1. Introduction
Cancer cachexia is a multifactorial syndrome characterized by persistent skeletal muscle wasting and weight loss [
1]. It can also be considered a metabolic syndrome, as it causes disorders and imbalances in glucose, amino acids, fatty acids, bile acids, ke-tone bodies, steroids, and mitochondrial energy metabolism [
2,
3,
4].
Cancer cachexia occurs in 80% of patients with advanced cancer and is associated with 40% of cancer deaths [
5,
6,
7,
8]. Cancer cachexia leads to decreased treatment resistance and poor disease prognosis [
9,
10,
11,
12]. Sarcopenia, defined as a loss of muscle mass and function, is the most important phenotype of cancer cachexia. Overcoming cancer sarcopenia has become the focus of cancer treatment in improving treatment response, patient prognosis, and quality of life [
1,
13,
14].
At the root of metabolic disorders in cancer sarcopenia is mitochondrial dysfunction [
15], and many fundamental cellular processes in muscle tissue, such as apoptosis, autophagy, reactive oxygen species signaling, and protein balance, maintained by mi-tochondria, are impaired [
15]. In particular, oxidative stress and imbalanced redox in mitochondria form a vicious cycle, leading to intensified mitochondrial separation, suppression of mitochondrial fusion/fission, inhibition of the electron transport chain, reduced ATP production, increased mitochondrial DNA damage, and impaired mitochondrial biogenesis, playing an important role in muscle tissue damage [
6,
16,
17]. Sat-ellite cells, which play a stem cell-like role in skeletal muscle metabolism, are impaired in function by inflammation, oxidative stress, and fibrosis [
18].
Loss of muscle mass and strength is common in older adults and is associated with increased dependency, frailty, and mortality [
19,
20]. Thus, aging is an important cause of sarcopenia [
21,
22], but the underlying molecular mechanisms have similarities with cancer sarcopenia. Skeletal muscle loss occurs due to an imbalance between protein synthesis and degradation, increased apoptosis of muscle cells, and reduced regenerative capacity [
22]. In terms of mitochondrial dysfunction, both share a common phenotype, including decreased OXPHOS, increased mitochondrial ROS, and impaired mitochondrial quality control [
6,
23]. Furthermore, mitochondrial DNA damage has been detected in aging, cancer, and neurodegenerative diseases, and has been noted as a common cause of tissue damage [
24]. Although the involvement of inflammatory cytokines plays an important role in cancer sarcopenia [
25,
26], increased levels of inflammatory cytokines have also been observed in older adults and are correlated with sarcopenia [
27].
Considering these findings, it is hypothesized that cancer-associated sarcopenia is a state of accelerated skeletal muscle aging. Therefore, in this study, we aimed to clari-fy the involvement of aging in cancer-associated sarcopenia.
3. Discussion
In this study, we investigated the relationship between cancer-related sarcopenia and skeletal muscle aging. We observed changes in various aging markers in skeletal muscles in both in vitro and mouse cachexia models, suggesting the promotion of cancer-related aging.
We evaluated markers related to aging in skeletal muscle tissue. A decline in differentiation potential is associated with decreased
PAX7 expression [
32] and increased
MYH8 expression [
33]. Aging increases collagen I, III, and VI levels in both slow and fast muscles [
34]. As indications of mitochondrial changes, decreased mitochondria levels in muscle cells; decreased ETC complexes I, III, IV, and V; decreased mitochondrial transcription factor A (TFAM) levels; OXPHOS inhibition; increased oxidative stress, decreased autophagy, and increased apoptosis have been reported [
35,
36,
37,
38]. Telomeres shorten with age and chronic stress promotes these changes [
39,
40]. AGE are produced non-enzymatically from glucose under oxidative stress but are difficult to degrade; therefore, levels increase with aging and chronic diseases such as diabetes [
41]. Although whether such changes in skeletal muscle phenotypes observed in aging were promoted in tumor-bearing models was previously unclear, the levels of all aging markers examined increased in skeletal muscle in in vitro and in vivo cancer cachexia models in the present study. Although determining whether many of these markers are age-specific is difficult, βGAL is the most widely used biomarker for aging and senescent cells [
42] and may be a cause of aging [
43]. These aging phenotypes may be promoted in cancer cachexia. Comparison of the aged and cachectic mouse models suggested that 8 weeks of age in the cachectic model was equivalent to 110 weeks of age in the aged model, suggesting that the cancerous environment promoted rapid aging of skeletal muscle.
