
Binding Affinity Determination in Drug Design: Insights from Lock and Key, Induced Fit, Conformational Selection, and Inhibitor Trapping Models


Abstract
1. Introduction
2. Binding Affinity, Binding and Dissociation Rate Constants
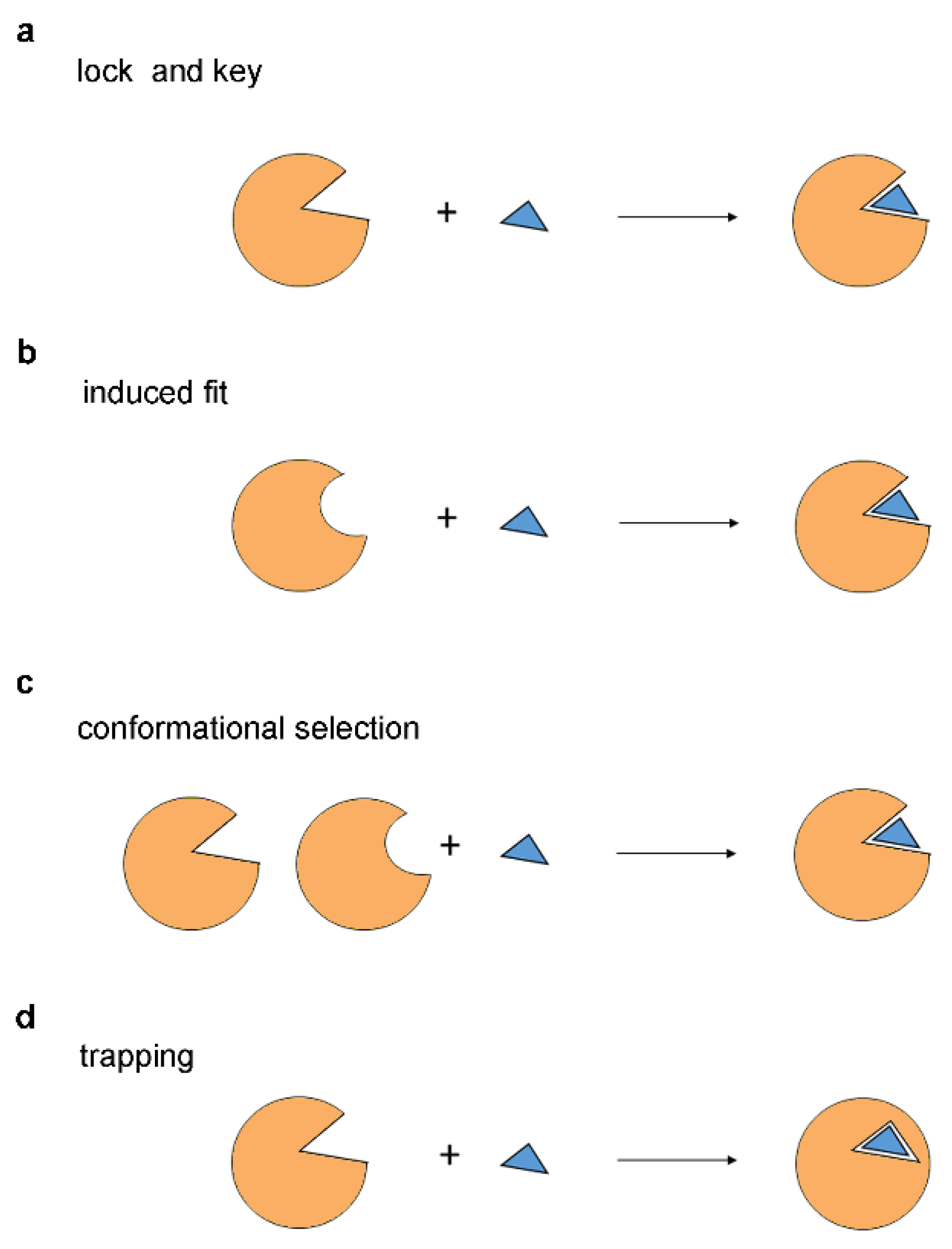
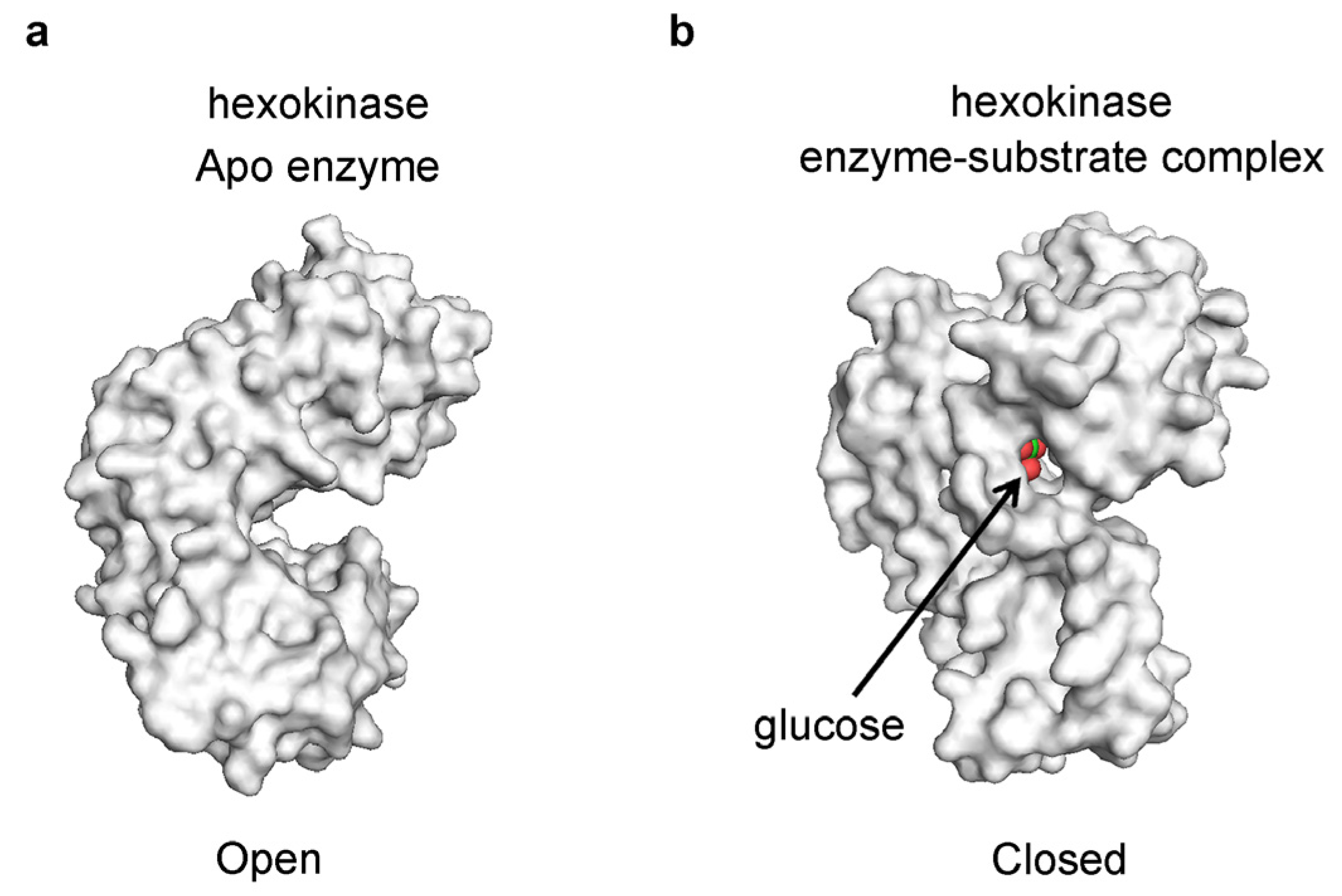
3. Models of Protein-Ligand Recognition
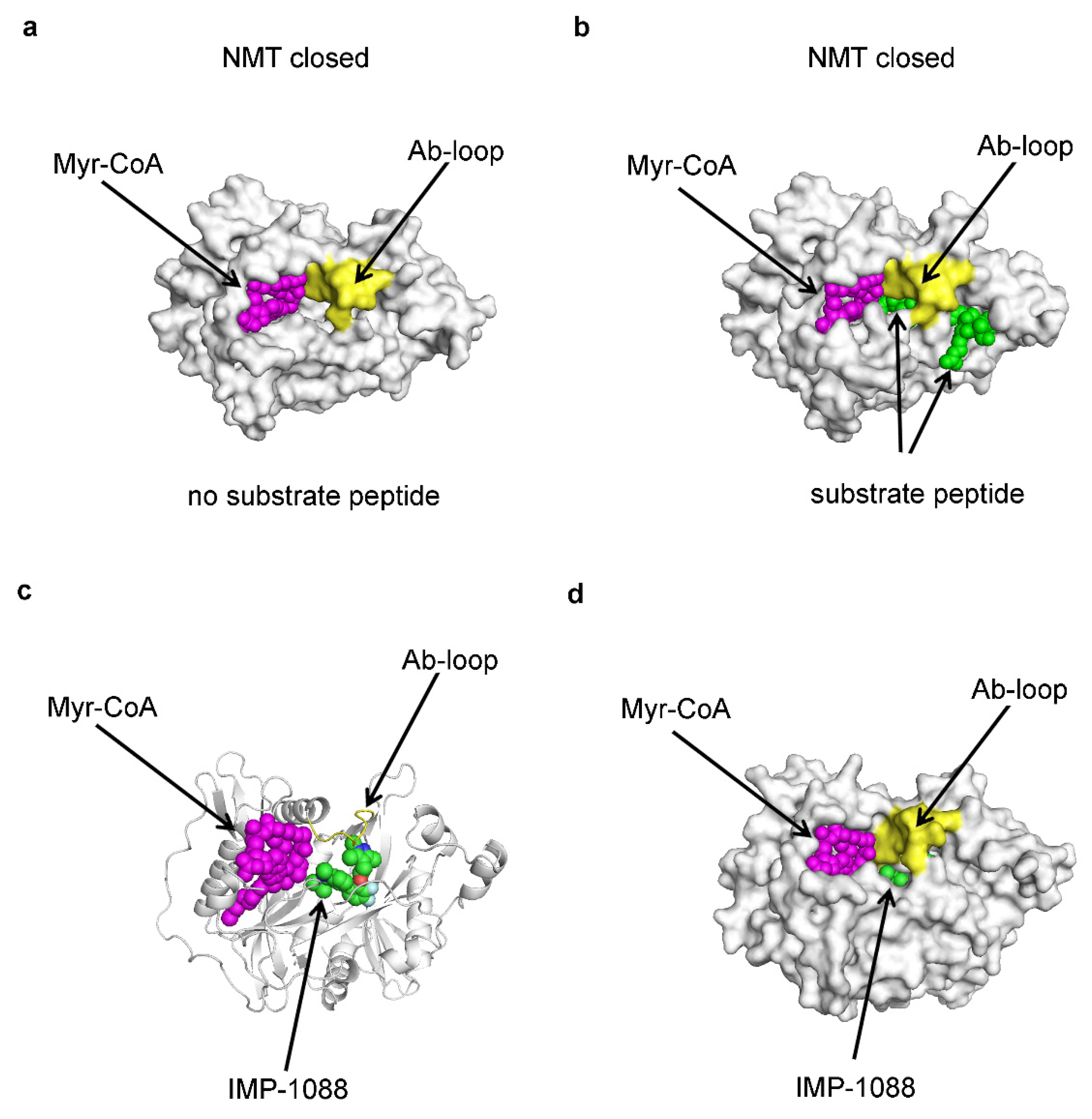
4. The Inhibitor Trapping Mechanism
5. Inhibitor Trapping in Drug Design
5.1. Inhibitor Trapping Can Dramatically Enhance the Binding Affinity
5.2. Trapping Enhances Drug Selectivity
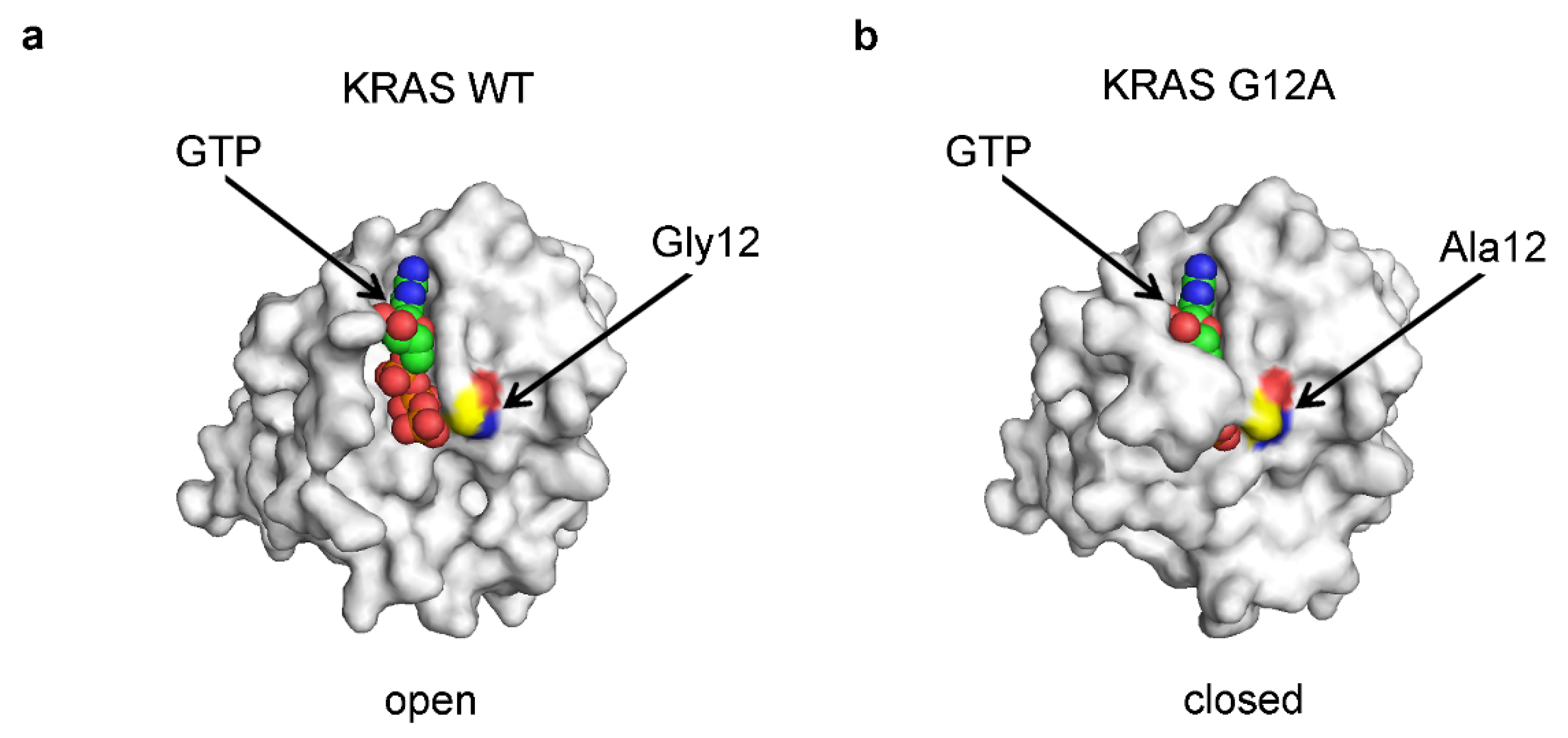
5.3. The Direct Interactions between the Protein and the Ligand Are Not the Sole Determinant of Binding Affinity
5.4. The Bonds Participating in the Entrapment Have a Dramatic Effect on the Binding Affinity
5.5. Effect of Water Exclusion on Binding Affinity
5.6. Conformational Changes and Protein Dynamics Are Critical for Binding Affinity
5.7. Inhibitor Trapping and Fragment-Based Drug Design
5.8. Inhibitor Trapping and Allostery
6. Predicting Binding Affinity
6.1. Models of Molecular Recognition in Computational Drug Design
6.2. A Unified Theory of Protein-Ligand Interaction
6.3. Predicting Binding Affinity
7. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Kairys, V.; Baranauskiene, L.; Kazlauskiene, M.; Matulis, D.; Kazlauskas, E. Binding affinity in drug design: Experimental and computational techniques. Expert Opin. Drug Discov. 2019, 14, 755–768. [Google Scholar] [CrossRef]
