
The Renoprotective Mechanisms of Sodium-Glucose Cotransporter-2 Inhibitors (SGLT2i)—A Narrative Review
 , ,
, ,
Abstract
:1. Introduction
1.1. Epidemiology of Type 2 Diabetes Mellitus
1.2. Epidemiology of Chronic Kidney Disease
1.3. CKD in Patients with DM
1.4. SGLT2i—An Emerging Treatment in Patients with DM
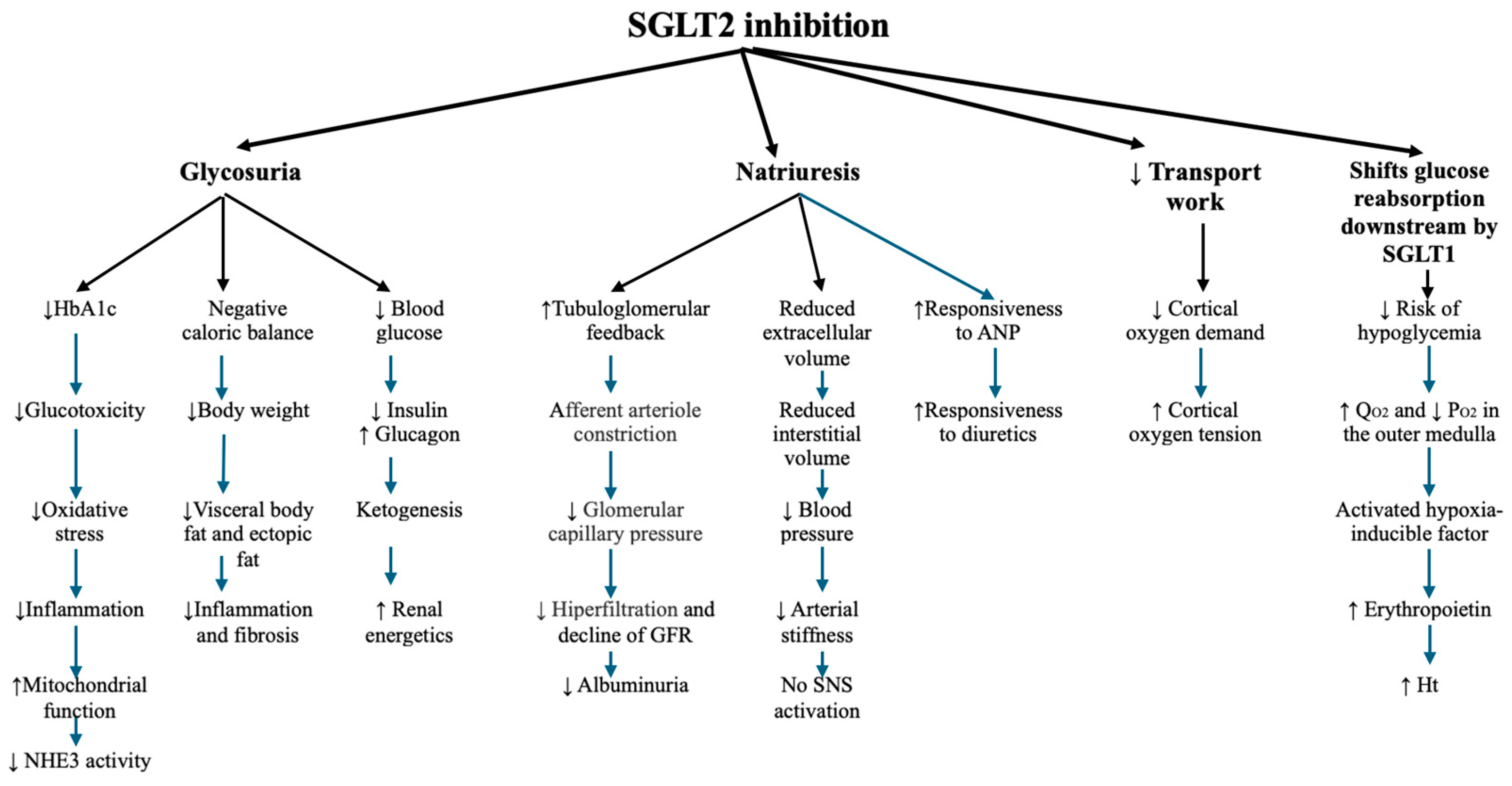
2. The Renal Effects of DM and the Protective Mechanisms of SGLT2i
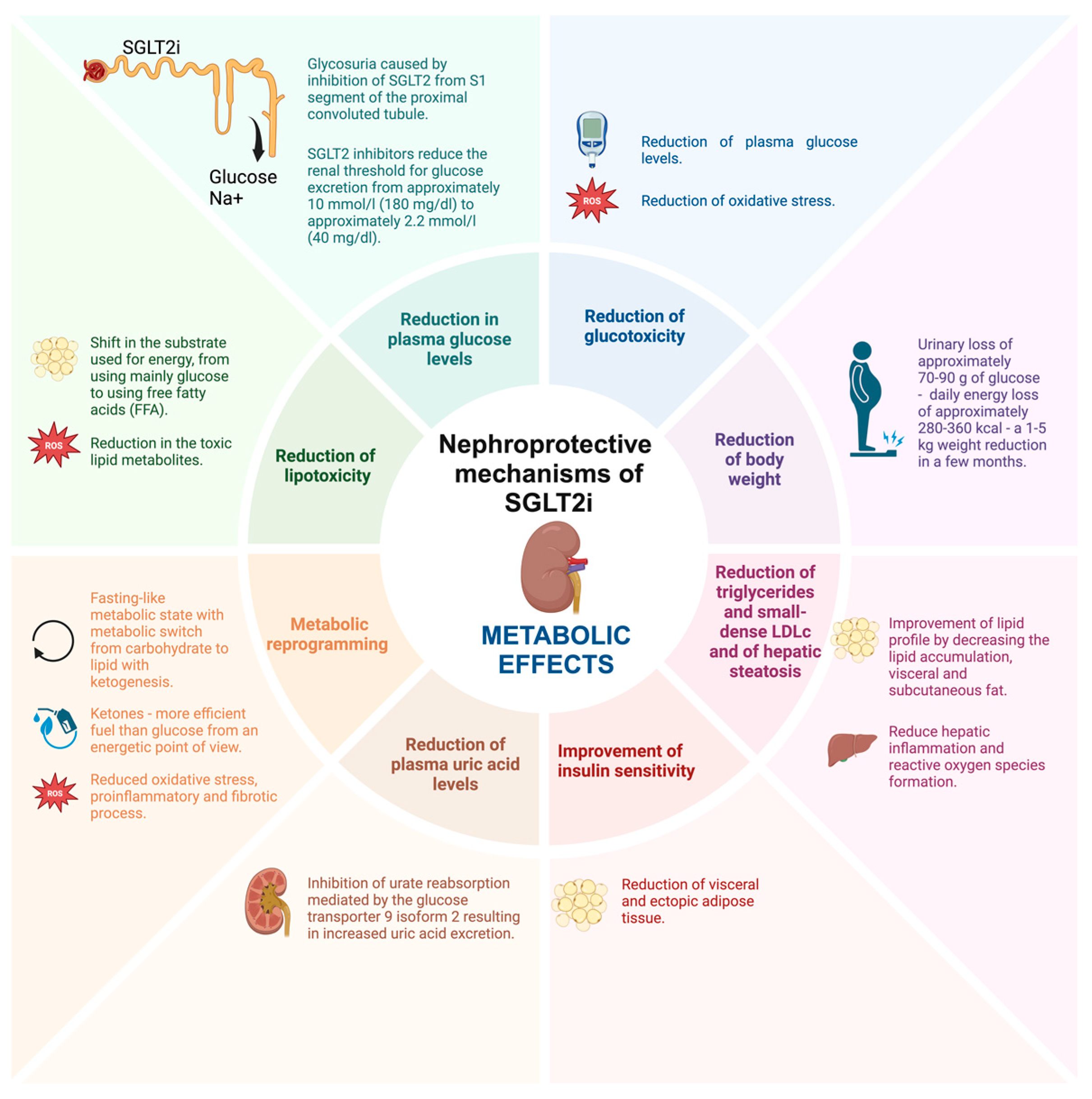
2.1. Metabolic Effects of SGLT2i
2.1.1. Reduction in Plasma Glucose Levels and Glucotoxicity
2.1.2. Reduction of Body Weight
2.1.3. Reduction of Lipotoxicity, Triglycerides, Small-Dense LDLc, and Insulin Sensitivity
2.1.4. Reduction of Hepatic Steatosis
2.1.5. Reduction of Plasma Uric Acid Levels
2.1.6. Metabolic Reprogramming—Restoration of Energy Efficiency by Switching Glucose to More Energy-Efficient Metabolites
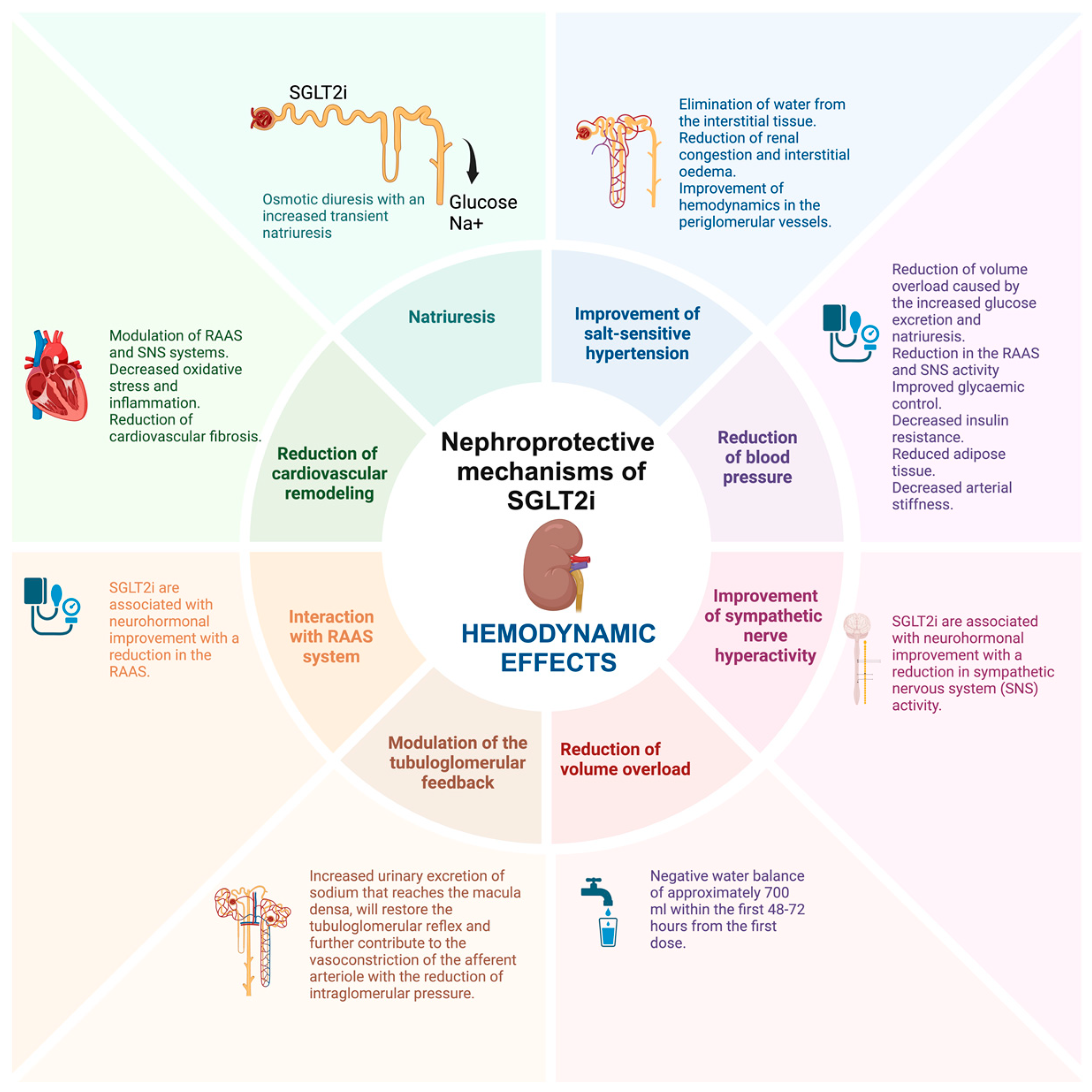
2.2. Hemodynamic Effects of SGLT2i
2.2.1. Natriuresis and Improvement of Salt-Sensitive Hypertension
2.2.2. Reduction of Blood Pressure, Improvement of Sympathetic Nerve Hyperactivity, and Reduction of Volume Overload
2.2.3. Modulation of the Tubuloglomerular Feedback
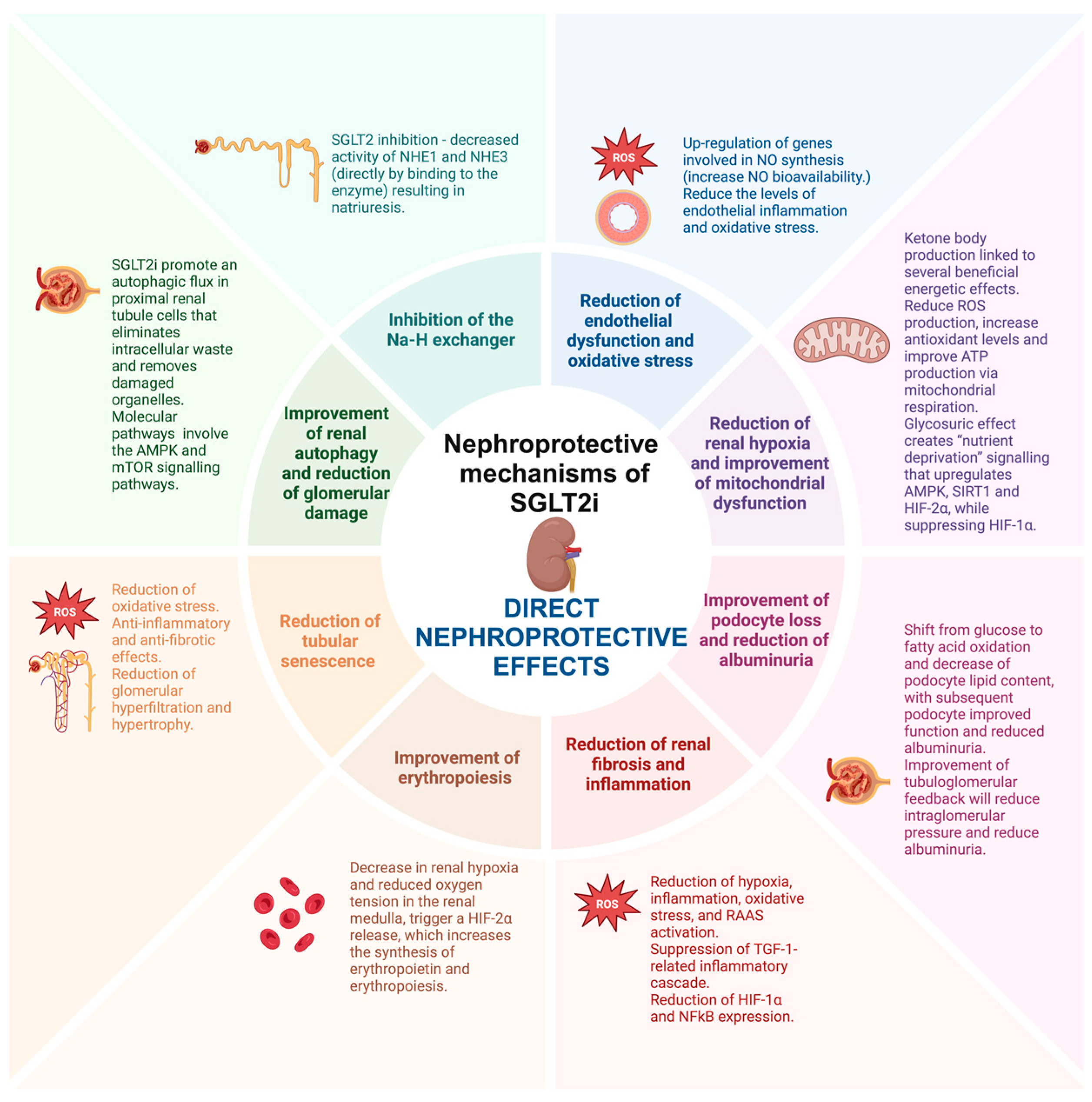
2.3. Direct Nephroprotective Effects
2.3.1. Inhibition of the Na-H Exchanger
2.3.2. Reduction of Endothelial Dysfunction and Oxidative Stress
2.3.3. Reduction of Renal Hypoxia
2.3.4. Improvement of Mitochondrial Dysfunction
2.3.5. Improvement of Podocyte Loss and Reduction of Albuminuria
2.3.6. Reduction of Renal Fibrosis and Inflammation
2.3.7. Improvement of Erythropoiesis
2.3.8. Reduction of Tubular Senescence
2.3.9. Improvement of Renal Autophagy and Reduction of Glomerular Damage
3. Clinical Trials and Guideline Recommendations
4. Conclusions
Funding
Conflicts of Interest
References
- Piovani, D.; Nikolopoulos, G.K.; Bonovas, S. Non-Communicable Diseases: The Invisible Epidemic. J. Clin. Med. 2022, 11, 5939. [Google Scholar] [CrossRef]
- Chan, J.C.N.; Lim, L.L.; Wareham, N.J.; Shaw, J.E.; Orchard, T.J.; Zhang, P.; Lau, E.S.H.; Eliasson, B.; Kong, A.P.S.; Ezzati, M.; et al. The Lancet Commission on diabetes: Using data to transform diabetes care and patient lives. Lancet 2021, 396, 2019–2082. [Google Scholar] [CrossRef]
- Unger, R.H.; Cherrington, A.D. Glucagonocentric restructuring of diabetes: A pathophysiologic and therapeutic makeover. J. Clin. Investig. 2012, 122, 4–12. [Google Scholar] [CrossRef] [PubMed]
- American Diabetes Association Professional Practice Committee. 1. Improving Care and Promoting Health in Populations: Standards of Medical Care in Diabetes-2022. Diabetes Care 2022, 45, S8–S16. [Google Scholar] [CrossRef] [PubMed]
- Davies, M.J.; Aroda, V.R.; Collins, B.S.; Gabbay, R.A.; Green, J.; Maruthur, N.M.; Rosas, S.E.; Del Prato, S.; Mathieu, C.; Mingrone, G.; et al. Management of hyperglycaemia in type 2 diabetes, 2022. A consensus report by the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetologia 2022, 65, 1925–1966. [Google Scholar] [CrossRef] [PubMed]
- Cho, N.H.; Shaw, J.E.; Karuranga, S.; Huang, Y.; da Rocha Fernandes, J.D.; Ohlrogge, A.W.; Malanda, B. IDF Diabetes Atlas: Global estimates of diabetes prevalence for 2017 and projections for 2045. Diabetes Res. Clin. Pract. 2018, 138, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Stratton, I.M.; Adler, A.I.; Neil, H.A.; Matthews, D.R.; Manley, S.E.; Cull, C.A.; Hadden, D.; Turner, R.C.; Holman, R.R. Association of glycaemia with macrovascular and microvascular complications of type 2 diabetes (UKPDS 35): Prospective observational study. BMJ 2000, 321, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.A.B.; Hashim, M.J.; King, J.K.; Govender, R.D.; Mustafa, H.; Al Kaabi, J. Epidemiology of Type 2 Diabetes—Global Burden of Disease and Forecasted Trends. J. Epidemiol. Glob. Health 2020, 10, 107–111. [Google Scholar] [CrossRef]
- Kumar, A.; Gangwar, R.; Zargar, A.A.; Kumar, R.; Sharma, A. Prevalence of Diabetes in India: A Review of IDF Diabetes Atlas 10th Edition. Curr. Diabetes Rev. 2024, 20, e130423215752. [Google Scholar] [CrossRef]
- Magliano, D.J.; Boyko, E.J. IDF Diabetes Atlas, 10th ed.; International Diabetes Federation: Brussels, Belgium, 2021. [Google Scholar]
- GBD 2021 Diabetes Collaborators. Global, regional, and national burden of diabetes from 1990 to 2021, with projections of prevalence to 2050: A systematic analysis for the Global Burden of Disease Study 2021. Lancet 2023, 402, 203–234. [Google Scholar] [CrossRef]
- Mirzaei, M.; Rahmaninan, M.; Mirzaei, M.; Nadjarzadeh, A.; Dehghani Tafti, A.A. Epidemiology of diabetes mellitus, pre-diabetes, undiagnosed and uncontrolled diabetes in Central Iran: Results from Yazd health study. BMC Public Health 2020, 20, 166. [Google Scholar] [CrossRef] [PubMed]
- The Emerging Risk Factors Collaboration; Sarwar, N.; Gao, P.; Seshasai, S.R.; Gobin, R.; Kaptoge, S.; Di Angelantonio, E.; Ingelsson, E.; Lawlor, D.A.; Selvin, E.; et al. Diabetes mellitus, fasting blood glucose concentration, and risk of vascular disease: A collaborative meta-analysis of 102 prospective studies. Lancet 2010, 375, 2215–2222. [Google Scholar] [CrossRef]
- Saran, R.; Li, Y.; Robinson, B.; Ayanian, J.; Balkrishnan, R.; Bragg-Gresham, J.; Chen, J.T.; Cope, E.; Gipson, D.; He, K.; et al. US Renal Data System 2014 Annual Data Report: Epidemiology of Kidney Disease in the United States. Am. J. Kidney Dis. 2015, 66, A7. [Google Scholar] [CrossRef]
- Kovesdy, C.P. Epidemiology of chronic kidney disease: An update 2022. Kidney Int. Suppl. 2022, 12, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Dehghani, A.; Korozhdehi, H.; Hossein Khalilzadeh, S.; Fallahzadeh, H.; Rahmanian, V. Prevalence of diabetes and its correlates among Iranian adults: Results of the first phase of Shahedieh cohort study. Health Sci. Rep. 2023, 6, e1170. [Google Scholar] [CrossRef] [PubMed]
- Kampmann, J.D.; Heaf, J.G.; Mogensen, C.B.; Mickley, H.; Wolff, D.L.; Brandt, F. Prevalence and incidence of chronic kidney disease stage 3-5—Results from KidDiCo. BMC Nephrol. 2023, 24, 17. [Google Scholar] [CrossRef]
- Sundstrom, J.; Bodegard, J.; Bollmann, A.; Vervloet, M.G.; Mark, P.B.; Karasik, A.; Taveira-Gomes, T.; Botana, M.; Birkeland, K.I.; Thuresson, M.; et al. Prevalence, outcomes, and cost of chronic kidney disease in a contemporary population of 2.4 million patients from 11 countries: The CaReMe CKD study. Lancet Reg. Health Eur. 2022, 20, 100438. [Google Scholar] [CrossRef]
- Lytvyn, Y.; Bjornstad, P.; van Raalte, D.H.; Heerspink, H.L.; Cherney, D.Z.I. The New Biology of Diabetic Kidney Disease-Mechanisms and Therapeutic Implications. Endocr. Rev. 2020, 41, 202–231. [Google Scholar] [CrossRef] [PubMed]
- Kidney Disease: Improving Global Outcomes (KDIGO) Diabetes Work Group. KDIGO 2022 Clinical Practice Guideline for Diabetes Management in Chronic Kidney Disease. Kidney Int. 2022, 102, S1–S127. [Google Scholar] [CrossRef]
- Navaneethan, S.D.; Zoungas, S.; Caramori, M.L.; Chan, J.C.N.; Heerspink, H.J.L.; Hurst, C.; Liew, A.; Michos, E.D.; Olowu, W.A.; Sadusky, T.; et al. Diabetes Management in Chronic Kidney Disease: Synopsis of the KDIGO 2022 Clinical Practice Guideline Update. Ann. Intern. Med. 2023, 176, 381–387. [Google Scholar] [CrossRef] [PubMed]
- Nordheim, E.; Geir Jenssen, T. Chronic kidney disease in patients with diabetes mellitus. Endocr. Connect. 2021, 10, R151–R159. [Google Scholar] [CrossRef] [PubMed]
- Saunders, M.; Laiteerapong, N. 2022 Clinical Practice Guideline Update for Diabetes Management of Chronic Kidney Disease: An Important First Step, More Work to Do. Ann. Intern. Med. 2023, 176, 417–418. [Google Scholar] [CrossRef]
