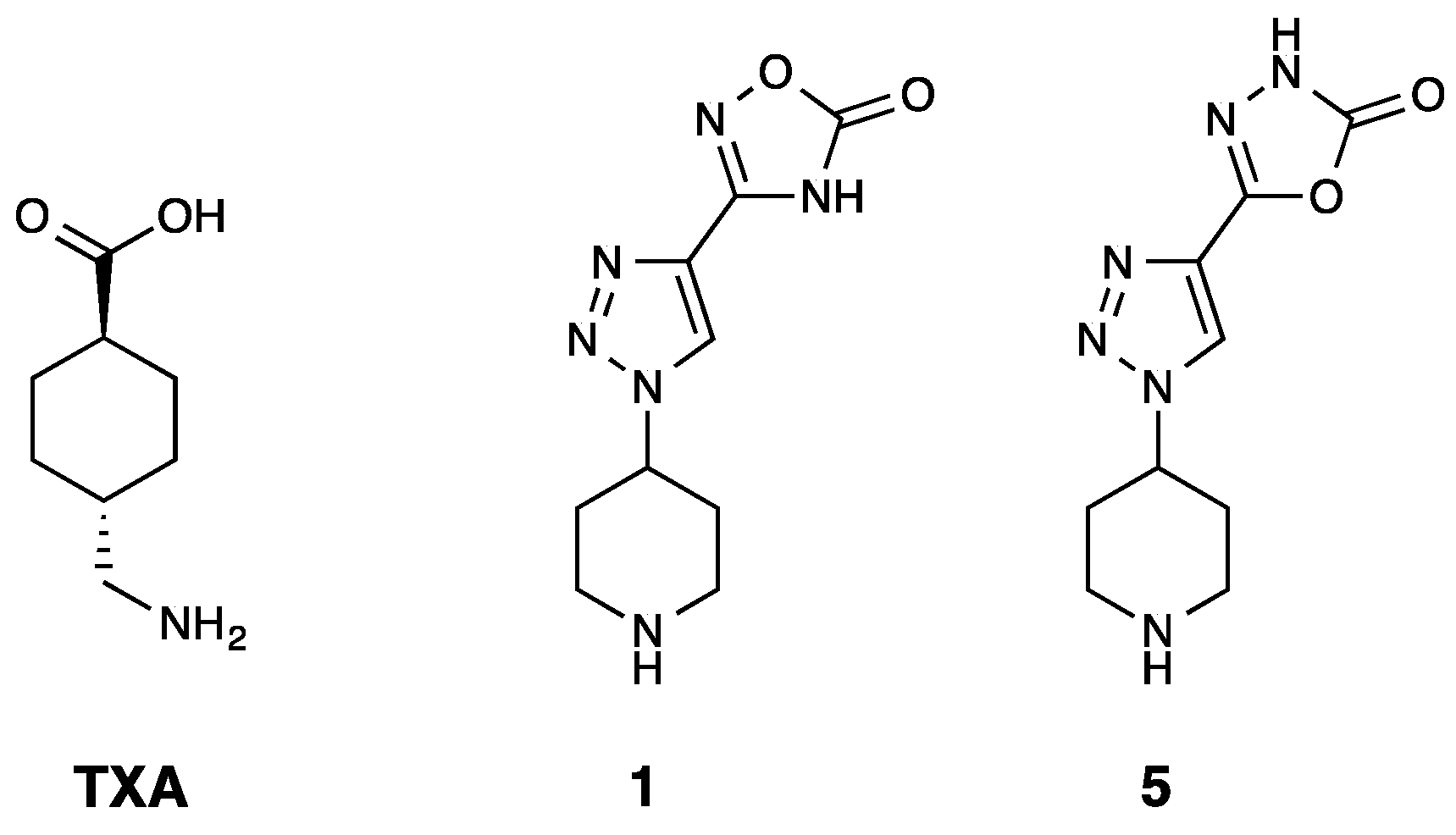
- Bosch-Sanz, O.; Rabadà, Y.; Biarnés, X.; Pedreño, J.; Caveda, L.; Balcells, M.; Martorell, J.; Sánchez-García, D. 1,2,3-Triazole Derivatives as Novel Antifibrinolytic Drugs. Int. J. Mol. Sci. 2022, 23, 14942. [Google Scholar] [CrossRef]
- Silva, M.M.C.G.; Thelwell, C.; Williams, S.C.; Longstaff, C. Regulation of fibrinolysis by C-terminal lysines operates through plasminogen and plasmin but not tissue-type plasminogen activator. J. Thromb. Haemost. 2012, 10, 2354–2360. [Google Scholar] [CrossRef] [PubMed]
- Takayasu, R.; Hasumi, K.; Shinohara, C.; Endo, A. Enhancement of fibrin binding and activation of plasminogen by staplabin through induction of a conformational change in plasminogen. FEBS Lett. 1997, 418, 58–62. [Google Scholar] [CrossRef] [PubMed]
- McEvoy, M.D.; Reeves, S.T.; Reves, J.G.; Spinale, F.G. Aprotinin in cardiac surgery: A review of conventional and novel mechanisms of action. Anesth. Analg. 2007, 105, 949–962. [Google Scholar] [CrossRef] [PubMed]
- Fritz, H.; Wunderer, G. Biochemistry and applications of aprotinin, the kallikrein inhibitor from bovine organs. Arzneimittelforschung 1983, 33, 479–494. [Google Scholar] [PubMed]
- Sperzel, M.; Huetter, J. Evaluation of aprotinin and tranexamic acid in different in vitro and in vivo models of fibrinolysis, coagulation and thrombus formation. J. Thromb. Haemost. 2007, 5, 2113–2118. [Google Scholar] [CrossRef]
- Ilich, A.; Bokarev, I.; Key, N.S. Global assays of fibrinolysis. Int. J. Lab. Hematol. 2017, 39, 441–447. [Google Scholar] [CrossRef] [PubMed]
- Bonnard, T.; Law, L.S.; Tennant, Z.; Hagemeyer, C.E. Development and validation of a high throughput whole blood thrombolysis plate assay. Sci. Rep. 2017, 7, 2346. [Google Scholar] [CrossRef]
- Richter, M.; Markwardt, F.; Klöcking, H.P. Pharmacokinetic studies with 3H-labelled synthetic antifibrinolytics. Pharmazie 1982, 37, 851–853. [Google Scholar]
- Sato, J.; Doi, T.; Wako, Y.; Hamamura, M.; Kanno, T.; Tsuchitani, M.; Narama, I. Histopathology of Incidental Findings in Beagles Used in Toxicity Studies. J. Toxicol. Pathol. 2012, 25, 103–134. [Google Scholar] [CrossRef] [PubMed]