- Tripathi, A.; Bankaitis, V.A. Molecular Docking: From Lock and Key to Combination Lock. J. Mol. Med. Clin. Appl. 2017, 2, 1. [Google Scholar] [CrossRef]
- Brooijmans, N.; Kuntz, I.D. Molecular recognition and docking algorithms. Annu. Rev. Biophys. Biomol. Struct. 2003, 32, 335–373. [Google Scholar] [CrossRef]
- de Ruyck, J.; Brysbaert, G.; Blossey, R.; Lensink, M.F. Molecular docking as a popular tool in drug design, an in silico travel. Adv. Appl. Bioinform. Chem. AABC 2016, 9, 1–11. [Google Scholar] [CrossRef]
- Kontoyianni, M.; McClellan, L.M.; Sokol, G.S. Evaluation of docking performance: Comparative data on docking algorithms. J. Med. Chem. 2004, 47, 558–565. [Google Scholar] [CrossRef]
- Meng, X.Y.; Zhang, H.X.; Mezei, M.; Cui, M. Molecular docking: A powerful approach for structure-based drug discovery. Curr. Comput.-Aided Drug Des. 2011, 7, 146–157. [Google Scholar] [CrossRef]
- Huang, S.Y.; Zou, X. Advances and challenges in protein-ligand docking. Int. J. Mol. Sci. 2010, 11, 3016–3034. [Google Scholar] [CrossRef]
- Li, J.; Fu, A.; Zhang, L. An Overview of Scoring Functions Used for Protein-Ligand Interactions in Molecular Docking. Interdiscip. Sci. Comput. Life Sci. 2019, 11, 320–328. [Google Scholar] [CrossRef]
- Chen, Y.C. Beware of docking! Trends Pharmacol. Sci. 2015, 36, 78–95. [Google Scholar] [CrossRef]
