
Mutational Landscape of Alzheimer’s Disease and Frontotemporal Dementia: Regional Variances in Northern, Central, and Southern Italy
 ,
,  , , , , , , ,
, , , , , , ,  , and
, and
Abstract
:1. Introduction
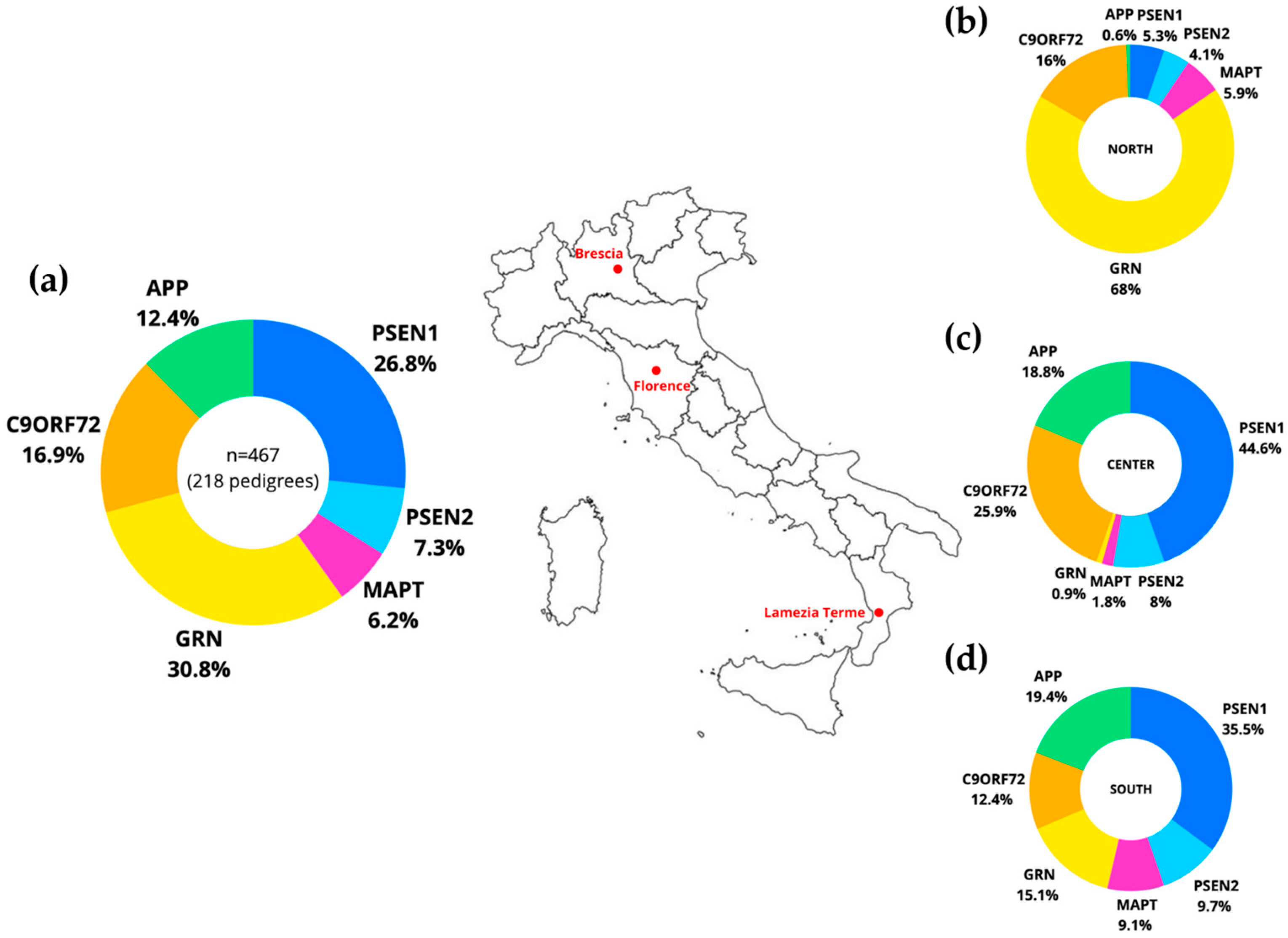
2. Results and Discussion
3. Materials and Methods
3.1. Study Design and Participants
3.2. Statistical Analysis
3.3. Ethics Committee
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rascovsky, K.; Salmon, D.P.; Lipton, A.M.; Leverenz, J.B.; DeCarli, C.; Jagust, W.J.; Clark, C.M.; Mendez, M.F.; Tang-Wai, D.F.; Graff-Radford, N.R.; et al. Rate of Progression Differs in Frontotemporal Dementia and Alzheimer Disease. Neurology 2005, 65, 397–403. [Google Scholar] [CrossRef] [PubMed]
- Bird, T.D. Alzheimer Disease Overview. In GeneReviews(®); Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; GeneReviews is a Registered Trademark of the University of Washington, Seattle; All Rights Reserved; University of Washington: SeattleSeattle, WA, USA, 1993. [Google Scholar]
- 2021 Alzheimer’s Disease Facts and Figures. Alzheimer’s Dement. 2021, 17, 327–406. [CrossRef] [PubMed]
- 2024 Alzheimer’s Disease Facts and Figures. Alzheimer’s Dement. 2024, 20, 3708–3821. [CrossRef] [PubMed]
- McKhann, G.; Drachman, D.; Folstein, M.; Katzman, R.; Price, D.; Stadlan, E.M. Clinical Diagnosis of Alzheimer’s Disease: Report of the NINCDS-ADRDA Work Group Under the Auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology 1984, 34, 939–944. [Google Scholar] [CrossRef] [PubMed]
- Lynch, C. World Alzheimer Report 2019: Attitudes to Dementia, a Global Survey. Alzheimer’s Dement. 2020, 16, e038255. [Google Scholar] [CrossRef]
- Villemagne, V.L.; Burnham, S.; Bourgeat, P.; Brown, B.; Ellis, K.A.; Salvado, O.; Szoeke, C.; Macaulay, S.L.; Martins, R.; Maruff, P.; et al. Amyloid Β Deposition, Neurodegeneration, and Cognitive Decline in Sporadic Alzheimer’s Disease: A Prospective Cohort Study. Lancet Neurol. 2013, 12, 357–367. [Google Scholar] [CrossRef] [PubMed]
- Bateman, R.J.; Xiong, C.; Benzinger, T.L.S.; Fagan, A.M.; Goate, A.; Fox, N.C.; Marcus, D.S.; Cairns, N.J.; Xie, X.; Blazey, T.M.; et al. Clinical and Biomarker Changes in Dominantly Inherited Alzheimer’s Disease. N. Engl. J. Med. 2012, 367, 795–804. [Google Scholar] [CrossRef] [PubMed]