- Shlipak, M.G.; Tummalapalli, S.L.; Boulware, L.E.; Grams, M.E.; Ix, J.H.; Jha, V.; Kengne, A.P.; Madero, M.; Mihaylova, B.; Tangri, N.; et al. The case for early identification and intervention of chronic kidney disease: Conclusions from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference. Kidney Int. 2021, 99, 34–47. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Mamza, J.B.; Morris, T.; Godfrey, G.; Asselbergs, F.W.; Denaxas, S.; Hemingway, H.; Banerjee, A. Lifetime risk of cardiovascular-renal disease in type 2 diabetes: A population-based study in 473,399 individuals. BMC Med. 2022, 20, 63. [Google Scholar] [CrossRef]
- Yau, K.; Dharia, A.; Alrowiyti, I.; Cherney, D.Z.I. Prescribing SGLT2 Inhibitors in Patients With CKD: Expanding Indications and Practical Considerations. Kidney Int. Rep. 2022, 7, 2546–2547. [Google Scholar] [CrossRef]
- Vallon, V.; Verma, S. Effects of SGLT2 Inhibitors on Kidney and Cardiovascular Function. Annu. Rev. Physiol. 2021, 83, 503–528. [Google Scholar] [CrossRef] [PubMed]
- Cowie, M.R.; Fisher, M. SGLT2 inhibitors: Mechanisms of cardiovascular benefit beyond glycaemic control. Nat. Rev. Cardiol. 2020, 17, 761–772. [Google Scholar] [CrossRef]
- Ghezzi, C.; Loo, D.D.F.; Wright, E.M. Physiology of renal glucose handling via SGLT1, SGLT2 and GLUT2. Diabetologia 2018, 61, 2087–2097. [Google Scholar] [CrossRef]
- Heerspink, H.J.L.; Stefansson, B.V.; Correa-Rotter, R.; Chertow, G.M.; Greene, T.; Hou, F.F.; Mann, J.F.E.; McMurray, J.J.V.; Lindberg, M.; Rossing, P.; et al. Dapagliflozin in Patients with Chronic Kidney Disease. N. Engl. J. Med. 2020, 383, 1436–1446. [Google Scholar] [CrossRef] [PubMed]
- Neal, B.; Perkovic, V.; Matthews, D.R. Canagliflozin and Cardiovascular and Renal Events in Type 2 Diabetes. N. Engl. J. Med. 2017, 377, 2099. [Google Scholar] [CrossRef]
- Wiviott, S.D.; Raz, I.; Bonaca, M.P.; Mosenzon, O.; Kato, E.T.; Cahn, A.; Silverman, M.G.; Zelniker, T.A.; Kuder, J.F.; Murphy, S.A.; et al. Dapagliflozin and Cardiovascular Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2019, 380, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Zinman, B.; Lachin, J.M.; Inzucchi, S.E. Empagliflozin, Cardiovascular Outcomes, and Mortality in Type 2 Diabetes. N. Engl. J. Med. 2016, 374, 1094. [Google Scholar] [CrossRef] [PubMed]
- DeFronzo, R.A.; Reeves, W.B.; Awad, A.S. Pathophysiology of diabetic kidney disease: Impact of SGLT2 inhibitors. Nat. Rev. Nephrol. 2021, 17, 319–334. [Google Scholar] [CrossRef]
- Giaccari, A. Expanding the Use of SGLT2i in Diabetes Beyond Type 2. Diabetes Care 2024, 47, 50–51. [Google Scholar] [CrossRef]
- Keller, D.M.; Ahmed, N.; Tariq, H.; Walgamage, M.; Walgamage, T.; Mohammed, A.; Chou, J.T.; Kaluzna-Oleksy, M.; Lesiak, M.; Straburzynska-Migaj, E. SGLT2 Inhibitors in Type 2 Diabetes Mellitus and Heart Failure-A Concise Review. J. Clin. Med. 2022, 11, 1470. [Google Scholar] [CrossRef] [PubMed]
- Nelinson, D.S.; Sosa, J.M.; Chilton, R.J. SGLT2 inhibitors: A narrative review of efficacy and safety. J. Osteopath. Med. 2021, 121, 229–239. [Google Scholar] [CrossRef]
- Saisho, Y. SGLT2 Inhibitors: The Star in the Treatment of Type 2 Diabetes? Diseases 2020, 8, 14. [Google Scholar] [CrossRef] [PubMed]
- Vallon, V. The mechanisms and therapeutic potential of SGLT2 inhibitors in diabetes mellitus. Annu. Rev. Med. 2015, 66, 255–270. [Google Scholar] [CrossRef]
- Wong, E.; Nguyen, T.V. Expanding the Role of SGLT2 Inhibitors Beyond Diabetes: A Case-Based Approach. Sr. Care Pharm. 2023, 38, 233–238. [Google Scholar] [CrossRef] [PubMed]
- Wright, E.M. SGLT2 Inhibitors: Physiology and Pharmacology. Kidney360 2021, 2, 2027–2037. [Google Scholar] [CrossRef]
- Kuriyama, S. A Potential Mechanism of Cardio-Renal Protection with Sodium-Glucose Cotransporter 2 Inhibitors: Amelioration of Renal Congestion. Kidney Blood Press. Res. 2019, 44, 449–456. [Google Scholar] [CrossRef] [PubMed]
- Zelniker, T.A.; Braunwald, E. Mechanisms of Cardiorenal Effects of Sodium-Glucose Cotransporter 2 Inhibitors: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2020, 75, 422–434. [Google Scholar] [CrossRef] [PubMed]
- Udell, J.A.; Jones, W.S.; Petrie, M.C.; Harrington, J.; Anker, S.D.; Bhatt, D.L.; Hernandez, A.F.; Butler, J. Sodium Glucose Cotransporter-2 Inhibition for Acute Myocardial Infarction: JACC Review Topic of the Week. J. Am. Coll. Cardiol. 2022, 79, 2058–2068. [Google Scholar] [CrossRef] [PubMed]
- Salvatore, T.; Galiero, R.; Caturano, A.; Rinaldi, L.; Di Martino, A.; Albanese, G.; Di Salvo, J.; Epifani, R.; Marfella, R.; Docimo, G.; et al. An Overview of the Cardiorenal Protective Mechanisms of SGLT2 Inhibitors. Int. J. Mol. Sci. 2022, 23, 3651. [Google Scholar] [CrossRef] [PubMed]
- Heerspink, H.J.; Perkins, B.A.; Fitchett, D.H.; Husain, M.; Cherney, D.Z. Sodium Glucose Cotransporter 2 Inhibitors in the Treatment of Diabetes Mellitus: Cardiovascular and Kidney Effects, Potential Mechanisms, and Clinical Applications. Circulation 2016, 134, 752–772. [Google Scholar] [CrossRef]
- Tomita, I.; Kume, S.; Sugahara, S.; Osawa, N.; Yamahara, K.; Yasuda-Yamahara, M.; Takeda, N.; Chin-Kanasaki, M.; Kaneko, T.; Mayoux, E.; et al. SGLT2 Inhibition Mediates Protection from Diabetic Kidney Disease by Promoting Ketone Body-Induced mTORC1 Inhibition. Cell Metab. 2020, 32, 404–419.e6. [Google Scholar] [CrossRef]
- Youm, Y.H.; Nguyen, K.Y.; Grant, R.W.; Goldberg, E.L.; Bodogai, M.; Kim, D.; D’Agostino, D.; Planavsky, N.; Lupfer, C.; Kanneganti, T.D.; et al. The ketone metabolite beta-hydroxybutyrate blocks NLRP3 inflammasome-mediated inflammatory disease. Nat. Med. 2015, 21, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Kubota, Y.; Shimizu, W. Clinical Benefits of Sodium-Glucose Cotransporter 2 Inhibitors and the Mechanisms Underlying Their Cardiovascular Effects. JACC Asia 2022, 2, 287–293. [Google Scholar] [CrossRef] [PubMed]
- Bilen, Y.; Almoushref, A.; Alkwatli, K.; Osman, O.; Mehdi, A.; Sawaf, H. Treatment and practical considerations of diabetic kidney disease. Front. Med. 2023, 10, 1264497. [Google Scholar] [CrossRef]
- Xie, Y.; Bowe, B.; Mokdad, A.H.; Xian, H.; Yan, Y.; Li, T.; Maddukuri, G.; Tsai, C.Y.; Floyd, T.; Al-Aly, Z. Analysis of the Global Burden of Disease study highlights the global, regional, and national trends of chronic kidney disease epidemiology from 1990 to 2016. Kidney Int. 2018, 94, 567–581. [Google Scholar] [CrossRef]
- Vasquez-Rios, G.; Nadkarni, G.N. SGLT2 Inhibitors: Emerging Roles in the Protection Against Cardiovascular and Kidney Disease Among Diabetic Patients. Int. J. Nephrol. Renovasc. Dis. 2020, 13, 281–296. [Google Scholar] [CrossRef] [PubMed]
- Vallon, V.; Platt, K.A.; Cunard, R.; Schroth, J.; Whaley, J.; Thomson, S.C.; Koepsell, H.; Rieg, T. SGLT2 mediates glucose reabsorption in the early proximal tubule. J. Am. Soc. Nephrol. 2011, 22, 104–112. [Google Scholar] [CrossRef] [PubMed]
- DeFronzo, R.A.; Hompesch, M.; Kasichayanula, S.; Liu, X.; Hong, Y.; Pfister, M.; Morrow, L.A.; Leslie, B.R.; Boulton, D.W.; Ching, A.; et al. Characterization of renal glucose reabsorption in response to dapagliflozin in healthy subjects and subjects with type 2 diabetes. Diabetes Care 2013, 36, 3169–3176. [Google Scholar] [CrossRef]
- Cherney, D.Z.I.; Cooper, M.E.; Tikkanen, I.; Pfarr, E.; Johansen, O.E.; Woerle, H.J.; Broedl, U.C.; Lund, S.S. Pooled analysis of Phase III trials indicate contrasting influences of renal function on blood pressure, body weight, and HbA1c reductions with empagliflozin. Kidney Int. 2018, 93, 231–244. [Google Scholar] [CrossRef] [PubMed]
- Brocco, E.; Velussi, M.; Cernigoi, A.M.; Abaterusso, C.; Bruseghin, M.; Carraro, A.; Sambataro, M.; Piarulli, F.; Sfriso, A.; Nosadini, R. Evidence of a threshold value of glycated hemoglobin to improve the course of renal function in type 2 diabetes with typical diabetic glomerulopathy. J. Nephrol. 2001, 14, 461–471. [Google Scholar]
- Ciavarella, A.; Vannini, P.; Flammini, M.; Bacci, L.; Forlani, G.; Borgnino, L.C. Effect of long-term near-normoglycemia on the progression of diabetic nephropathy. Diabete Metab. 1985, 11, 3–8. [Google Scholar] [PubMed]