- Pantsar, T.; Poso, A. Binding Affinity via Docking: Fact and Fiction. Molecules 2018, 23, 1899. [Google Scholar] [CrossRef]
- Macip, G.; Garcia-Segura, P.; Mestres-Truyol, J.; Saldivar-Espinoza, B.; Ojeda-Montes, M.J.; Gimeno, A.; Cereto-Massague, A.; Garcia-Vallve, S.; Pujadas, G. Haste makes waste: A critical review of docking-based virtual screening in drug repurposing for SARS-CoV-2 main protease (M-pro) inhibition. Med. Res. Rev. 2022, 42, 744–769. [Google Scholar] [CrossRef]
- Mikra, C.; Rossos, G.; Hadjikakou, S.K.; Kourkoumelis, N. Molecular Docking and Structure Activity Relationship Studies of NSAIDs. What do they Reveal about IC50? Lett. Drug Des. Discov. 2017, 14, 949–958. [Google Scholar] [CrossRef]
- Spassov, D.S.; Atanasova, M.; Doytchinova, I. Novel Hits for N-Myristoyltransferase Inhibition Discovered by Docking-Based Screening. Molecules 2022, 27, 5478. [Google Scholar] [CrossRef]
- Spassov, D.S.; Atanasova, M.; Doytchinova, I. Inhibitor Trapping in N-Myristoyltransferases as a Mechanism for Drug Potency. Int. J. Mol. Sci. 2023, 24, 11610. [Google Scholar] [CrossRef]
- Meli, R.; Morris, G.M.; Biggin, P.C. Scoring Functions for Protein-Ligand Binding Affinity Prediction using Structure-Based Deep Learning: A Review. Front. Bioinform. 2022, 2, 885983. [Google Scholar] [CrossRef]