- Elnager, A.; Abdullah, W.Z.; Hassan, R.; Idris, Z.; Wan Arfah, N.; Sulaiman, S.A.; Mustafa, Z. In Vitro whole blood clot lysis for fibrinolytic activity study using D-dimer and confocal microscopy. Adv. Hematol. 2014, 2014, 814684. [Google Scholar] [PubMed]
- Wang, X.; Friis, T.E.; Masci, P.P.; Crawford, R.W.; Liao, W.; Xiao, Y. Alteration of blood clot structures by interleukin-1 beta in association with bone defects healing. Sci. Rep. 2016, 6, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Malde, A.K.; Zuo, L.; Breeze, M.; Stroet, M.; Poger, D.; Nair, P.C.; Oostenbrink, C.; Mark, A.E. An Automated Force Field Topology Builder (ATB) and Repository: Version 1.0. J. Chem. Theory Comput. 2011, 7, 4026–4037. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | IC50 (mM) | p Value |
|---|---|---|
| TXA | 30.87 ± 5.50 | |
| 1 | 20.64 ± 0.78 | 0.0332 |
| 5 | 17.92 ± 1.24 | 0.0164 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rabadà, Y.; Bosch-Sanz, O.; Biarnés, X.; Pedreño, J.; Caveda, L.; Sánchez-García, D.; Martorell, J.; Balcells, M. Unravelling the Antifibrinolytic Mechanism of Action of the 1,2,3-Triazole Derivatives. Int. J. Mol. Sci. 2024, 25, 7002. https://doi.org/10.3390/ijms25137002
Rabadà Y, Bosch-Sanz O, Biarnés X, Pedreño J, Caveda L, Sánchez-García D, Martorell J, Balcells M. Unravelling the Antifibrinolytic Mechanism of Action of the 1,2,3-Triazole Derivatives. International Journal of Molecular Sciences. 2024; 25(13):7002. https://doi.org/10.3390/ijms25137002
Chicago/Turabian StyleRabadà, Yvette, Oriol Bosch-Sanz, Xevi Biarnés, Javier Pedreño, Luis Caveda, David Sánchez-García, Jordi Martorell, and Mercedes Balcells. 2024. "Unravelling the Antifibrinolytic Mechanism of Action of the 1,2,3-Triazole Derivatives" International Journal of Molecular Sciences 25, no. 13: 7002. https://doi.org/10.3390/ijms25137002
APA StyleRabadà, Y., Bosch-Sanz, O., Biarnés, X., Pedreño, J., Caveda, L., Sánchez-García, D., Martorell, J., & Balcells, M. (2024). Unravelling the Antifibrinolytic Mechanism of Action of the 1,2,3-Triazole Derivatives. International Journal of Molecular Sciences, 25(13), 7002. https://doi.org/10.3390/ijms25137002