- Barthélemy, N.R.; Li, Y.; Joseph-Mathurin, N.; Gordon, B.A.; Hassenstab, J.; Benzinger, T.L.S.; Buckles, V.; Fagan, A.M.; Perrin, R.J.; Goate, A.M.; et al. A Soluble Phosphorylated Tau Signature Links Tau, Amyloid and the Evolution of Stages of Dominantly Inherited Alzheimer’s Disease. Nat. Med. 2020, 26, 398–407. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Thal, D.R.; Ghebremedhin, E.; Del Tredici, K. Stages of the Pathologic Process in Alzheimer Disease: Age Categories from 1 to 100 Years. J. Neuropathol. Exp. Neurol. 2011, 70, 960–969. [Google Scholar] [CrossRef]
- Gouras, G.K.; Tsai, J.; Naslund, J.; Vincent, B.; Edgar, M.; Checler, F.; Greenfield, J.P.; Haroutunian, V.; Buxbaum, J.D.; Xu, H.; et al. Intraneuronal Abeta42 Accumulation in Human Brain. Am. J. Pathol. 2000, 156, 15–20. [Google Scholar] [CrossRef]
- Lee, V.M.; Goedert, M.; Trojanowski, J.Q. Neurodegenerative Tauopathies. Annu. Rev. Neurosci. 2001, 24, 1121–1159. [Google Scholar] [CrossRef] [PubMed]
- Brion, J.P. Neurofibrillary Tangles and Alzheimer’s Disease. Eur. Neurol. 1998, 40, 130–140. [Google Scholar] [CrossRef] [PubMed]
- Querfurth, H.W.; LaFerla, F.M. Alzheimer’s Disease. N. Engl. J. Med. 2010, 362, 329–344. [Google Scholar] [CrossRef] [PubMed]
- Harman, D. Alzheimer’s Disease Pathogenesis: Role of Aging. Ann. N. Y. Acad. Sci. 2006, 1067, 454–460. [Google Scholar] [CrossRef] [PubMed]
- Prasher, V.P.; Farrer, M.J.; Kessling, A.M.; Fisher, E.M.; West, R.J.; Barber, P.C.; Butler, A.C. Molecular Mapping of Alzheimer-Type Dementia in Down’s Syndrome. Ann. Neurol. 1998, 43, 380–383. [Google Scholar] [CrossRef] [PubMed]
- Tanzi, R.E.; Gusella, J.F.; Watkins, P.C.; Bruns, G.A.; St George-Hyslop, P.; Van Keuren, M.L.; Patterson, D.; Pagan, S.; Kurnit, D.M.; Neve, R.L. Amyloid Beta Protein Gene: cDNA, mRNA Distribution, and Genetic Linkage Near the Alzheimer Locus. Science 1987, 235, 880–884. [Google Scholar] [CrossRef] [PubMed]
- Goate, A.; Chartier-Harlin, M.C.; Mullan, M.; Brown, J.; Crawford, F.; Fidani, L.; Giuffra, L.; Haynes, A.; Irving, N.; James, L. Segregation of a Missense Mutation in the Amyloid Precursor Protein Gene with Familial Alzheimer’s Disease. Nature 1991, 349, 704–706. [Google Scholar] [CrossRef]
- Pilotto, A.; Padovani, A.; Borroni, B. Clinical, Biological, and Imaging Features of Monogenic Alzheimer’s Disease. Biomed. Res. Int. 2013, 2013, 689591. [Google Scholar] [CrossRef] [PubMed]
- Sherrington, R.; Rogaev, E.I.; Liang, Y.; Rogaeva, E.A.; Levesque, G.; Ikeda, M.; Chi, H.; Lin, C.; Li, G.; Holman, K.; et al. Cloning of a Gene Bearing Missense Mutations in Early-Onset Familial Alzheimer’s Disease. Nature 1995, 375, 754–760. [Google Scholar] [CrossRef]