- Hatanaka, T.; Ogawa, D.; Tachibana, H.; Eguchi, J.; Inoue, T.; Yamada, H.; Takei, K.; Makino, H.; Wada, J. Inhibition of SGLT2 alleviates diabetic nephropathy by suppressing high glucose-induced oxidative stress in type 1 diabetic mice. Pharmacol. Res. Perspect. 2016, 4, e00239. [Google Scholar] [CrossRef]
- Al-Sofiani, M.E.; Ganji, S.S.; Kalyani, R.R. Body composition changes in diabetes and aging. J. Diabetes Complicat. 2019, 33, 451–459. [Google Scholar] [CrossRef]
- Han, T.S.; Al-Gindan, Y.Y.; Govan, L.; Hankey, C.R.; Lean, M.E.J. Associations of BMI, waist circumference, body fat, and skeletal muscle with type 2 diabetes in adults. Acta Diabetol. 2019, 56, 947–954. [Google Scholar] [CrossRef] [PubMed]
- Preis, S.R.; Massaro, J.M.; Robins, S.J.; Hoffmann, U.; Vasan, R.S.; Irlbeck, T.; Meigs, J.B.; Sutherland, P.; D’Agostino, R.B., Sr.; O’Donnell, C.J.; et al. Abdominal subcutaneous and visceral adipose tissue and insulin resistance in the Framingham heart study. Obesity 2010, 18, 2191–2198. [Google Scholar] [CrossRef] [PubMed]
- The, E.-K.C.G.; Herrington, W.G.; Staplin, N.; Wanner, C.; Green, J.B.; Hauske, S.J.; Emberson, J.R.; Preiss, D.; Judge, P.; Mayne, K.J.; et al. Empagliflozin in Patients with Chronic Kidney Disease. N. Engl. J. Med. 2023, 388, 117–127. [Google Scholar] [CrossRef]
- Bolinder, J.; Ljunggren, O.; Kullberg, J.; Johansson, L.; Wilding, J.; Langkilde, A.M.; Sugg, J.; Parikh, S. Effects of dapagliflozin on body weight, total fat mass, and regional adipose tissue distribution in patients with type 2 diabetes mellitus with inadequate glycemic control on metformin. J. Clin. Endocrinol. Metab. 2012, 97, 1020–1031. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Yang, W.; Gao, X.; Chen, Y.; Zhou, L.; Zhang, S.; Han, X.; Ji, L. The Association Between the Dosage of SGLT2 Inhibitor and Weight Reduction in Type 2 Diabetes Patients: A Meta-Analysis. Obesity 2018, 26, 70–80. [Google Scholar] [CrossRef] [PubMed]
- Pereira, M.J.; Eriksson, J.W. Emerging Role of SGLT-2 Inhibitors for the Treatment of Obesity. Drugs 2019, 79, 219–230. [Google Scholar] [CrossRef] [PubMed]
- Perkovic, V.; Jardine, M.J.; Neal, B.; Bompoint, S.; Heerspink, H.J.L.; Charytan, D.M.; Edwards, R.; Agarwal, R.; Bakris, G.; Bull, S.; et al. Canagliflozin and Renal Outcomes in Type 2 Diabetes and Nephropathy. N. Engl. J. Med. 2019, 380, 2295–2306. [Google Scholar] [CrossRef]
- Cefalu, W.T.; Leiter, L.A.; Yoon, K.H.; Arias, P.; Niskanen, L.; Xie, J.; Balis, D.A.; Canovatchel, W.; Meininger, G. Efficacy and safety of canagliflozin versus glimepiride in patients with type 2 diabetes inadequately controlled with metformin (CANTATA-SU): 52 week results from a randomised, double-blind, phase 3 non-inferiority trial. Lancet 2013, 382, 941–950. [Google Scholar] [CrossRef]
- Kurinami, N.; Sugiyama, S.; Nishimura, H.; Morita, A.; Yoshida, A.; Hieshima, K.; Miyamoto, F.; Kajiwara, K.; Jinnouchi, K.; Jinnouchi, T.; et al. Clinical Factors Associated with Initial Decrease in Body-Fat Percentage Induced by Add-on Sodium-Glucose Co-transporter 2 Inhibitors in Patient with Type 2 Diabetes Mellitus. Clin. Drug Investig. 2018, 38, 19–27. [Google Scholar] [CrossRef]
- Lee, P.C.; Ganguly, S.; Goh, S.Y. Weight loss associated with sodium-glucose cotransporter-2 inhibition: A review of evidence and underlying mechanisms. Obes. Rev. 2018, 19, 1630–1641. [Google Scholar] [CrossRef] [PubMed]
- Nishiyama, A.; Kitada, K. Possible renoprotective mechanisms of SGLT2 inhibitors. Front. Med. 2023, 10, 1115413. [Google Scholar] [CrossRef] [PubMed]
- Pan, R.; Zhang, Y.; Wang, R.; Xu, Y.; Ji, H.; Zhao, Y. Effect of SGLT-2 inhibitors on body composition in patients with type 2 diabetes mellitus: A meta-analysis of randomized controlled trials. PLoS ONE 2022, 17, e0279889. [Google Scholar] [CrossRef]
- Opazo-Rios, L.; Mas, S.; Marin-Royo, G.; Mezzano, S.; Gomez-Guerrero, C.; Moreno, J.A.; Egido, J. Lipotoxicity and Diabetic Nephropathy: Novel Mechanistic Insights and Therapeutic Opportunities. Int. J. Mol. Sci. 2020, 21, 2632. [Google Scholar] [CrossRef] [PubMed]
- Del Prato, S. Role of glucotoxicity and lipotoxicity in the pathophysiology of Type 2 diabetes mellitus and emerging treatment strategies. Diabet. Med. 2009, 26, 1185–1192. [Google Scholar] [CrossRef]
- Song, J.; Li, X.; Ni, J. A Role for Sodium-Glucose Cotransporter 2 Inhibitors in the Treatment of Chronic Kidney Disease: A Mini Review. Kidney Blood Press. Res. 2023, 48, 599–610. [Google Scholar] [CrossRef] [PubMed]
- Gaita, L.; Timar, B.; Timar, R.; Fras, Z.; Gaita, D.; Banach, M. Lipid Disorders Management Strategies (2024) in Prediabetic and Diabetic Patients. Pharmaceuticals 2024, 17, 219. [Google Scholar] [CrossRef]
- Cai, T.; Ke, Q.; Fang, Y.; Wen, P.; Chen, H.; Yuan, Q.; Luo, J.; Zhang, Y.; Sun, Q.; Lv, Y.; et al. Sodium-glucose cotransporter 2 inhibition suppresses HIF-1alpha-mediated metabolic switch from lipid oxidation to glycolysis in kidney tubule cells of diabetic mice. Cell Death Dis. 2020, 11, 390. [Google Scholar] [CrossRef] [PubMed]
- Szekeres, Z.; Toth, K.; Szabados, E. The Effects of SGLT2 Inhibitors on Lipid Metabolism. Metabolites 2021, 11, 87. [Google Scholar] [CrossRef]
- Zaccardi, F.; Webb, D.R.; Htike, Z.Z.; Youssef, D.; Khunti, K.; Davies, M.J. Efficacy and safety of sodium-glucose co-transporter-2 inhibitors in type 2 diabetes mellitus: Systematic review and network meta-analysis. Diabetes Obes. Metab. 2016, 18, 783–794. [Google Scholar] [CrossRef] [PubMed]
- Heda, R.; Yazawa, M.; Shi, M.; Bhaskaran, M.; Aloor, F.Z.; Thuluvath, P.J.; Satapathy, S.K. Non-alcoholic fatty liver and chronic kidney disease: Retrospect, introspect, and prospect. World J. Gastroenterol. 2021, 27, 1864–1882. [Google Scholar] [CrossRef] [PubMed]
- Kiapidou, S.; Liava, C.; Kalogirou, M.; Akriviadis, E.; Sinakos, E. Chronic kidney disease in patients with non-alcoholic fatty liver disease: What the Hepatologist should know? Ann. Hepatol. 2020, 19, 134–144. [Google Scholar] [CrossRef]
- Musso, G.; Gambino, R.; Tabibian, J.H.; Ekstedt, M.; Kechagias, S.; Hamaguchi, M.; Hultcrantz, R.; Hagstrom, H.; Yoon, S.K.; Charatcharoenwitthaya, P.; et al. Association of non-alcoholic fatty liver disease with chronic kidney disease: A systematic review and meta-analysis. PLoS Med. 2014, 11, e1001680. [Google Scholar] [CrossRef]
- Raj, D.; Tomar, B.; Lahiri, A.; Mulay, S.R. The gut-liver-kidney axis: Novel regulator of fatty liver associated chronic kidney disease. Pharmacol. Res. 2020, 152, 104617. [Google Scholar] [CrossRef] [PubMed]
- Gofton, C.; Upendran, Y.; Zheng, M.H.; George, J. MAFLD: How is it different from NAFLD? Clin. Mol. Hepatol. 2023, 29, S17–S31. [Google Scholar] [CrossRef] [PubMed]
- Targher, G.; Pichiri, I.; Zoppini, G.; Trombetta, M.; Bonora, E. Increased prevalence of chronic kidney disease in patients with Type 1 diabetes and non-alcoholic fatty liver. Diabet. Med. 2012, 29, 220–226. [Google Scholar] [CrossRef] [PubMed]
- Leiter, L.A.; Forst, T.; Polidori, D.; Balis, D.A.; Xie, J.; Sha, S. Effect of canagliflozin on liver function tests in patients with type 2 diabetes. Diabetes Metab. 2016, 42, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, M.; Suzuki, K.; Kato, K.; Jojima, T.; Iijima, T.; Murohisa, T.; Iijima, M.; Takekawa, H.; Usui, I.; Hiraishi, H.; et al. Evaluation of the effects of dapagliflozin, a sodium-glucose co-transporter-2 inhibitor, on hepatic steatosis and fibrosis using transient elastography in patients with type 2 diabetes and non-alcoholic fatty liver disease. Diabetes Obes. Metab. 2019, 21, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Angulo, P.; Lindor, K.D. Non-alcoholic fatty liver disease. J. Gastroenterol. Hepatol. 2002, 17, S186–S190. [Google Scholar] [CrossRef] [PubMed]
- Scheen, A.J. Beneficial effects of SGLT2 inhibitors on fatty liver in type 2 diabetes: A common comorbidity associated with severe complications. Diabetes Metab. 2019, 45, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Gaita, L.; Timar, R.; Lupascu, N.; Roman, D.; Albai, A.; Potre, O.; Timar, B. The Impact Of Hyperuricemia On Cardiometabolic Risk Factors In Patients With Diabetes Mellitus: A Cross-Sectional Study. Diabetes Metab. Syndr. Obes. 2019, 12, 2003–2010. [Google Scholar] [CrossRef]