- Verdonk, M.L.; Cole, J.C.; Hartshorn, M.J.; Murray, C.W.; Taylor, R.D. Improved protein-ligand docking using GOLD. Proteins 2003, 52, 609–623. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Attique, S.A.; Hassan, M.; Usman, M.; Atif, R.M.; Mahboob, S.; Al-Ghanim, K.A.; Bilal, M.; Nawaz, M.Z. A Molecular Docking Approach to Evaluate the Pharmacological Properties of Natural and Synthetic Treatment Candidates for Use against Hypertension. Int. J. Environ. Res. Public Health 2019, 16, 923. [Google Scholar] [CrossRef]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef]
- Guedes, I.A.; Barreto, A.M.S.; Marinho, D.; Krempser, E.; Kuenemann, M.A.; Sperandio, O.; Dardenne, L.E.; Miteva, M.A. New machine learning and physics-based scoring functions for drug discovery. Sci. Rep. 2021, 11, 3198. [Google Scholar] [CrossRef]
- Illingworth, C.J.; Morris, G.M.; Parkes, K.E.; Snell, C.R.; Reynolds, C.A. Assessing the role of polarization in docking. J. Phys. Chem. A 2008, 112, 12157–12163. [Google Scholar] [CrossRef]
- Ruvinsky, A.M. Role of binding entropy in the refinement of protein-ligand docking predictions: Analysis based on the use of 11 scoring functions. J. Comput. Chem. 2007, 28, 1364–1372. [Google Scholar] [CrossRef]
- Bienstock, R.J. Solvation methods for protein-ligand docking. Methods Mol. Biol. 2015, 1289, 3–12. [Google Scholar] [CrossRef]
- Spassov, D.S.; Atanasova, M.; Doytchinova, I. Inhibitor Trapping in Kinases. Int. J. Mol. Sci. 2024, 25, 3249. [Google Scholar] [CrossRef]