- Van Broeckhoven, C.; Backhovens, H.; Cruts, M.; De Winter, G.; Bruyland, M.; Cras, P.; Martin, J.J. Mapping of a Gene Predisposing to Early-Onset Alzheimer’s Disease to Chromosome 14q24.3. Nat. Genet. 1992, 2, 335–339. [Google Scholar] [CrossRef]
- Levy-Lahad, E.; Wasco, W.; Poorkaj, P.; Romano, D.M.; Oshima, J.; Pettingell, W.H.; Yu, C.E.; Jondro, P.D.; Schmidt, S.D.; Wang, K. Candidate Gene for the Chromosome 1 Familial Alzheimer’s Disease Locus. Science 1995, 269, 973–977. [Google Scholar] [CrossRef] [PubMed]
- Jayadev, S.; Leverenz, J.B.; Steinbart, E.; Stahl, J.; Klunk, W.; Yu, C.; Bird, T.D. Alzheimer’s Disease Phenotypes and Genotypes Associated with Mutations in Presenilin 2. Brain 2010, 133, 1143–1154. [Google Scholar] [CrossRef] [PubMed]
- Schellenberg, G.D.; Anderson, L.; O’dahl, S.; Wisjman, E.M.; Sadovnick, A.D.; Ball, M.J.; Larson, E.B.; Kukull, W.A.; Martin, G.M.; Roses, A.D. APP717, APP693, and PRIP Gene Mutations are Rare in Alzheimer Disease. Am. J. Hum. Genet. 1991, 49, 511–517. [Google Scholar] [PubMed]
- Tanzi, R.E.; Vaula, G.; Romano, D.M.; Mortilla, M.; Huang, T.L.; Tupler, R.G.; Wasco, W.; Hyman, B.T.; Haines, J.L.; Jenkins, B.J. Assessment of Amyloid Beta-Protein Precursor Gene Mutations in a Large Set of Familial and Sporadic Alzheimer Disease Cases. Am. J. Hum. Genet. 1992, 51, 273–282. [Google Scholar] [PubMed]
- Bertram, L.; Tanzi, R.E. The Genetic Epidemiology of Neurodegenerative Disease. J. Clin. Investig. 2005, 115, 1449–1457. [Google Scholar] [CrossRef] [PubMed]
- Rademakers, R.; Cruts, M.; Van Broeckhoven, C. Genetics of Early-Onset Alzheimer Dementia. Sci. World J. 2003, 3, 497–519. [Google Scholar] [CrossRef] [PubMed]
- Haass, C.; Kaether, C.; Thinakaran, G.; Sisodia, S. Trafficking and Proteolytic Processing of APP. Cold Spring Harb Perspect. Med. 2012, 2, a006270. [Google Scholar] [CrossRef] [PubMed]
- Cole, S.L.; Vassar, R. The Role of Amyloid Precursor Protein Processing by BACE1, the Beta-Secretase, in Alzheimer Disease Pathophysiology. J. Biol. Chem. 2008, 283, 29621–29625. [Google Scholar] [CrossRef] [PubMed]
- Bang, J.; Spina, S.; Miller, B.L. Frontotemporal Dementia. Lancet 2015, 386, 1672–1682. [Google Scholar] [CrossRef] [PubMed]
- Ratnavalli, E.; Brayne, C.; Dawson, K.; Hodges, J.R. The Prevalence of Frontotemporal Dementia. Neurology 2002, 58, 1615–1621. [Google Scholar] [CrossRef]