- Chino, Y.; Samukawa, Y.; Sakai, S.; Nakai, Y.; Yamaguchi, J.; Nakanishi, T.; Tamai, I. SGLT2 inhibitor lowers serum uric acid through alteration of uric acid transport activity in renal tubule by increased glycosuria. Biopharm. Drug Dispos. 2014, 35, 391–404. [Google Scholar] [CrossRef]
- Koike, Y.; Shirabe, S.I.; Maeda, H.; Yoshimoto, A.; Arai, K.; Kumakura, A.; Hirao, K.; Terauchi, Y. Effect of canagliflozin on the overall clinical state including insulin resistance in Japanese patients with type 2 diabetes mellitus. Diabetes Res. Clin. Pract. 2019, 149, 140–146. [Google Scholar] [CrossRef]
- Dong, M.; Chen, H.; Wen, S.; Yuan, Y.; Yang, L.; Xu, D.; Zhou, L. The Mechanism of Sodium-Glucose Cotransporter-2 Inhibitors in Reducing Uric Acid in Type 2 Diabetes Mellitus. Diabetes Metab. Syndr. Obes. 2023, 16, 437–445. [Google Scholar] [CrossRef] [PubMed]
- Darshi, M.; Van Espen, B.; Sharma, K. Metabolomics in Diabetic Kidney Disease: Unraveling the Biochemistry of a Silent Killer. Am. J. Nephrol. 2016, 44, 92–103. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.M.; Feng, S.T.; Wen, Y.; Tang, T.T.; Wang, B.; Liu, B.C. Cardiorenal protection of SGLT2 inhibitors-Perspectives from metabolic reprogramming. EBioMedicine 2022, 83, 104215. [Google Scholar] [CrossRef] [PubMed]
- Sas, K.M.; Kayampilly, P.; Byun, J.; Nair, V.; Hinder, L.M.; Hur, J.; Zhang, H.; Lin, C.; Qi, N.R.; Michailidis, G.; et al. Tissue-specific metabolic reprogramming drives nutrient flux in diabetic complications. JCI Insight 2016, 1, e86976. [Google Scholar] [CrossRef]
- Kappel, B.A.; Lehrke, M.; Schutt, K.; Artati, A.; Adamski, J.; Lebherz, C.; Marx, N. Effect of Empagliflozin on the Metabolic Signature of Patients With Type 2 Diabetes Mellitus and Cardiovascular Disease. Circulation 2017, 136, 969–972. [Google Scholar] [CrossRef]
- Lupsa, B.C.; Kibbey, R.G.; Inzucchi, S.E. Ketones: The double-edged sword of SGLT2 inhibitors? Diabetologia 2023, 66, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Kashiwagi, A.; Maegawa, H. Metabolic and hemodynamic effects of sodium-dependent glucose cotransporter 2 inhibitors on cardio-renal protection in the treatment of patients with type 2 diabetes mellitus. J. Diabetes Investig. 2017, 8, 416–427. [Google Scholar] [CrossRef] [PubMed]
- Vallon, V.; Thomson, S.C. Targeting renal glucose reabsorption to treat hyperglycaemia: The pleiotropic effects of SGLT2 inhibition. Diabetologia 2017, 60, 215–225. [Google Scholar] [CrossRef]
- Hattori, Y. Beneficial effects on kidney during treatment with sodium-glucose cotransporter 2 inhibitors: Proposed role of ketone utilization. Heart Fail. Rev. 2021, 26, 947–952. [Google Scholar] [CrossRef]
- Roberts, L.D.; Bostrom, P.; O’Sullivan, J.F.; Schinzel, R.T.; Lewis, G.D.; Dejam, A.; Lee, Y.K.; Palma, M.J.; Calhoun, S.; Georgiadi, A.; et al. beta-Aminoisobutyric acid induces browning of white fat and hepatic beta-oxidation and is inversely correlated with cardiometabolic risk factors. Cell Metab. 2014, 19, 96–108. [Google Scholar] [CrossRef]
- Sulyok, E.; Farkas, B.; Nagy, B.; Varnagy, A.; Kovacs, K.; Bodis, J. Tissue Sodium Accumulation: Pathophysiology and Clinical Implications. Antioxidants 2022, 11, 750. [Google Scholar] [CrossRef]
- Hallow, K.M.; Helmlinger, G.; Greasley, P.J.; McMurray, J.J.V.; Boulton, D.W. Why do SGLT2 inhibitors reduce heart failure hospitalization? A differential volume regulation hypothesis. Diabetes Obes. Metab. 2018, 20, 479–487. [Google Scholar] [CrossRef]
- Schork, A.; Saynisch, J.; Vosseler, A.; Jaghutriz, B.A.; Heyne, N.; Peter, A.; Haring, H.U.; Stefan, N.; Fritsche, A.; Artunc, F. Effect of SGLT2 inhibitors on body composition, fluid status and renin-angiotensin-aldosterone system in type 2 diabetes: A prospective study using bioimpedance spectroscopy. Cardiovasc. Diabetol. 2019, 18, 46. [Google Scholar] [CrossRef]
- Maxson, R.; Starr, J.; Sewell, J.; Lyas, C. SGLT2 Inhibitors to Slow Chronic Kidney Disease Progression: A Review. Clin. Ther. 2024, 46, e23–e28. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, A.; Shankar, M.; Lerma, E.V.; Wiegley, N.; GlomCon Editorial, T. Sodium Glucose Cotransporter 2 (SGLT2) Inhibitors and CKD: Are You a #Flozinator? Kidney Med. 2023, 5, 100608. [Google Scholar] [CrossRef] [PubMed]
- Ruggenenti, P.; Cravedi, P.; Remuzzi, G. The RAAS in the pathogenesis and treatment of diabetic nephropathy. Nat. Rev. Nephrol. 2010, 6, 319–330. [Google Scholar] [CrossRef] [PubMed]
- Warren, A.M.; Knudsen, S.T.; Cooper, M.E. Diabetic nephropathy: An insight into molecular mechanisms and emerging therapies. Expert. Opin. Ther. Targets 2019, 23, 579–591. [Google Scholar] [CrossRef] [PubMed]
- DeFronzo, R.A.; Norton, L.; Abdul-Ghani, M. Renal, metabolic and cardiovascular considerations of SGLT2 inhibition. Nat. Rev. Nephrol. 2017, 13, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.C.; Cherney, D.Z.I. The actions of SGLT2 inhibitors on metabolism, renal function and blood pressure. Diabetologia 2018, 61, 2098–2107. [Google Scholar] [CrossRef] [PubMed]
- Busch, R.S.; Kane, M.P. Combination SGLT2 inhibitor and GLP-1 receptor agonist therapy: A complementary approach to the treatment of type 2 diabetes. Postgrad. Med. 2017, 129, 686–697. [Google Scholar] [CrossRef]
- Tsuboi, N.; Sasaki, T.; Okabayashi, Y.; Haruhara, K.; Kanzaki, G.; Yokoo, T. Assessment of nephron number and single-nephron glomerular filtration rate in a clinical setting. Hypertens. Res. 2021, 44, 605–617. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, W.; Kubota, Y.; Hoshika, Y.; Mozawa, K.; Tara, S.; Tokita, Y.; Yodogawa, K.; Iwasaki, Y.K.; Yamamoto, T.; Takano, H.; et al. Effects of empagliflozin versus placebo on cardiac sympathetic activity in acute myocardial infarction patients with type 2 diabetes mellitus: The EMBODY trial. Cardiovasc. Diabetol. 2020, 19, 148. [Google Scholar] [CrossRef] [PubMed]
- Wan, N.; Fujisawa, Y.; Kobara, H.; Masaki, T.; Nakano, D.; Rahman, A.; Nishiyama, A. Effects of an SGLT2 inhibitor on the salt sensitivity of blood pressure and sympathetic nerve activity in a nondiabetic rat model of chronic kidney disease. Hypertens. Res. 2020, 43, 492–499. [Google Scholar] [CrossRef] [PubMed]
- Gueguen, C.; Burke, S.L.; Barzel, B.; Eikelis, N.; Watson, A.M.D.; Jha, J.C.; Jackson, K.L.; Sata, Y.; Lim, K.; Lambert, G.W.; et al. Empagliflozin modulates renal sympathetic and heart rate baroreflexes in a rabbit model of diabetes. Diabetologia 2020, 63, 1424–1434. [Google Scholar] [CrossRef] [PubMed]
- Rahman, A.; Hitomi, H.; Nishiyama, A. Cardioprotective effects of SGLT2 inhibitors are possibly associated with normalization of the circadian rhythm of blood pressure. Hypertens. Res. 2017, 40, 535–540. [Google Scholar] [CrossRef]
- Takeshige, Y.; Fujisawa, Y.; Rahman, A.; Kittikulsuth, W.; Nakano, D.; Mori, H.; Masaki, T.; Ohmori, K.; Kohno, M.; Ogata, H.; et al. A sodium-glucose co-transporter 2 inhibitor empagliflozin prevents abnormality of circadian rhythm of blood pressure in salt-treated obese rats. Hypertens. Res. 2016, 39, 415–422. [Google Scholar] [CrossRef]
- Cherney, D.Z.; Perkins, B.A.; Soleymanlou, N.; Har, R.; Fagan, N.; Johansen, O.E.; Woerle, H.J.; von Eynatten, M.; Broedl, U.C. The effect of empagliflozin on arterial stiffness and heart rate variability in subjects with uncomplicated type 1 diabetes mellitus. Cardiovasc. Diabetol. 2014, 13, 28. [Google Scholar] [CrossRef]
- Lambers Heerspink, H.J.; de Zeeuw, D.; Wie, L.; Leslie, B.; List, J. Dapagliflozin a glucose-regulating drug with diuretic properties in subjects with type 2 diabetes. Diabetes Obes. Metab. 2013, 15, 853–862. [Google Scholar] [CrossRef]
- Dekkers, C.C.J.; Petrykiv, S.; Laverman, G.D.; Cherney, D.Z.; Gansevoort, R.T.; Heerspink, H.J.L. Effects of the SGLT-2 inhibitor dapagliflozin on glomerular and tubular injury markers. Diabetes Obes. Metab. 2018, 20, 1988–1993. [Google Scholar] [CrossRef]