The results of the present study revealed that cachexia causes DNA deletion and dysfunction of skeletal muscle mitochondria. Recent reports have demonstrated the association of mitochondrial DNA deletion with aging. Mitochondrial DNA deletions can be as large as several kilobases and are associated with human pathologies including cancer, aging, and mitochondrial diseases [
44]. Mitochondrial DNA deletions lead to the loss of ETC complexes and a general decline in mitochondrial function, including OXPHOS [
44]. High-definition sequencing has revealed that various mitochondrial DNA deletions frequently occur with age [
31]. In the present study, we observed extensive deletions in the major arc of mitochondrial DNA in cachectic skeletal muscle, leading to mutations in ETC complexes I, IV, and V and suppression of OXPHOS. Simultaneously, mitochondrial ROS levels increased and mitophagy was suppressed. The fact that doxorubicin, which damages mitochondrial DNA, accelerates aging in mice also suggests a relationship between mitochondrial DNA abnormalities and aging [
45,
46].
Our results also showed that large-scale deletions of mitochondrial DNA increase with age; however, normal mitochondrial DNA signals were also present, indicating mitochondrial DNA heteroplasmy. Heteroplasmy is the phenomenon of mutations coexisting with normal alleles [
47]. In this case, OXPHOS disorders are only observed when the proportion of abnormal mitochondrial DNA exceeds a certain threshold [
48]. The relationships between mitochondrial DNA heteroplasmy and aging, cancer, and neurodegenerative diseases have attracted considerable attention [
49,
50]. In the present study, mitochondrial DNA deletions occurred within a relatively short period in the cachexia model, which may be related to a decrease in mitochondrial quality control, leading to the establishment of abnormal mitochondrial DNA. Although we were unable to examine this, mitochondrial DNA point mutations, base modifications, and copy number changes are also mitochondrial DNA changes associated with aging. Future studies examining these changes in cachexia are required.
We showed that increased mitochondrial oxidative stress caused by inflammatory cytokines is involved in the formation of mitochondrial DNA deletions. The causes of large-scale mitochondrial DNA deletions remain largely unknown. Abnormalities in mitochondrial DNA replication, repair, and crosstalk between these pathways are thought to be involved in the generation of large-scale deletions [
44]. Mitochondrial DNA is vulnerable to oxidative stress, which induces mitochondrial DNA alterations [
50]. Our data also suggest that the generation of mitochondrial ROS by inflammatory cytokines is involved in the development of mitochondrial DNA deletions.
Mitochondrial DNA deletions increase exponentially with age and maps to a wider region of the mitochondrial genome than previously reported [
31]. From 50 to 86 years of age, the mitochondrial DNA deletion frequency increases from 0.008% to 0.15% [
30]. Furthermore, in skeletal muscle, increasing mitochondrial DNA deletion frequency is correlated with age-related muscle fiber loss and accelerated mortality [
51]. Mitochondrial dysfunction induces systemic inflammation by excreting mitochondrial DNA into the cytoplasm or extracellular space and by promoting the secretion of inflammatory cytokines through the nucleotide-binding domain, leucine-rich-containing family pyrin domain-containing-3 (NLRP3) inflammasome in non-immuneand immune cells [
52,
53,
54,
55]. In contrast, our data suggest that inflammatory cytokines may increase mitochondrial ROS levels and induce mitochondrial DNA damage. Recent reports have shown that the inhibition of the inflammatory cytokine pathway reduces mitochondrial dysfunction [
56]. Thus, mitochondrial dysfunction and inflammatory cytokines may lead to a vicious cycle that exacerbates systemic inflammation and mitochondrial damage. Furthermore, the results of the present study suggest that this cycle promotes tissue aging. In the future, controlling damage-related molecular patterns, including those of inflammatory cytokines and mitochondrial DNA, may help suppress tissue aging.