- Neumann, M.; Mackenzie, I.R.A. Review: Neuropathology of Non-Tau Frontotemporal Lobar Degeneration. Neuropathol. Appl. Neurobiol. 2019, 45, 19–40. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, I.R.A.; Neumann, M. Molecular Neuropathology of Frontotemporal Dementia: Insights into Disease Mechanisms from Postmortem Studies. J. Neurochem. 2016, 138 (Suppl. S1), 54–70. [Google Scholar] [CrossRef] [PubMed]
- Gorno-Tempini, M.L.; Hillis, A.E.; Weintraub, S.; Kertesz, A.; Mendez, M.; Cappa, S.F.; Ogar, J.M.; Rohrer, J.D.; Black, S.; Boeve, B.F.; et al. Classification of Primary Progressive Aphasia and its Variants. Neurology 2011, 76, 1006–1014. [Google Scholar] [CrossRef] [PubMed]
- Rascovsky, K.; Hodges, J.R.; Knopman, D.; Mendez, M.F.; Kramer, J.H.; Neuhaus, J.; van Swieten, J.C.; Seelaar, H.; Dopper, E.G.P.; Onyike, C.U.; et al. Sensitivity of Revised Diagnostic Criteria for the Behavioural Variant of Frontotemporal Dementia. Brain 2011, 134, 2456–2477. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, R.; Manzoni, C.; Hardy, J. Genetics and Molecular Mechanisms of Frontotemporal Lobar Degeneration: An Update and Future Avenues. Neurobiol. Aging 2019, 78, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Rohrer, J.D.; Guerreiro, R.; Vandrovcova, J.; Uphill, J.; Reiman, D.; Beck, J.; Isaacs, A.M.; Authier, A.; Ferrari, R.; Fox, N.C.; et al. The Heritability and Genetics of Frontotemporal Lobar Degeneration. Neurology 2009, 73, 1451–1456. [Google Scholar] [CrossRef] [PubMed]
- Wood, E.M.; Falcone, D.; Suh, E.; Irwin, D.J.; Chen-Plotkin, A.S.; Lee, E.B.; Xie, S.X.; Van Deerlin, V.M.; Grossman, M. Development and Validation of Pedigree Classification Criteria for Frontotemporal Lobar Degeneration. JAMA Neurol. 2013, 70, 1411–1417. [Google Scholar] [CrossRef]
- Fostinelli, S.; Ciani, M.; Zanardini, R.; Zanetti, O.; Binetti, G.; Ghidoni, R.; Benussi, L. The Heritability of Frontotemporal Lobar Degeneration: Validation of Pedigree Classification Criteria in a Northern Italy Cohort. J. Alzheimer’s Dis. 2018, 61, 753–760. [Google Scholar] [CrossRef] [PubMed]
- Hutton, M.; Lendon, C.L.; Rizzu, P.; Baker, M.; Froelich, S.; Houlden, H.; Pickering-Brown, S.; Chakraverty, S.; Isaacs, A.; Grover, A.; et al. Association of Missense and 5′-Splice-Site Mutations in Tau with the Inherited Dementia FTDP-17. Nature 1998, 393, 702–705. [Google Scholar] [CrossRef]
- Poorkaj, P.; Bird, T.D.; Wijsman, E.; Nemens, E.; Garruto, R.M.; Anderson, L.; Andreadis, A.; Wiederholt, W.C.; Raskind, M.; Schellenberg, G.D. Tau is a Candidate Gene for Chromosome 17 Frontotemporal Dementia. Ann. Neurol. 1998, 43, 815–825. [Google Scholar] [CrossRef]