- Mazidi, M.; Rezaie, P.; Gao, H.K.; Kengne, A.P. Effect of Sodium-Glucose Cotransport-2 Inhibitors on Blood Pressure in People with Type 2 Diabetes Mellitus: A Systematic Review and Meta-Analysis of 43 Randomized Control Trials with 22 528 Patients. J. Am. Heart Assoc. 2017, 6, e004007. [Google Scholar] [CrossRef]
- Scheen, A.J.; Delanaye, P. Effects of reducing blood pressure on renal outcomes in patients with type 2 diabetes: Focus on SGLT2 inhibitors and EMPA-REG OUTCOME. Diabetes Metab. 2017, 43, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Kidokoro, K.; Cherney, D.Z.I.; Bozovic, A.; Nagasu, H.; Satoh, M.; Kanda, E.; Sasaki, T.; Kashihara, N. Evaluation of Glomerular Hemodynamic Function by Empagliflozin in Diabetic Mice Using In Vivo Imaging. Circulation 2019, 140, 303–315. [Google Scholar] [CrossRef] [PubMed]
- Nishiyama, A.; Rahman, M.; Inscho, E.W. Role of interstitial ATP and adenosine in the regulation of renal hemodynamics and microvascular function. Hypertens. Res. 2004, 27, 791–804. [Google Scholar] [CrossRef] [PubMed]
- Sen, T.; Heerspink, H.J.L. A kidney perspective on the mechanism of action of sodium glucose co-transporter 2 inhibitors. Cell Metab. 2021, 33, 732–739. [Google Scholar] [CrossRef]
- Kashihara, N.; Kidokoro, K.; Kanda, E. Renoprotective effects of sodium-glucose cotransporter-2 inhibitors and underlying mechanisms. Curr. Opin. Nephrol. Hypertens. 2020, 29, 112–118. [Google Scholar] [CrossRef] [PubMed]
- Vallon, V.; Thomson, S.C. The tubular hypothesis of nephron filtration and diabetic kidney disease. Nat. Rev. Nephrol. 2020, 16, 317–336. [Google Scholar] [CrossRef]
- Cravedi, P.; Remuzzi, G. Pathophysiology of proteinuria and its value as an outcome measure in chronic kidney disease. Br. J. Clin. Pharmacol. 2013, 76, 516–523. [Google Scholar] [CrossRef]
- Cherney, D.Z.I.; Zinman, B.; Inzucchi, S.E.; Koitka-Weber, A.; Mattheus, M.; von Eynatten, M.; Wanner, C. Effects of empagliflozin on the urinary albumin-to-creatinine ratio in patients with type 2 diabetes and established cardiovascular disease: An exploratory analysis from the EMPA-REG OUTCOME randomised, placebo-controlled trial. Lancet Diabetes Endocrinol. 2017, 5, 610–621. [Google Scholar] [CrossRef]
- Amemiya, M.; Loffing, J.; Lotscher, M.; Kaissling, B.; Alpern, R.J.; Moe, O.W. Expression of NHE-3 in the apical membrane of rat renal proximal tubule and thick ascending limb. Kidney Int. 1995, 48, 1206–1215. [Google Scholar] [CrossRef]
- Beloto-Silva, O.; Machado, U.F.; Oliveira-Souza, M. Glucose-induced regulation of NHEs activity and SGLTs expression involves the PKA signaling pathway. J. Membr. Biol. 2011, 239, 157–165. [Google Scholar] [CrossRef]
- Ganz, M.B.; Hawkins, K.; Reilly, R.F. High glucose induces the activity and expression of Na(+)/H(+) exchange in glomerular mesangial cells. Am. J. Physiol. Renal Physiol. 2000, 278, F91–F96. [Google Scholar] [CrossRef] [PubMed]
- Klisic, J.; Nief, V.; Reyes, L.; Ambuhl, P.M. Acute and chronic regulation of the renal Na/H+ exchanger NHE3 in rats with STZ-induced diabetes mellitus. Nephron Physiol. 2006, 102, p27–p35. [Google Scholar] [CrossRef]
- Nakamura, N.; Tanaka, S.; Teko, Y.; Mitsui, K.; Kanazawa, H. Four Na+/H+ exchanger isoforms are distributed to Golgi and post-Golgi compartments and are involved in organelle pH regulation. J. Biol. Chem. 2005, 280, 1561–1572. [Google Scholar] [CrossRef] [PubMed]
- Nikolovska, K.; Seidler, U.E.; Stock, C. The Role of Plasma Membrane Sodium/Hydrogen Exchangers in Gastrointestinal Functions: Proliferation and Differentiation, Fluid/Electrolyte Transport and Barrier Integrity. Front. Physiol. 2022, 13, 899286. [Google Scholar] [CrossRef]
- Slepkov, E.R.; Rainey, J.K.; Sykes, B.D.; Fliegel, L. Structural and functional analysis of the Na+/H+ exchanger. Biochem. J. 2007, 401, 623–633. [Google Scholar] [CrossRef]
- Zerbini, G.; Roth, T.; Podesta, F.; Cagliero, E.; Doria, A.; Canessa, M.; Lorenzi, M. Activity and expression of the Na+/H+ exchanger in human endothelial cells cultured in high glucose. Diabetologia 1995, 38, 785–791. [Google Scholar] [CrossRef]
- Ishizawa, K.; Wang, Q.; Li, J.; Xu, N.; Nemoto, Y.; Morimoto, C.; Fujii, W.; Tamura, Y.; Fujigaki, Y.; Tsukamoto, K.; et al. Inhibition of Sodium Glucose Cotransporter 2 Attenuates the Dysregulation of Kelch-Like 3 and NaCl Cotransporter in Obese Diabetic Mice. J. Am. Soc. Nephrol. 2019, 30, 782–794. [Google Scholar] [CrossRef] [PubMed]
- Onishi, A.; Fu, Y.; Patel, R.; Darshi, M.; Crespo-Masip, M.; Huang, W.; Song, P.; Freeman, B.; Kim, Y.C.; Soleimani, M.; et al. A role for tubular Na(+)/H(+) exchanger NHE3 in the natriuretic effect of the SGLT2 inhibitor empagliflozin. Am. J. Physiol. Renal Physiol. 2020, 319, F712–F728. [Google Scholar] [CrossRef] [PubMed]
- Packer, M. Interplay of adenosine monophosphate-activated protein kinase/sirtuin-1 activation and sodium influx inhibition mediates the renal benefits of sodium-glucose co-transporter-2 inhibitors in type 2 diabetes: A novel conceptual framework. Diabetes Obes. Metab. 2020, 22, 734–742. [Google Scholar] [CrossRef]
- Hadi, H.A.; Carr, C.S.; Al Suwaidi, J. Endothelial dysfunction: Cardiovascular risk factors, therapy, and outcome. Vasc. Health Risk Manag. 2005, 1, 183–198. [Google Scholar]
- King, G.L.; Shiba, T.; Oliver, J.; Inoguchi, T.; Bursell, S.E. Cellular and molecular abnormalities in the vascular endothelium of diabetes mellitus. Annu. Rev. Med. 1994, 45, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Russo, E.; Bussalino, E.; Maccio, L.; Verzola, D.; Saio, M.; Esposito, P.; Leoncini, G.; Pontremoli, R.; Viazzi, F. Non-Haemodynamic Mechanisms Underlying Hypertension-Associated Damage in Target Kidney Components. Int. J. Mol. Sci. 2023, 24, 9422. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Ni, Y.Q.; Zhan, J.K.; Liu, Y.S. The Role of SGLT2 Inhibitors in Vascular Aging. Aging Dis. 2021, 12, 1323–1336. [Google Scholar] [CrossRef]
- Ishibashi, Y.; Matsui, T.; Yamagishi, S. Tofogliflozin, A Highly Selective Inhibitor of SGLT2 Blocks Proinflammatory and Proapoptotic Effects of Glucose Overload on Proximal Tubular Cells Partly by Suppressing Oxidative Stress Generation. Horm. Metab. Res. 2016, 48, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Tian, J.; Sodhi, K.; Shapiro, J.I. The Na/K-ATPase Signaling and SGLT2 Inhibitor-Mediated Cardiorenal Protection: A Crossed Road? J. Membr. Biol. 2021, 254, 513–529. [Google Scholar] [CrossRef]
- Takiyama, Y.; Haneda, M. Hypoxia in diabetic kidneys. Biomed. Res. Int. 2014, 2014, 837421. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Li, Z.L.; Zhang, Y.L.; Wen, Y.; Gao, Y.M.; Liu, B.C. Hypoxia and chronic kidney disease. EBioMedicine 2022, 77, 103942. [Google Scholar] [CrossRef]
- Packer, M. Mechanisms Leading to Differential Hypoxia-Inducible Factor Signaling in the Diabetic Kidney: Modulation by SGLT2 Inhibitors and Hypoxia Mimetics. Am. J. Kidney Dis. 2021, 77, 280–286. [Google Scholar] [CrossRef] [PubMed]
- Hesp, A.C.; Schaub, J.A.; Prasad, P.V.; Vallon, V.; Laverman, G.D.; Bjornstad, P.; van Raalte, D.H. The role of renal hypoxia in the pathogenesis of diabetic kidney disease: A promising target for newer renoprotective agents including SGLT2 inhibitors? Kidney Int. 2020, 98, 579–589. [Google Scholar] [CrossRef]
- Bessho, R.; Takiyama, Y.; Takiyama, T.; Kitsunai, H.; Takeda, Y.; Sakagami, H.; Ota, T. Hypoxia-inducible factor-1alpha is the therapeutic target of the SGLT2 inhibitor for diabetic nephropathy. Sci. Rep. 2019, 9, 14754. [Google Scholar] [CrossRef]
- Sasako, T.; Tanaka, T.; Yamauchi, T. Adaptive Response as a Potential Key Link Between SGLT2 Inhibition and Renoprotection. Kidney Int. Rep. 2021, 6, 2022–2024. [Google Scholar] [CrossRef] [PubMed]