- Du, X.; Li, Y.; Xia, Y.L.; Ai, S.M.; Liang, J.; Sang, P.; Ji, X.L.; Liu, S.Q. Insights into Protein-Ligand Interactions: Mechanisms, Models, and Methods. Int. J. Mol. Sci. 2016, 17, 144. [Google Scholar] [CrossRef]
- Kakkar, T.; Boxenbaum, H.; Mayersohn, M. Estimation of Ki in a competitive enzyme-inhibition model: Comparisons among three methods of data analysis. Drug Metab. Dispos. Biol. Fate Chem. 1999, 27, 756–762. [Google Scholar]
- Fischer, E. Einfluss der Configuration auf die Wirkung der Enzyme. Berichte Der Dtsch. Chem. Ges. 1894, 27, 2985–2993. [Google Scholar] [CrossRef]
- Teilum, K.; Olsen, J.G.; Kragelund, B.B. Functional aspects of protein flexibility. Cell. Mol. Life Sci. CMLS 2009, 66, 2231–2247. [Google Scholar] [CrossRef]
- Koshland, D.E. The Key-Lock Theory and the Induced Fit Theory. Angew. Chem. 1994, 33, 2375–2378. [Google Scholar] [CrossRef]
- Koshland, D.E. Application of a Theory of Enzyme Specificity to Protein Synthesis. Proc. Natl. Acad. Sci. USA 1958, 44, 98–104. [Google Scholar] [CrossRef]
- Gerstein, M.; Lesk, A.M.; Chothia, C. Structural mechanisms for domain movements in proteins. Biochemistry 1994, 33, 6739–6749. [Google Scholar] [CrossRef]
- Boehr, D.D.; Nussinov, R.; Wright, P.E. The role of dynamic conformational ensembles in biomolecular recognition. Nat. Chem. Biol. 2009, 5, 789–796. [Google Scholar] [CrossRef]