- Baker, M.; Mackenzie, I.R.; Pickering-Brown, S.M.; Gass, J.; Rademakers, R.; Lindholm, C.; Snowden, J.; Adamson, J.; Sadovnick, A.D.; Rollinson, S.; et al. Mutations in Progranulin Cause Tau-Negative Frontotemporal Dementia Linked to Chromosome 17. Nature 2006, 442, 916–919. [Google Scholar] [CrossRef] [PubMed]
- Cruts, M.; Gijselinck, I.; van der Zee, J.; Engelborghs, S.; Wils, H.; Pirici, D.; Rademakers, R.; Vandenberghe, R.; Dermaut, B.; Martin, J.; et al. Null Mutations in Progranulin Cause Ubiquitin-Positive Frontotemporal Dementia Linked to Chromosome 17q21. Nature 2006, 442, 920–924. [Google Scholar] [CrossRef] [PubMed]
- DeJesus-Hernandez, M.; Mackenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J.; Nicholson, A.M.; Finch, N.A.; Flynn, H.; Adamson, J.; et al. Expanded GGGGCC Hexanucleotide Repeat in Noncoding Region of C9ORF72 Causes Chromosome 9p-Linked FTD and ALS. Neuron 2011, 72, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Renton, A.E.; Majounie, E.; Waite, A.; Simón-Sánchez, J.; Rollinson, S.; Gibbs, J.R.; Schymick, J.C.; Laaksovirta, H.; van Swieten, J.C.; Myllykangas, L.; et al. A Hexanucleotide Repeat Expansion in C9ORF72 is the Cause of Chromosome 9p21-Linked ALS-FTD. Neuron 2011, 72, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Spillantini, M.G.; Goedert, M. Tau Pathology and Neurodegeneration. Lancet Neurol. 2013, 12, 609–622. [Google Scholar] [CrossRef] [PubMed]
- Benussi, L.; Ghidoni, R.; Binetti, G. Progranulin Mutations are a Common Cause of FTLD in Northern Italy. Alzheimer Dis. Assoc. Disord. 2010, 24, 308–309. [Google Scholar] [CrossRef] [PubMed]
- Benussi, L.; Ghidoni, R.; Pegoiani, E.; Moretti, D.V.; Zanetti, O.; Binetti, G. Progranulin Leu271LeufsX10 is One of the most Common FTLD and CBS Associated Mutations Worldwide. Neurobiol. Dis. 2009, 33, 379–385. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Van Damme, P.; Cruchaga, C.; Gitcho, M.A.; Vidal, J.M.; Seijo-Martínez, M.; Wang, L.; Wu, J.Y.; Robberecht, W.; Goate, A. Pathogenic Cysteine Mutations Affect Progranulin Function and Production of Mature Granulins. J. Neurochem. 2010, 112, 1305–1315. [Google Scholar] [CrossRef]
- Pottier, C.; Ravenscroft, T.A.; Sanchez-Contreras, M.; Rademakers, R. Genetics of FTLD: Overview and what Else we can Expect from Genetic Studies. J. Neurochem. 2016, 138 (Suppl. S1), 32–53. [Google Scholar] [CrossRef]
- Seelaar, H.; Rohrer, J.D.; Pijnenburg, Y.A.L.; Fox, N.C.; van Swieten, J.C. Clinical, Genetic and Pathological Heterogeneity of Frontotemporal Dementia: A Review. J. Neurol. Neurosurg. Psychiatry 2011, 82, 476–486. [Google Scholar] [CrossRef]