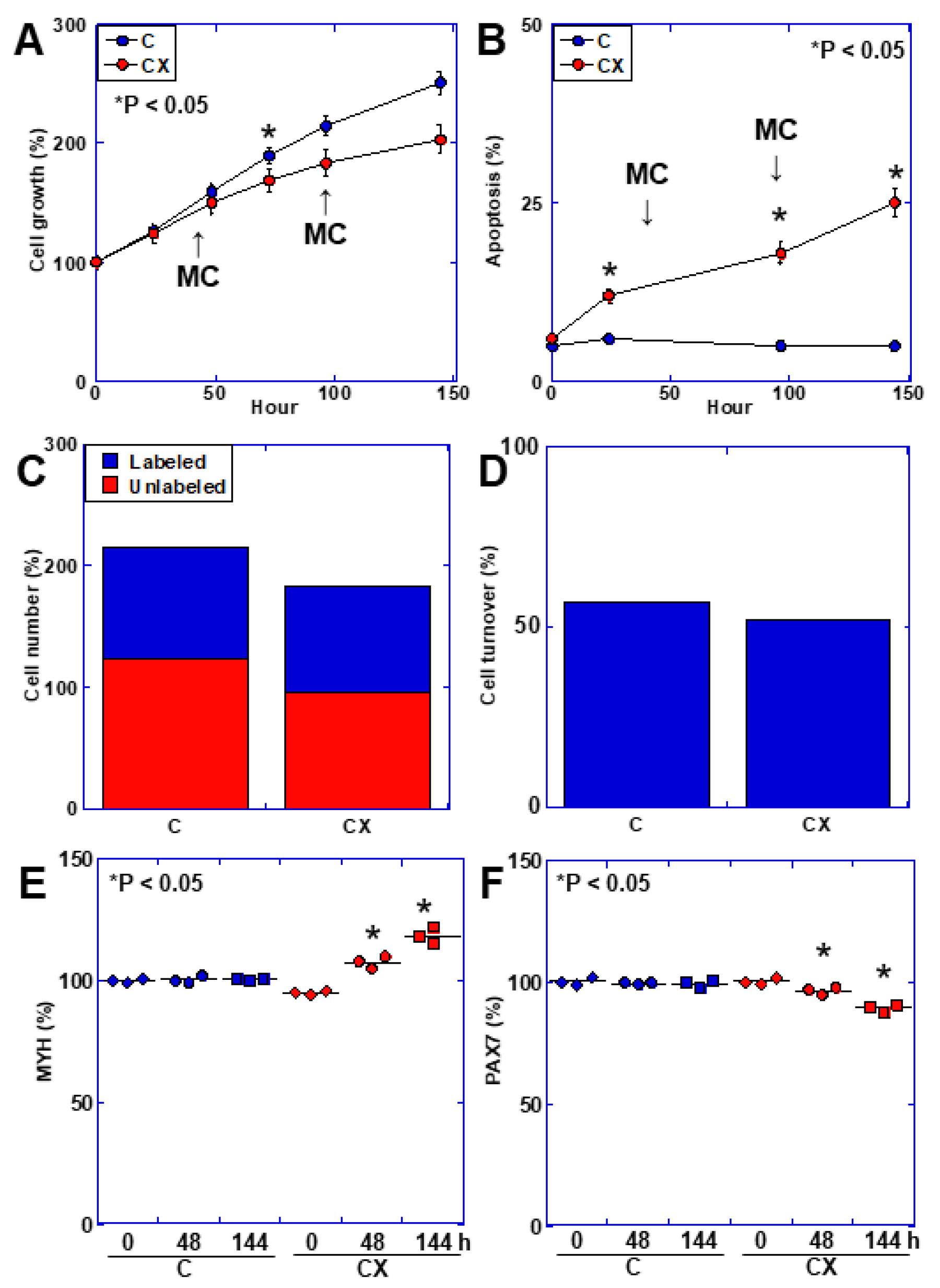
Our data showed that both in vitro cachexia models and HMGB1 treatment reduced autophagy and the expression of mitophagy-related proteins such as PINK1 and Parkin. Autophagy is an important mechanism for mitochondrial quality control, and its reduction leads to the abnormal accumulation of injured mitochondria in cancer and aging [
22,
57]. In this study, autophagy was suppressed in cancer cachexia and aging, despite the occurrence of mitochondrial injury that would normally promote mitophagy, such as increased mitochondrial ROS production, decreased MMP, decreased OXPHOS, and mitochondrial DNA deletion. This is thought to perpetuate mitochondrial abnormalities and establish the phenotype of sarcopenia. Furthermore, it has been suggested that mitochondrial dysfunction and inflammation mutually exacerbate each other, leading to the inhibition of autophagy [
15]. However, the details of the mechanism have not yet been fully elucidated [
15]. Our results show that HMGB1 not only causes mitochondrial dysfunction but also inhibits autophagy and reduces mitochondrial turnover.