- Sen, T.; Li, J.; Neuen, B.L.; Neal, B.; Arnott, C.; Parikh, C.R.; Coca, S.G.; Perkovic, V.; Mahaffey, K.W.; Yavin, Y.; et al. Effects of the SGLT2 inhibitor canagliflozin on plasma biomarkers TNFR-1, TNFR-2 and KIM-1 in the CANVAS trial. Diabetologia 2021, 64, 2147–2158. [Google Scholar] [CrossRef] [PubMed]
- Zannad, F.; Ferreira, J.P.; Butler, J.; Filippatos, G.; Januzzi, J.L.; Sumin, M.; Zwick, M.; Saadati, M.; Pocock, S.J.; Sattar, N.; et al. Effect of empagliflozin on circulating proteomics in heart failure: Mechanistic insights into the EMPEROR programme. Eur. Heart J. 2022, 43, 4991–5002. [Google Scholar] [CrossRef] [PubMed]
- Detmer, S.A.; Chan, D.C. Functions and dysfunctions of mitochondrial dynamics. Nat. Rev. Mol. Cell Biol. 2007, 8, 870–879. [Google Scholar] [CrossRef] [PubMed]
- Aluksanasuwan, S.; Plumworasawat, S.; Malaitad, T.; Chaiyarit, S.; Thongboonkerd, V. High glucose induces phosphorylation and oxidation of mitochondrial proteins in renal tubular cells: A proteomics approach. Sci. Rep. 2020, 10, 5843. [Google Scholar] [CrossRef]
- Puchalska, P.; Crawford, P.A. Multi-dimensional Roles of Ketone Bodies in Fuel Metabolism, Signaling, and Therapeutics. Cell Metab. 2017, 25, 262–284. [Google Scholar] [CrossRef] [PubMed]
- Selvaraj, S.; Kelly, D.P.; Margulies, K.B. Implications of Altered Ketone Metabolism and Therapeutic Ketosis in Heart Failure. Circulation 2020, 141, 1800–1812. [Google Scholar] [CrossRef]
- Zhou, H.; Wang, S.; Zhu, P.; Hu, S.; Chen, Y.; Ren, J. Empagliflozin rescues diabetic myocardial microvascular injury via AMPK-mediated inhibition of mitochondrial fission. Redox Biol. 2018, 15, 335–346. [Google Scholar] [CrossRef] [PubMed]
- Levey, A.S.; Gansevoort, R.T.; Coresh, J.; Inker, L.A.; Heerspink, H.L.; Grams, M.E.; Greene, T.; Tighiouart, H.; Matsushita, K.; Ballew, S.H.; et al. Change in Albuminuria and GFR as End Points for Clinical Trials in Early Stages of CKD: A Scientific Workshop Sponsored by the National Kidney Foundation in Collaboration With the US Food and Drug Administration and European Medicines Agency. Am. J. Kidney Dis. 2020, 75, 84–104. [Google Scholar] [CrossRef]
- Pagtalunan, M.E.; Miller, P.L.; Jumping-Eagle, S.; Nelson, R.G.; Myers, B.D.; Rennke, H.G.; Coplon, N.S.; Sun, L.; Meyer, T.W. Podocyte loss and progressive glomerular injury in type II diabetes. J. Clin. Investig. 1997, 99, 342–348. [Google Scholar] [CrossRef]
- Steffes, M.W.; Schmidt, D.; McCrery, R.; Basgen, J.M.; International Diabetic Nephropathy Study Group. Glomerular cell number in normal subjects and in type 1 diabetic patients. Kidney Int. 2001, 59, 2104–2113. [Google Scholar] [CrossRef]
- Tamsma, J.T.; van den Born, J.; Bruijn, J.A.; Assmann, K.J.; Weening, J.J.; Berden, J.H.; Wieslander, J.; Schrama, E.; Hermans, J.; Veerkamp, J.H.; et al. Expression of glomerular extracellular matrix components in human diabetic nephropathy: Decrease of heparan sulphate in the glomerular basement membrane. Diabetologia 1994, 37, 313–320. [Google Scholar] [CrossRef]
- Satchell, S. The role of the glomerular endothelium in albumin handling. Nat. Rev. Nephrol. 2013, 9, 717–725. [Google Scholar] [CrossRef] [PubMed]
- de Vries, A.P.; Ruggenenti, P.; Ruan, X.Z.; Praga, M.; Cruzado, J.M.; Bajema, I.M.; D’Agati, V.D.; Lamb, H.J.; Pongrac Barlovic, D.; Hojs, R.; et al. Fatty kidney: Emerging role of ectopic lipid in obesity-related renal disease. Lancet Diabetes Endocrinol. 2014, 2, 417–426. [Google Scholar] [CrossRef]
- Nishi, H.; Nangaku, M. Podocyte lipotoxicity in diabetic kidney disease. Kidney Int. 2019, 96, 809–812. [Google Scholar] [CrossRef]
- Ilatovskaya, D.V.; Levchenko, V.; Lowing, A.; Shuyskiy, L.S.; Palygin, O.; Staruschenko, A. Podocyte injury in diabetic nephropathy: Implications of angiotensin II-dependent activation of TRPC channels. Sci. Rep. 2015, 5, 17637. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.S.; Susztak, K. Podocytes: The Weakest Link in Diabetic Kidney Disease? Curr. Diabetes Rep. 2016, 16, 45. [Google Scholar] [CrossRef] [PubMed]
- Russo, L.M.; Sandoval, R.M.; Campos, S.B.; Molitoris, B.A.; Comper, W.D.; Brown, D. Impaired tubular uptake explains albuminuria in early diabetic nephropathy. J. Am. Soc. Nephrol. 2009, 20, 489–494. [Google Scholar] [CrossRef]
- Yuan, Q.; Tang, B.; Zhang, C. Signaling pathways of chronic kidney diseases, implications for therapeutics. Signal Transduct. Target. Ther. 2022, 7, 182. [Google Scholar] [CrossRef]
- Afsar, B.; Afsar, R.E. Sodium-glucose cotransporter inhibitors and kidney fibrosis: Review of the current evidence and related mechanisms. Pharmacol. Rep. 2023, 75, 44–68. [Google Scholar] [CrossRef]
- Watanabe, K.; Sato, E.; Mishima, E.; Miyazaki, M.; Tanaka, T. What’s New in the Molecular Mechanisms of Diabetic Kidney Disease: Recent Advances. Int. J. Mol. Sci. 2022, 24, 570. [Google Scholar] [CrossRef] [PubMed]
- La Grotta, R.; de Candia, P.; Olivieri, F.; Matacchione, G.; Giuliani, A.; Rippo, M.R.; Tagliabue, E.; Mancino, M.; Rispoli, F.; Ferroni, S.; et al. Anti-inflammatory effect of SGLT-2 inhibitors via uric acid and insulin. Cell. Mol. Life Sci. 2022, 79, 273. [Google Scholar] [CrossRef] [PubMed]
- Endo, A.; Hirose, T.; Sato, S.; Ito, H.; Takahashi, C.; Ishikawa, R.; Kamada, A.; Oba-Yabana, I.; Kimura, T.; Takahashi, K.; et al. Sodium glucose cotransporter 2 inhibitor suppresses renal injury in rats with renal congestion. Hypertens. Res. 2024, 47, 33–45. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Guo, X.; Yan, G.; Zhang, Y.; Yao, Y.; Qiao, Y.; Wang, D.; Chen, G.; Zhang, W.; Tang, C.; et al. Dapagliflozin Attenuates Contrast-induced Acute Kidney Injury by Regulating the HIF-1alpha/HE4/NF-kappaB Pathway. J. Cardiovasc. Pharmacol. 2022, 79, 904–913. [Google Scholar] [CrossRef] [PubMed]
- Shakour, N.; Karami, S.; Iranshahi, M.; Butler, A.E.; Sahebkar, A. Antifibrotic effects of sodium-glucose cotransporter-2 inhibitors: A comprehensive review. Diabetes Metab. Syndr. 2024, 18, 102934. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Li, T.; Xian, J.; Chen, J.; Huang, Y.; Zhang, Q.; Lin, X.; Lu, H.; Lin, Y. SGLT2 inhibitor dapagliflozin attenuates cardiac fibrosis and inflammation by reverting the HIF-2alpha signaling pathway in arrhythmogenic cardiomyopathy. FASEB J. 2022, 36, e22410. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.H.; Kim, S.H.; Kang, J.M.; Heo, J.H.; Kim, D.J.; Park, S.H.; Sung, M.; Kim, J.; Oh, J.; Yang, D.H.; et al. Empagliflozin attenuates diabetic tubulopathy by improving mitochondrial fragmentation and autophagy. Am. J. Physiol. Renal Physiol. 2019, 317, F767–F780. [Google Scholar] [CrossRef]
- Heerspink, H.J.L.; Kosiborod, M.; Inzucchi, S.E.; Cherney, D.Z.I. Renoprotective effects of sodium-glucose cotransporter-2 inhibitors. Kidney Int. 2018, 94, 26–39. [Google Scholar] [CrossRef]
- Panchapakesan, U.; Pegg, K.; Gross, S.; Komala, M.G.; Mudaliar, H.; Forbes, J.; Pollock, C.; Mather, A. Effects of SGLT2 inhibition in human kidney proximal tubular cells--renoprotection in diabetic nephropathy? PLoS ONE 2013, 8, e54442. [Google Scholar] [CrossRef]
- Sano, M.; Takei, M.; Shiraishi, Y.; Suzuki, Y. Increased Hematocrit During Sodium-Glucose Cotransporter 2 Inhibitor Therapy Indicates Recovery of Tubulointerstitial Function in Diabetic Kidneys. J. Clin. Med. Res. 2016, 8, 844–847. [Google Scholar] [CrossRef]
- Singh, D.K.; Winocour, P.; Farrington, K. Erythropoietic stress and anemia in diabetes mellitus. Nat. Rev. Endocrinol. 2009, 5, 204–210. [Google Scholar] [CrossRef]
- Baker, W.L.; Smyth, L.R.; Riche, D.M.; Bourret, E.M.; Chamberlin, K.W.; White, W.B. Effects of sodium-glucose co-transporter 2 inhibitors on blood pressure: A systematic review and meta-analysis. J. Am. Soc. Hypertens. 2014, 8, 262–275. [Google Scholar] [CrossRef]
- Sano, M.; Goto, S. Possible Mechanism of Hematocrit Elevation by Sodium Glucose Cotransporter 2 Inhibitors and Associated Beneficial Renal and Cardiovascular Effects. Circulation 2019, 139, 1985–1987. [Google Scholar] [CrossRef] [PubMed]