- Vogt, A.D.; Di Cera, E. Conformational selection is a dominant mechanism of ligand binding. Biochemistry 2013, 52, 5723–5729. [Google Scholar] [CrossRef]
- Jura, N.; Zhang, X.; Endres, N.F.; Seeliger, M.A.; Schindler, T.; Kuriyan, J. Catalytic control in the EGF receptor and its connection to general kinase regulatory mechanisms. Mol. Cell 2011, 42, 9–22. [Google Scholar] [CrossRef]
- Berezhkovskii, A.M.; Szabo, A.; Rotbart, T.; Urbakh, M.; Kolomeisky, A.B. Dependence of the Enzymatic Velocity on the Substrate Dissociation Rate. J. Phys. Chem. B 2017, 121, 3437–3442. [Google Scholar] [CrossRef]
- Nishimasu, H.; Fushinobu, S.; Shoun, H.; Wakagi, T. Crystal structures of an ATP-dependent hexokinase with broad substrate specificity from the hyperthermophilic archaeon Sulfolobus tokodaii. J. Biol. Chem. 2007, 282, 9923–9931. [Google Scholar] [CrossRef]
- Cardenas, M.L.; Cornish-Bowden, A.; Ureta, T. Evolution and regulatory role of the hexokinases. Biochim. Et Biophys. Acta 1998, 1401, 242–264. [Google Scholar] [CrossRef]
- Rodriguez-Saavedra, C.; Morgado-Martinez, L.E.; Burgos-Palacios, A.; King-Diaz, B.; Lopez-Coria, M.; Sanchez-Nieto, S. Moonlighting Proteins: The Case of the Hexokinases. Front. Mol. Biosci. 2021, 8, 701975. [Google Scholar] [CrossRef]
- Schrödinger, L.; DeLano, W. PyMOL. 2020. Available online: http://www.pymol.org/pymol (accessed on 25 June 2024).