- van der Zee, J.; Gijselinck, I.; Dillen, L.; Van Langenhove, T.; Theuns, J.; Engelborghs, S.; Philtjens, S.; Vandenbulcke, M.; Sleegers, K.; Sieben, A.; et al. A Pan-European Study of the C9orf72 Repeat Associated with FTLD: Geographic Prevalence, Genomic Instability, and Intermediate Repeats. Hum. Mutat. 2013, 34, 363–373. [Google Scholar] [CrossRef] [PubMed]
- Takada, L.T. The Genetics of Monogenic Frontotemporal Dementia. Dement. Neuropsychol. 2015, 9, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Giardina, E.; Mandich, P.; Ghidoni, R.; Ticozzi, N.; Rossi, G.; Fenoglio, C.; Tiziano, F.D.; Esposito, F.; Capellari, S.; Nacmias, B.; et al. Distribution of the C9orf72 Hexanucleotide Repeat Expansion in Healthy Subjects: A Multicenter Study Promoted by the Italian IRCCS Network of Neuroscience and Neurorehabilitation. Front. Neurol. 2024, 15, 1284459. [Google Scholar] [CrossRef] [PubMed]
- Benussi, L.; Rossi, G.; Glionna, M.; Tonoli, E.; Piccoli, E.; Fostinelli, S.; Paterlini, A.; Flocco, R.; Albani, D.; Pantieri, R.; et al. C9ORF72 Hexanucleotide Repeat Number in Frontotemporal Lobar Degeneration: A Genotype-Phenotype Correlation Study. J. Alzheimer’s Dis. 2014, 38, 799–808. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.M.; Nicholas, J.; Grossman, M.; McMillan, C.T.; Irwin, D.J.; Massimo, L.; Van Deerlin, V.M.; Warren, J.D.; Fox, N.C.; Rossor, M.N.; et al. Age at Symptom Onset and Death and Disease Duration in Genetic Frontotemporal Dementia: An International Retrospective Cohort Study. Lancet Neurol. 2020, 19, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Sherrington, R.; Froelich, S.; Sorbi, S.; Campion, D.; Chi, H.; Rogaeva, E.A.; Levesque, G.; Rogaev, E.I.; Lin, C.; Liang, Y.; et al. Alzheimer’s Disease Associated with Mutations in Presenilin 2 is Rare and Variably Penetrant. Hum. Mol. Genet. 1996, 5, 985–988. [Google Scholar] [CrossRef] [PubMed]
- Binetti, G.; Signorini, S.; Squitti, R.; Alberici, A.; Benussi, L.; Cassetta, E.; Frisoni, G.B.; Barbiero, L.; Feudatari, E.; Nicosia, F.; et al. Atypical Dementia Associated with a Novel Presenilin-2 Mutation. Ann. Neurol. 2003, 54, 832–836. [Google Scholar] [CrossRef] [PubMed]
- Grossman, M. The Non-Fluent/Agrammatic Variant of Primary Progressive Aphasia. Lancet Neurol. 2012, 11, 545–555. [Google Scholar] [CrossRef]