- Papaetis, G.S. SGLT2 inhibitors, intrarenal hypoxia and the diabetic kidney: Insights into pathophysiological concepts and current evidence. Arch. Med. Sci. Atheroscler. Dis. 2023, 8, e155–e168. [Google Scholar] [CrossRef] [PubMed]
- Heyman, S.N.; Armaly, Z.; Hamo-Giladi, D.B.; Abassi, Z. Novel perspectives regarding the physiologic mechanisms by which gliflozins induce reticulocytosis and erythrocytosis. Am. J. Physiol. Endocrinol. Metab. 2023, 325, E621–E623. [Google Scholar] [CrossRef]
- Hodrea, J.; Balogh, D.B.; Hosszu, A.; Lenart, L.; Besztercei, B.; Koszegi, S.; Sparding, N.; Genovese, F.; Wagner, L.J.; Szabo, A.J.; et al. Reduced O-GlcNAcylation and tubular hypoxia contribute to the antifibrotic effect of SGLT2 inhibitor dapagliflozin in the diabetic kidney. Am. J. Physiol. Renal Physiol. 2020, 318, F1017–F1029. [Google Scholar] [CrossRef]
- Chiarelli, F.; Gaspari, S.; Marcovecchio, M.L. Role of growth factors in diabetic kidney disease. Horm. Metab. Res. 2009, 41, 585–593. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.L.; Pan, J.S.; Lu, Y.P.; Sun, P.; Han, J. Inflammatory signaling and cellular senescence. Cell. Signal. 2009, 21, 378–383. [Google Scholar] [CrossRef] [PubMed]
- Rigalleau, V.; Garcia, M.; Lasseur, C.; Laurent, F.; Montaudon, M.; Raffaitin, C.; Barthe, N.; Beauvieux, M.C.; Vendrely, B.; Chauveau, P.; et al. Large kidneys predict poor renal outcome in subjects with diabetes and chronic kidney disease. BMC Nephrol. 2010, 11, 3. [Google Scholar] [CrossRef] [PubMed]
- Satriano, J.; Mansoury, H.; Deng, A.; Sharma, K.; Vallon, V.; Blantz, R.C.; Thomson, S.C. Transition of kidney tubule cells to a senescent phenotype in early experimental diabetes. Am. J. Physiol. Cell Physiol. 2010, 299, C374–C380. [Google Scholar] [CrossRef]
- Thomas, M.C.; Burns, W.C.; Cooper, M.E. Tubular changes in early diabetic nephropathy. Adv. Chronic Kidney Dis. 2005, 12, 177–186. [Google Scholar] [CrossRef]
- Thomson, S.C.; Deng, A.; Bao, D.; Satriano, J.; Blantz, R.C.; Vallon, V. Ornithine decarboxylase, kidney size, and the tubular hypothesis of glomerular hyperfiltration in experimental diabetes. J. Clin. Investig. 2001, 107, 217–224. [Google Scholar] [CrossRef]
- Vallon, V.; Komers, R. Pathophysiology of the diabetic kidney. Compr. Physiol. 2011, 1, 1175–1232. [Google Scholar] [CrossRef]
- Kim, M.N.; Moon, J.H.; Cho, Y.M. Sodium-glucose cotransporter-2 inhibition reduces cellular senescence in the diabetic kidney by promoting ketone body-induced NRF2 activation. Diabetes Obes. Metab. 2021, 23, 2561–2571. [Google Scholar] [CrossRef]
- La Grotta, R.; Frige, C.; Matacchione, G.; Olivieri, F.; de Candia, P.; Ceriello, A.; Prattichizzo, F. Repurposing SGLT-2 Inhibitors to Target Aging: Available Evidence and Molecular Mechanisms. Int. J. Mol. Sci. 2022, 23, 12325. [Google Scholar] [CrossRef] [PubMed]
- Ge, X.; Wang, L.; Fei, A.; Ye, S.; Zhang, Q. Research progress on the relationship between autophagy and chronic complications of diabetes. Front. Physiol. 2022, 13, 956344. [Google Scholar] [CrossRef] [PubMed]
- Tseng, C.H.; Shah, K.M.; Chiu, I.J.; Hsiao, L.L. The Role of Autophagy in Type 2 Diabetic Kidney Disease Management. Cells 2023, 12, 2691. [Google Scholar] [CrossRef] [PubMed]
- Juszczak, F.; Caron, N.; Mathew, A.V.; Decleves, A.E. Critical Role for AMPK in Metabolic Disease-Induced Chronic Kidney Disease. Int. J. Mol. Sci. 2020, 21, 7994. [Google Scholar] [CrossRef]
- Saad, R.; Tadmor, H.; Ertracht, O.; Nakhoul, N.; Nakhoul, F.; Evgeny, F.; Atar, S. The Molecular Effects of SGLT2i Empagliflozin on the Autophagy Pathway in Diabetes Mellitus Type 2 and Its Complications. J. Diabetes Res. 2022, 2022, 8337823. [Google Scholar] [CrossRef]
- Xu, J.; Kitada, M.; Ogura, Y.; Liu, H.; Koya, D. Dapagliflozin Restores Impaired Autophagy and Suppresses Inflammation in High Glucose-Treated HK-2 Cells. Cells 2021, 10, 1457. [Google Scholar] [CrossRef]
- Hawley, S.A.; Ford, R.J.; Smith, B.K.; Gowans, G.J.; Mancini, S.J.; Pitt, R.D.; Day, E.A.; Salt, I.P.; Steinberg, G.R.; Hardie, D.G. The Na+/Glucose Cotransporter Inhibitor Canagliflozin Activates AMPK by Inhibiting Mitochondrial Function and Increasing Cellular AMP Levels. Diabetes 2016, 65, 2784–2794. [Google Scholar] [CrossRef] [PubMed]
- Korbut, A.I.; Taskaeva, I.S.; Bgatova, N.P.; Muraleva, N.A.; Orlov, N.B.; Dashkin, M.V.; Khotskina, A.S.; Zavyalov, E.L.; Konenkov, V.I.; Klein, T.; et al. SGLT2 Inhibitor Empagliflozin and DPP4 Inhibitor Linagliptin Reactivate Glomerular Autophagy in db/db Mice, a Model of Type 2 Diabetes. Int. J. Mol. Sci. 2020, 21, 2987. [Google Scholar] [CrossRef]
- Li, X.; Gao, L.; Li, X.; Xia, J.; Pan, Y.; Bai, C. Autophagy, Pyroptosis and Ferroptosis are Rising Stars in the Pathogenesis of Diabetic Nephropathy. Diabetes Metab. Syndr. Obes. 2024, 17, 1289–1299. [Google Scholar] [CrossRef] [PubMed]
- Cannon, C.P.; Pratley, R.; Dagogo-Jack, S.; Mancuso, J.; Huyck, S.; Masiukiewicz, U.; Charbonnel, B.; Frederich, R.; Gallo, S.; Cosentino, F.; et al. Cardiovascular Outcomes with Ertugliflozin in Type 2 Diabetes. N. Engl. J. Med. 2020, 383, 1425–1435. [Google Scholar] [CrossRef] [PubMed]
- The Nuffield Department of Population Health Renal Studies Group; SGLT2 inhibitor Meta-Analysis Cardio-Renal Trialists’ Consortium. Impact of diabetes on the effects of sodium glucose co-transporter-2 inhibitors on kidney outcomes: Collaborative meta-analysis of large placebo-controlled trials. Lancet 2022, 400, 1788–1801. [Google Scholar] [CrossRef]
- Podesta, M.A.; Sabiu, G.; Galassi, A.; Ciceri, P.; Cozzolino, M. SGLT2 Inhibitors in Diabetic and Non-Diabetic Chronic Kidney Disease. Biomedicines 2023, 11, 279. [Google Scholar] [CrossRef] [PubMed]
- Tian, L.; Ai, S.; Zheng, H.; Yang, H.; Zhou, M.; Tang, J.; Liu, W.; Zhao, W.; Wang, Y. Cardiovascular and renal outcomes with sodium glucose co-transporter 2 inhibitors in patients with type 2 diabetes mellitus: A system review and network meta-analysis. Front. Pharmacol. 2022, 13, 986186. [Google Scholar] [CrossRef]
- Heerspink, H.J.L.; Karasik, A.; Thuresson, M.; Melzer-Cohen, C.; Chodick, G.; Khunti, K.; Wilding, J.P.H.; Garcia Rodriguez, L.A.; Cea-Soriano, L.; Kohsaka, S.; et al. Kidney outcomes associated with use of SGLT2 inhibitors in real-world clinical practice (CVD-REAL 3): A multinational observational cohort study. Lancet Diabetes Endocrinol. 2020, 8, 27–35. [Google Scholar] [CrossRef]
- Cahn, A.; Melzer-Cohen, C.; Pollack, R.; Chodick, G.; Shalev, V. Acute renal outcomes with sodium-glucose co-transporter-2 inhibitors: Real-world data analysis. Diabetes Obes. Metab. 2019, 21, 340–348. [Google Scholar] [CrossRef]
- Kurki, S.; Halla-Aho, V.; Haussmann, M.; Lahdesmaki, H.; Leinonen, J.V.; Koskinen, M. A comparative study of clinical trial and real-world data in patients with diabetic kidney disease. Sci. Rep. 2024, 14, 1731. [Google Scholar] [CrossRef]
- Anders, H.J.; Davis, J.M.; Thurau, K. Nephron Protection in Diabetic Kidney Disease. N. Engl. J. Med. 2016, 375, 2096–2098. [Google Scholar] [CrossRef] [PubMed]
- Lim, B.J.; Yang, J.W.; Zou, J.; Zhong, J.; Matsusaka, T.; Pastan, I.; Zhang, M.Z.; Harris, R.C.; Yang, H.C.; Fogo, A.B. Tubulointerstitial fibrosis can sensitize the kidney to subsequent glomerular injury. Kidney Int. 2017, 92, 1395–1403. [Google Scholar] [CrossRef] [PubMed]
- Rajasekeran, H.; Cherney, D.Z.; Lovshin, J.A. Do effects of sodium-glucose cotransporter-2 inhibitors in patients with diabetes give insight into potential use in non-diabetic kidney disease? Curr. Opin. Nephrol. Hypertens. 2017, 26, 358–367. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Garvin, J.L.; Liu, R.; Carretero, O.A. Role of macula densa adenosine triphosphate (ATP) in tubuloglomerular feedback. Kidney Int. 2004, 66, 1479–1485. [Google Scholar] [CrossRef]
- van Bommel, E.J.M.; Muskiet, M.H.A.; van Baar, M.J.B.; Tonneijck, L.; Smits, M.M.; Emanuel, A.L.; Bozovic, A.; Danser, A.H.J.; Geurts, F.; Hoorn, E.J.; et al. The renal hemodynamic effects of the SGLT2 inhibitor dapagliflozin are caused by post-glomerular vasodilatation rather than pre-glomerular vasoconstriction in metformin-treated patients with type 2 diabetes in the randomized, double-blind RED trial. Kidney Int. 2020, 97, 202–212. [Google Scholar] [CrossRef]