In this study, we clarified the commonality of phenotypes between cancer sarcopenia and aging sarcopenia and the underlying mitochondrial disorder. From these results, we considered that cancer sarcopenia is a state in which skeletal muscle aging is accelerated. However, considering that the aging phenotypes are multimodal and cannot be determined solely by limited phenotype, our conclusions are considered to be limited. However, this study was able to shed light on the molecular basis of both aging and cancer, such as the fact that mitochondrial DNA deletion is common to both aging and cancer. In the future, a more extensive comparison of the two conditions will be necessary using various omics analyses. Another limitation of this study is that it was conducted using a culture system and a mouse model, but human analysis was lacking. To prove that the findings of this study can be extrapolated to humans, studies using human samples are essential. By approaching the nature of cancer sarcopenia from the perspective of aging, it is hoped that we can deepen our understanding of not only cancer sarcopenia but also aging itself, and develop treatments for it.
4. Materials and Methods
4.1. Animals
The aged mouse models were five male BALB/c mice aged 4, 55, and 110 weeks purchased from SLC Japan (Shizuoka, Japan). The animals were maintained in a pathogen-free animal facility under a 12/12 h light/dark cycle in a temperature (22 °C)- and humidity-controlled environment, in accordance with the institutional guidelines approved by the Committee for Animal Experimentation of Nara Medical University, Kashihara, Japan, following current regulations and standards of the Japanese Ministry of Health, Labor and Welfare (approval no. 12777, 20 April 2020). The animals were acclimated to their housing for 7 days before the start of the experiment.
The mouse cachexia model was based on previous reports of the intraperitoneal inoculation of CT26 cancer cells (5 × 10
6 cells) into syngeneic BALB/c mice (
n = 5) [
28]. Five mice were used as starting controls, and another five were used as non-tumor controls at the time of euthanasia. The mice were euthanized under anesthesia 4 weeks after inoculation. The skeletal muscles were prepared as we previously described [
28], and the quadriceps femoris muscle (QFM) was separated from the bones.
4.2. Cell Lines and Reagents
The CT26 mouse colon cancer cell line was a gift from Professor I. J. Fidler (MD Anderson Cancer Center, Houston, TX, USA) [
58]. C2C12 mouse myoblasts were purchased from Dainihon Pharmacy Co. (Tokyo, Japan). The cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Wako Pure Chemical Industries, Ltd., Osaka, Japan) supplemented with 10% fetal bovine serum (Sigma-Aldrich Chemical Co., St. Louis, MO, USA). Human recombinant HMGB1 (Biolegend, San Diego, CA, USA), mouse TNFα (Cell Signaling, Danvers, MA, USA), anti-HMGB1 antibody (clone 3E8, Biolegend), and anti-mouse TNFα antibody (Neutrakine, Proteintech, Rosemont, IL, USA) were purchased from commercial sources.
4.3. In Vitro Cachexia Model
For the in vitro cachexia model [
59], ascites of CT26 cell-inoculated BALB/c mice were added to supplemented regular medium at 20%
v/
v. As a control, regular medium was added to the culture medium of CT26 cells at 20%
v/
v.
4.4. Protein Extraction
Proteins were extracted from QCM stored at −8 °C as we previously described [
28]. Whole-cell lysates were prepared as previously described using radio-immunoprecipitation assay (RIPA) buffer containing 0.1% SDS (Thermo Fisher Scientific, Tokyo, Japan) [
60]. Protein assays were performed using a Protein Assay Rapid Kit (Wako Pure Chemical Corporation, Osaka, Japan).
4.5. Western Blot Analysis
Protein lysates (25 μg) were separated on 12.5% sodium dodecyl sulfate-polyacrylamide gels, followed by electrotransfer onto a nitrocellulose filter. The membranes were then incubated with primary and peroxidase-conjugated immunoglobulin G (IgG) antibodies (Agilent Technologies, Santa Clara, CA, USA). Immune complexes were detected using an enhanced chemiluminescence (ECL) Western blot detection system (Amersham, Aylesbury, UK). The primary antibodies used in this analysis are provided in
Table 1 and were used at a dilution of 1:1000 for immunoblot analysis.