- Barbier, M.; Camuzat, A.; Hachimi, K.E.; Guegan, J.; Rinaldi, D.; Lattante, S.; Houot, M.; Sánchez-Valle, R.; Sabatelli, M.; Antonell, A.; et al. SLITRK2, an X-Linked Modifier of the Age at Onset in C9orf72 Frontotemporal Lobar Degeneration. Brain 2021, 144, 2798–2811. [Google Scholar] [CrossRef]
- Zhang, M.; Xi, Z.; Ghani, M.; Jia, P.; Pal, M.; Werynska, K.; Moreno, D.; Sato, C.; Liang, Y.; Robertson, J.; et al. Genetic and Epigenetic Study of ALS-Discordant Identical Twins with Double Mutations in SOD1 and ARHGEF28. J. Neurol. Neurosurg. Psychiatry. 2016, 87, 1268–1270. [Google Scholar] [CrossRef]
- Chouliaras, L.; Rutten, B.P.F.; Kenis, G.; Peerbooms, O.; Visser, P.J.; Verhey, F.; van Os, J.; Steinbusch, H.W.M.; van den Hove, D.L.A. Epigenetic Regulation in the Pathophysiology of Alzheimer’s Disease. Prog. Neurobiol. 2010, 90, 498–510. [Google Scholar] [CrossRef] [PubMed]
- Bradley-Whitman, M.A.; Lovell, M.A. Epigenetic Changes in the Progression of Alzheimer’s Disease. Mech. Ageing Dev. 2013, 134, 486–495. [Google Scholar] [CrossRef] [PubMed]
- Benussi, L.; Binetti, G.; Sina, E.; Gigola, L.; Bettecken, T.; Meitinger, T.; Ghidoni, R. A Novel Deletion in Progranulin Gene is Associated with FTDP-17 and CBS. Neurobiol. Aging 2008, 29, 427–435. [Google Scholar] [CrossRef] [PubMed]
- Tremolizzo, L.; Gelosa, G.; Galbussera, A.; Isella, V.; Arosio, C.; Bertola, F.; Casati, G.; Piperno, A.; Ferrarese, C.; Appollonio, I. Higher than Expected Progranulin Mutation Rate in a Case Series of Italian FTLD Patients. Alzheimer Dis. Assoc. Disord. 2009, 23, 301. [Google Scholar] [CrossRef] [PubMed]
- Benussi, L.; Rademakers, R.; Rutherford, N.J.; Wojtas, A.; Glionna, M.; Paterlini, A.; Albertini, V.; Bettecken, T.; Binetti, G.; Ghidoni, R. Estimating the Age of the most Common Italian GRN Mutation: Walking Back to Canossa Times. J. Alzheimer’s Dis. 2013, 33, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Bruni, A.C.; Bernardi, L.; Colao, R.; Rubino, E.; Smirne, N.; Frangipane, F.; Terni, B.; Curcio, S.A.M.; Mirabelli, M.; Clodomiro, A.; et al. Worldwide Distribution of PSEN1 Met146Leu Mutation: A Large Variability for a Founder Mutation. Neurology 2010, 74, 798–806. [Google Scholar] [CrossRef] [PubMed]
- Bruno, F.; Laganà, V.; Di Lorenzo, R.; Bruni, A.C.; Maletta, R. Calabria as a Genetic Isolate: A Model for the Study of Neurodegenerative Diseases. Biomedicines 2022, 10, 2288. [Google Scholar] [CrossRef] [PubMed]
- Gallo, M.; Tomaino, C.; Puccio, G.; Frangipane, F.; Curcio, S.A.M.; Bernardi, L.; Geracitano, S.; Anfossi, M.; Mirabelli, M.; Colao, R.; et al. Novel MAPT Val75Ala Mutation and PSEN2 Arg62Hys in Two Siblings with Frontotemporal Dementia. Neurol. Sci. 2010, 31, 65–70. [Google Scholar] [CrossRef] [PubMed]