- DelaFuente, G. Specific inactivation of yeast hexokinase induced by xylose in the presence of a phosphoryl donor substrate. Eur. J. Biochem. 1970, 16, 240–243. [Google Scholar] [CrossRef]
- Kuser, P.; Cupri, F.; Bleicher, L.; Polikarpov, I. Crystal structure of yeast hexokinase PI in complex with glucose: A classical “induced fit” example revised. Proteins 2008, 72, 731–740. [Google Scholar] [CrossRef]
- Bennett, W.S., Jr.; Steitz, T.A. Glucose-induced conformational change in yeast hexokinase. Proc. Natl. Acad. Sci. USA 1978, 75, 4848–4852. [Google Scholar] [CrossRef] [PubMed]
- DelaFuente, G.; Lagunas, R.; Sols, A. Induced fit in yeast hexokinase. Eur. J. Biochem. 1970, 16, 226–233. [Google Scholar] [CrossRef]
- Dian, C.; Perez-Dorado, I.; Riviere, F.; Asensio, T.; Legrand, P.; Ritzefeld, M.; Shen, M.; Cota, E.; Meinnel, T.; Tate, E.W.; et al. High-resolution snapshots of human N-myristoyltransferase in action illuminate a mechanism promoting N-terminal Lys and Gly myristoylation. Nat. Commun. 2020, 11, 1132. [Google Scholar] [CrossRef]
- Frearson, J.A.; Brand, S.; McElroy, S.P.; Cleghorn, L.A.; Smid, O.; Stojanovski, L.; Price, H.P.; Guther, M.L.; Torrie, L.S.; Robinson, D.A.; et al. N-myristoyltransferase inhibitors as new leads to treat sleeping sickness. Nature 2010, 464, 728–732. [Google Scholar] [CrossRef] [PubMed]
- Mousnier, A.; Bell, A.S.; Swieboda, D.P.; Morales-Sanfrutos, J.; Perez-Dorado, I.; Brannigan, J.A.; Newman, J.; Ritzefeld, M.; Hutton, J.A.; Guedan, A.; et al. Fragment-derived inhibitors of human N-myristoyltransferase block capsid assembly and replication of the common cold virus. Nat. Chem. 2018, 10, 599–606. [Google Scholar] [CrossRef]
- Huang, L.; Guo, Z.; Wang, F.; Fu, L. KRAS mutation: From undruggable to druggable in cancer. Signal Transduct. Target. Ther. 2021, 6, 386. [Google Scholar] [CrossRef] [PubMed]
- Bos, J.L. ras oncogenes in human cancer: A review. Cancer Res. 1989, 49, 4682–4689. [Google Scholar]
- Murugan, A.K.; Grieco, M.; Tsuchida, N. RAS mutations in human cancers: Roles in precision medicine. Semin. Cancer Biol. 2019, 59, 23–35. [Google Scholar] [CrossRef]
- Simanshu, D.K.; Nissley, D.V.; McCormick, F. RAS Proteins and Their Regulators in Human Disease. Cell 2017, 170, 17–33. [Google Scholar] [CrossRef]
- Xu, S.; Long, B.N.; Boris, G.H.; Chen, A.; Ni, S.; Kennedy, M.A. Structural insight into the rearrangement of the switch I region in GTP-bound G12A K-Ras. Acta Crystallographica. Sect. D Struct. Biol. 2017, 73, 970–984. [Google Scholar] [CrossRef]
- Singhal, A.; Li, B.T.; O’Reilly, E.M. Targeting KRAS in cancer. Nat. Med. 2024, 30, 969–983. [Google Scholar] [CrossRef] [PubMed]
- Arter, C.; Trask, L.; Ward, S.; Yeoh, S.; Bayliss, R. Structural features of the protein kinase domain and targeted binding by small-molecule inhibitors. J. Biol. Chem. 2022, 298, 102247. [Google Scholar] [CrossRef] [PubMed]
- Attwood, M.M.; Fabbro, D.; Sokolov, A.V.; Knapp, S.; Schioth, H.B. Trends in kinase drug discovery: Targets, indications and inhibitor design. Nat. Rev. Drug Discov. 2021, 20, 839–861. [Google Scholar] [CrossRef] [PubMed]