4.6. Mitochondrial Imaging
Mitochondrial function was examined using fluorescent probes. After treatment with or without BBR (25 μM), cells were incubated with the probes for 30 min at 37 °C and then photographed using an All-in-One fluorescence microscope (KEYENCE). We used MitoROS (mitochondrial superoxide) (10 μM, AAT Bioquest Inc., Sunnyvale, CA, USA) to assess oxidative stress, mitoGreen (100 nM, PromoCell GmbH, Heidelberg, Germany) to assess mitochondrial volume, and tetramethylrhodamine ethyl ester (TMRE) (200 nM, Sigma-Aldrich) to assess MMP. Mitophagy was detected using a Mitophagy Detection Kit (Dojindo) according to the manufacturer’s instructions.
4.7. Enzyme-Linked Immunosorbent Assay (ELISA) and Fluorometric Assay
Whole-cell lysates and mitochondrial fractions were prepared as previously described using RIPA buffer containing 0.1% SDS (Thermo Fisher) [
59] and a mitochondrial isolation kit for cultured cells (Thermo Fisher), respectively. ELISA kits were used to measure the protein levels (
Table 6). The assay was performed using whole-cell lysates and mitochondrial fractions according to the manufacturer’s instructions.
4.8. DNA Isolation
Mitochondrial DNA was isolated from the extracted mitochondria using TRIzol reagent (Invitrogen, Waltham, MA, USA) and purified using an RNeasy Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer’s protocols. Purified DNA was quantified using a NanoDrop ND-1000 spectrophotometer (Thermo Fisher Scientific).
4.9. Telomere Quantification
Telomere quantification was performed using a Relative Mouse Telomere Length Quantification qPCR Assay Kit (#M8908, Sciencells Research Lab, Carlsbad, CA, USA) according to the manufacturer’s instructions. DNA sample (2 μg in 1 μL) was mixed with primer stock solution (2 μL), 2 × GoldNStart TaqGreen qPCR master mix (10 μL), and Nuclease-free H2O (7 μL). The PCR conditions were as follows: 95℃ for 10 min, followed by 32 cycles of 95 °C for 20 s, 52 °C for 20 s, and 72 °C for 45 s. PCR was performed using an Applied Biosystems QuantStudio Absolute Q digital PCR system (Thermo Fisher Scientific). The comparative ∆∆Cq (quantification cycle value) method was applied to calculate the relative amount of telomeres using the following series of equations:
∆Cq (telomere, TEL) = Cq (TEL, sample 2) − Cq (TEL, sample 1), where TEL = telomere and ∆Cq is the difference in quantification cycles between two samples.
∆Cq (single copy reference, SCR) = Cq (SCR, sample 2) − Cq (SCR, sample 1).
∆∆Cq = ∆Cq (TEL) − ∆Cq (SCR).
Relative telomere length of sample 2 to sample 1 (fold) = 2−∆∆Cq.
4.10. Mitochondrial DNA Mutations
PCR was performed with 0.5 µg DNA extracted from mitochondria. The primer sets used are listed in
Table 6 and were synthesized by Sigma Genosys (St. Louis, MO, USA). To amplify long DNA fragments, TAKARA Ex Premier DNA polymerase (TAKARA Bio, Kusatsu, Japan) was used according to the manufacturer’s instructions. The PCR conditions were as follows: 94 °C for 1 min, followed by 30 cycles of 98 °C for 10 s and 68 °C for 5 min. PCR was performed using an Applied Biosystems QuantStudio Absolute Q digital PCR system (Thermo Fisher Scientific). The PCR products were electrophoresed on a 1% agarose gel and stained with ethidium bromide.
4.11. Mitochondrial Stress Test (Seahorse Assay)
Mitochondrial and glycolytic stress tests were performed as described previously [
60]. The oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) of 1 × 10
4 viable C2C12 cells per well were measured using a Seahorse XFe24 Extracellular Flux Analyzer with Seahorse XF24 FluxPaks (Agilent Technologies, Chicopee, ON, Canada).
4.12. Statistical Analysis
Statistical significances were calculated using unpaired Student’s t-tests using InStat, version 3.0 (GraphPad Software, Inc., La Jolla, CA, USA). Data are expressed as the mean ± standard deviation of three independent experiments. Two-sided p < 0.05 was considered to indicate statistical significance.





{kind=link}
{kind=link}
{kind=link}