- McKhann, G.M.; Knopman, D.S.; Chertkow, H.; Hyman, B.T.; Jack, C.R.J.; Kawas, C.H.; Klunk, W.E.; Koroshetz, W.J.; Manly, J.J.; Mayeux, R.; et al. The Diagnosis of Dementia due to Alzheimer’s Disease: Recommendations from the National Institute on Aging-Alzheimer’s Association Workgroups on Diagnostic Guidelines for Alzheimer’s Disease. Alzheimer’s Dement. 2011, 7, 263–269. [Google Scholar] [CrossRef]
- Neary, D.; Snowden, J.S.; Gustafson, L.; Passant, U.; Stuss, D.; Black, S.; Freedman, M.; Kertesz, A.; Robert, P.H.; Albert, M.; et al. Frontotemporal Lobar Degeneration: A Consensus on Clinical Diagnostic Criteria. Neurology 1998, 51, 1546–1554. [Google Scholar] [CrossRef]
- Ghidoni, R. Rawdata AD_FTD Italian Cohorts; [Dataset]; Zenodo: Geneve, Switzerland, 2024. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| APP (n = 58) | PSEN1 (n = 125) | PSEN2 (n = 34) | MAPT (n = 29) | GRN (n = 144) | C9orf72 (n = 79) | p-Value | |
|---|---|---|---|---|---|---|---|
| Number of families | 21 | 32 | 19 | 16 | 79 | 57 | |
| Sex (% female) | 34.5 | 52.8 | 50.0 | 51.7 | 47.2 | 53.2 | 0.2676 a |
| Age at onset | 59.9 ± 10.7 | 44.9 ± 9.7 | 59.3 ± 15.2 | 48.0 ± 11.0 | 61.4 ± 8.9 | 57.4 ± 8.7 | <0.0001 b |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saraceno, C.; Pagano, L.; Laganà, V.; Geviti, A.; Bagnoli, S.; Ingannato, A.; Mazzeo, S.; Longobardi, A.; Fostinelli, S.; Bellini, S.; et al. Mutational Landscape of Alzheimer’s Disease and Frontotemporal Dementia: Regional Variances in Northern, Central, and Southern Italy. Int. J. Mol. Sci. 2024, 25, 7035. https://doi.org/10.3390/ijms25137035
Saraceno C, Pagano L, Laganà V, Geviti A, Bagnoli S, Ingannato A, Mazzeo S, Longobardi A, Fostinelli S, Bellini S, et al. Mutational Landscape of Alzheimer’s Disease and Frontotemporal Dementia: Regional Variances in Northern, Central, and Southern Italy. International Journal of Molecular Sciences. 2024; 25(13):7035. https://doi.org/10.3390/ijms25137035
Chicago/Turabian StyleSaraceno, Claudia, Lorenzo Pagano, Valentina Laganà, Andrea Geviti, Silvia Bagnoli, Assunta Ingannato, Salvatore Mazzeo, Antonio Longobardi, Silvia Fostinelli, Sonia Bellini, and et al. 2024. "Mutational Landscape of Alzheimer’s Disease and Frontotemporal Dementia: Regional Variances in Northern, Central, and Southern Italy" International Journal of Molecular Sciences 25, no. 13: 7035. https://doi.org/10.3390/ijms25137035