- Castoldi, G.; Carletti, R.; Barzaghi, F.; Meani, M.; Zatti, G.; Perseghin, G.; Di Gioia, C.R.T.; Zerbini, G. Sodium Glucose Cotransporter-2 Inhibitors in Non-Diabetic Kidney Disease: Evidence in Experimental Models. Pharmaceuticals 2024, 17, 362. [Google Scholar] [CrossRef] [PubMed]
- Brunner, H.R.; Gavras, H.; Waeber, B.; Kershaw, G.R.; Turini, G.A.; Vukovich, R.A.; McKinstry, D.N.; Gavras, I. Oral angiotensin-converting enzyme inhibitor in long-term treatment of hypertensive patients. Ann. Intern. Med. 1979, 90, 19–23. [Google Scholar] [CrossRef]
- Burnier, M. Angiotensin II type 1 receptor blockers. Circulation 2001, 103, 904–912. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, D.; Kuwabara, T.; Mukoyama, M. Need to continue or discontinue RAS inhibitors as CKD stage advances? Any alternative? Hypertens. Res. 2023, 46, 2048–2050. [Google Scholar] [CrossRef] [PubMed]
- Werner, C.; Baumhakel, M.; Teo, K.K.; Schmieder, R.; Mann, J.; Unger, T.; Yusuf, S.; Bohm, M. RAS blockade with ARB and ACE inhibitors: Current perspective on rationale and patient selection. Clin. Res. Cardiol. 2008, 97, 418–431. [Google Scholar] [CrossRef]
- Burrell, L.M.; Johnston, C.I.; Tikellis, C.; Cooper, M.E. ACE2, a new regulator of the renin-angiotensin system. Trends Endocrinol. Metab. 2004, 15, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Stanton, R.C. Sodium glucose transport 2 (SGLT2) inhibition decreases glomerular hyperfiltration: Is there a role for SGLT2 inhibitors in diabetic kidney disease? Circulation 2014, 129, 542–544. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.S.; Chang, S.H.; Chang, G.J.; Liu, J.R.; Chan, Y.H.; Lee, H.F.; Wen, M.S.; Chen, W.J.; Yeh, Y.H.; Kuo, C.T.; et al. A comparison between angiotensin converting enzyme inhibitors and angiotensin receptor blockers on end stage renal disease and major adverse cardiovascular events in diabetic patients: A population-based dynamic cohort study in Taiwan. Cardiovasc. Diabetol. 2016, 15, 56. [Google Scholar] [CrossRef]
- Zou, H.; Zhou, B.; Xu, G. SGLT2 inhibitors: A novel choice for the combination therapy in diabetic kidney disease. Cardiovasc. Diabetol. 2017, 16, 65. [Google Scholar] [CrossRef] [PubMed]
- Gerard, A.O.; Laurain, A.; Sicard, A.; Merino, D.; Pathak, A.; Drici, M.D.; Favre, G.; Esnault, V.L.M. New Strategies for Volume Control in Patients with Diabetes Mellitus, a Narrative Review. Pharmaceutics 2022, 14, 1569. [Google Scholar] [CrossRef] [PubMed]
- Unnikrishnan, A.G.; Kalra, S.; Purandare, V.; Vasnawala, H. Genital Infections with Sodium Glucose Cotransporter-2 Inhibitors: Occurrence and Management in Patients with Type 2 Diabetes Mellitus. Indian. J. Endocrinol. Metab. 2018, 22, 837–842. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Lee, S.H.; Cho, J.H.; Lee, H.; Yim, H.W.; Yoon, K.H.; Kim, H.S. Discontinuation rate and reason for discontinuation after sodium-glucose cotransporter 2 inhibitor prescription in real clinical practice. J. Clin. Pharm. Ther. 2020, 45, 1271–1277. [Google Scholar] [CrossRef]
- Tsai, W.C.; Hsu, S.P.; Chiu, Y.L.; Yang, J.Y.; Pai, M.F.; Ko, M.J.; Tu, Y.K.; Hung, K.Y.; Chien, K.L.; Peng, Y.S.; et al. Cardiovascular and renal efficacy and safety of sodium-glucose cotransporter-2 inhibitors in patients without diabetes: A systematic review and meta-analysis of randomised placebo-controlled trials. BMJ Open 2022, 12, e060655. [Google Scholar] [CrossRef]
- Mascolo, A.; Di Napoli, R.; Balzano, N.; Cappetta, D.; Urbanek, K.; De Angelis, A.; Scisciola, L.; Di Meo, I.; Sullo, M.G.; Rafaniello, C.; et al. Safety profile of sodium glucose co-transporter 2 (SGLT2) inhibitors: A brief summary. Front. Cardiovasc. Med. 2022, 9, 1010693. [Google Scholar] [CrossRef]
- Kidney Disease: Improving Global Outcomes (KDIGO) CKD Work Group. KDIGO 2024 Clinical Practice Guideline for the Evaluation and Management of Chronic Kidney Disease. Kidney Int. 2024, 105, S117–S314. [Google Scholar] [CrossRef]
- Gong, C.; Shen, S.C.; Zhang, K.; Zhou, L.; Shen, J.J.; Zhao, J.Y.; Ding, S.G.; Ma, L.K.; Gao, H. Association of sodium-glucose cotransporter 2 inhibitors with cardiovascular outcome and safety events: A meta-analysis of randomized controlled clinical trials. Front. Cardiovasc. Med. 2022, 9, 926979. [Google Scholar] [CrossRef] [PubMed]
- Milder, T.Y.; Stocker, S.L.; Day, R.O.; Greenfield, J.R. Potential Safety Issues with Use of Sodium-Glucose Cotransporter 2 Inhibitors, Particularly in People with Type 2 Diabetes and Chronic Kidney Disease. Drug Saf. 2020, 43, 1211–1221. [Google Scholar] [CrossRef]
- Kani, R.; Watanabe, A.; Miyamoto, Y.; Ejiri, K.; Iwagami, M.; Takagi, H.; Slipczuk, L.; Tsugawa, Y.; Aikawa, T.; Kuno, T. Comparison of Effectiveness Among Different Sodium-Glucose Cotransoporter-2 Inhibitors According to Underlying Conditions: A Network Meta-Analysis of Randomized Controlled Trials. J. Am. Heart Assoc. 2024, 13, e031805. [Google Scholar] [CrossRef]
- American Diabetes Association Professional Practice Committee. 11. Chronic Kidney Disease and Risk Management: Standards of Care in Diabetes-2024. Diabetes Care 2024, 47, S219–S230. [Google Scholar] [CrossRef] [PubMed]
- American Diabetes Association Professional Practice Committee. 9. Pharmacologic Approaches to Glycemic Treatment: Standards of Care in Diabetes-2024. Diabetes Care 2024, 47, S158–S178. [Google Scholar] [CrossRef] [PubMed]
- Marx, N.; Federici, M.; Schutt, K.; Muller-Wieland, D.; Ajjan, R.A.; Antunes, M.J.; Christodorescu, R.M.; Crawford, C.; Di Angelantonio, E.; Eliasson, B.; et al. 2023 ESC Guidelines for the management of cardiovascular disease in patients with diabetes. Eur. Heart J. 2023, 44, 4043–4140. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
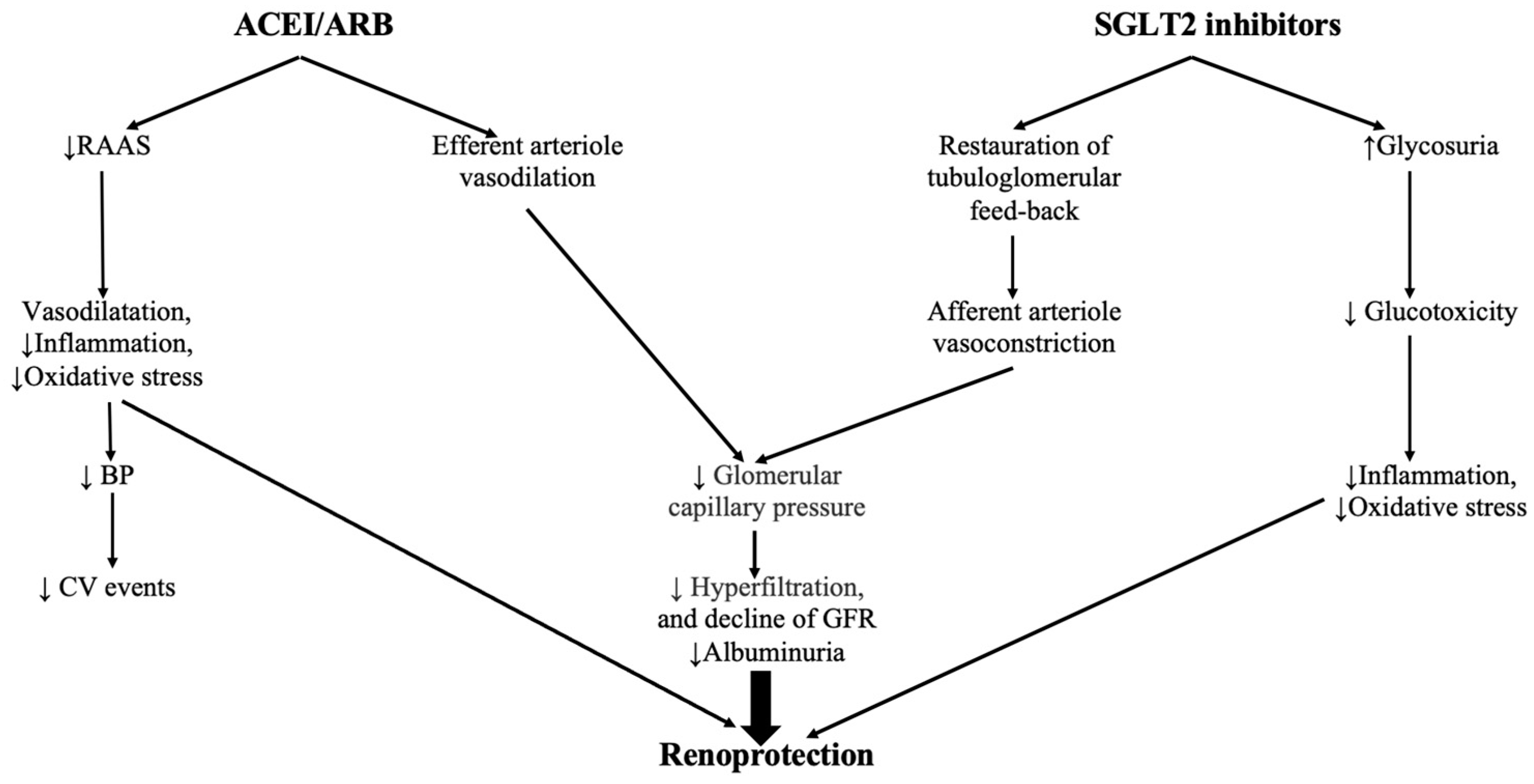
| Category | Nephroprotective Effects of SGLT2i |
|---|---|
| Metabolic effects | Reduction in plasma glucose levels Reduction of glucotoxicity Reduction of body weight Reduction of lipotoxicity Reduction of triglycerides and small-dense LDLc Improvement of insulin sensitivity Reduction of hepatic steatosis Reduction of plasma uric acid levels Metabolic reprogramming (restoration of energy efficiency by switching glucose to more energy efficient metabolites) |
| Hemodynamic effects | Natriuresis Improvement of salt-sensitive hypertension Reduction of blood pressure Improvement of sympathetic nerve hyperactivity Reduction of volume overload Modulation of the tubuloglomerular feedback |
| Direct nephroprotective effects | Inhibition of the Na-H exchanger Reduction of endothelial dysfunction and oxidative stress Reduction of renal hypoxia Improvement of mitochondrial dysfunction Improvement of podocyte loss Reduction of albuminuria Reduction of renal fibrosis Reduction of inflammation Improvement of erythropoiesis Reduction of tubular senescence Improvement of renal autophagy Reduction of glomerular damage |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iordan, L.; Gaita, L.; Timar, R.; Avram, V.; Sturza, A.; Timar, B. The Renoprotective Mechanisms of Sodium-Glucose Cotransporter-2 Inhibitors (SGLT2i)—A Narrative Review. Int. J. Mol. Sci. 2024, 25, 7057. https://doi.org/10.3390/ijms25137057
Iordan L, Gaita L, Timar R, Avram V, Sturza A, Timar B. The Renoprotective Mechanisms of Sodium-Glucose Cotransporter-2 Inhibitors (SGLT2i)—A Narrative Review. International Journal of Molecular Sciences. 2024; 25(13):7057. https://doi.org/10.3390/ijms25137057
Chicago/Turabian StyleIordan, Liana, Laura Gaita, Romulus Timar, Vlad Avram, Adrian Sturza, and Bogdan Timar. 2024. "The Renoprotective Mechanisms of Sodium-Glucose Cotransporter-2 Inhibitors (SGLT2i)—A Narrative Review" International Journal of Molecular Sciences 25, no. 13: 7057. https://doi.org/10.3390/ijms25137057