- Cohen, P.; Cross, D.; Janne, P.A. Kinase drug discovery 20 years after imatinib: Progress and future directions. Nat. Rev. Drug Discov. 2021, 20, 551–569. [Google Scholar] [CrossRef]
- Saraste, M.; Sibbald, P.R.; Wittinghofer, A. The P-loop--a common motif in ATP- and GTP-binding proteins. Trends Biochem. Sci. 1990, 15, 430–434. [Google Scholar] [CrossRef] [PubMed]
- Nagar, B.; Bornmann, W.G.; Pellicena, P.; Schindler, T.; Veach, D.R.; Miller, W.T.; Clarkson, B.; Kuriyan, J. Crystal structures of the kinase domain of c-Abl in complex with the small molecule inhibitors PD173955 and imatinib (STI-571). Cancer Res. 2002, 62, 4236–4243. [Google Scholar] [PubMed]
- Seeliger, M.A.; Nagar, B.; Frank, F.; Cao, X.; Henderson, M.N.; Kuriyan, J. c-Src binds to the cancer drug imatinib with an inactive Abl/c-Kit conformation and a distributed thermodynamic penalty. Structure 2007, 15, 299–311. [Google Scholar] [CrossRef]
- Wilson, C.; Agafonov, R.V.; Hoemberger, M.; Kutter, S.; Zorba, A.; Halpin, J.; Buosi, V.; Otten, R.; Waterman, D.; Theobald, D.L.; et al. Kinase dynamics. Using ancient protein kinases to unravel a modern cancer drug’s mechanism. Science 2015, 347, 882–886. [Google Scholar] [CrossRef]
- Agafonov, R.V.; Wilson, C.; Otten, R.; Buosi, V.; Kern, D. Energetic dissection of Gleevec’s selectivity toward human tyrosine kinases. Nat. Struct. Mol. Biol. 2014, 21, 848–853. [Google Scholar] [CrossRef]
- Kaleem, B.; Shahab, S.; Zaidi, U.; Shamsi, T.S. P-Loop mutations-Negative prognosticators in tyrosine kinase inhibitors resistant chronic myeloid leukemia patients. Int. J. Lab. Hematol. 2022, 44, 538–546. [Google Scholar] [CrossRef]
- Roumiantsev, S.; Shah, N.P.; Gorre, M.E.; Nicoll, J.; Brasher, B.B.; Sawyers, C.L.; Van Etten, R.A. Clinical resistance to the kinase inhibitor STI-571 in chronic myeloid leukemia by mutation of Tyr-253 in the Abl kinase domain P-loop. Proc. Natl. Acad. Sci. USA 2002, 99, 10700–10705. [Google Scholar] [CrossRef]
- Angell, R.M.; Bamborough, P.; Cleasby, A.; Cockerill, S.G.; Jones, K.L.; Mooney, C.J.; Somers, D.O.; Walker, A.L. Biphenyl amide p38 kinase inhibitors 1: Discovery and binding mode. Bioorg. Med. Chem. Lett. 2008, 18, 318–323. [Google Scholar] [CrossRef] [PubMed]
- Schonherr, H.; Cernak, T. Profound methyl effects in drug discovery and a call for new C-H methylation reactions. Angew. Chem. 2013, 52, 12256–12267. [Google Scholar] [CrossRef]
- Marangoni, J.M.; Wu, S.C.; Fogen, D.; Wong, S.L.; Ng, K.K.S. Engineering a disulfide-gated switch in streptavidin enables reversible binding without sacrificing binding affinity. Sci. Rep. 2020, 10, 12483. [Google Scholar] [CrossRef]
- Weber, P.C.; Ohlendorf, D.H.; Wendoloski, J.J.; Salemme, F.R. Structural origins of high-affinity biotin binding to streptavidin. Science 1989, 243, 85–88. [Google Scholar] [CrossRef] [PubMed]
- Chu, V.; Freitag, S.; Le Trong, I.; Stenkamp, R.E.; Stayton, P.S. Thermodynamic and structural consequences of flexible loop deletion by circular permutation in the streptavidin-biotin system. Protein Sci. A Publ. Protein Soc. 1998, 7, 848–859. [Google Scholar] [CrossRef]
- Pinheiro, P.S.M.; Franco, L.S.; Fraga, C.A.M. The Magic Methyl and Its Tricks in Drug Discovery and Development. Pharmaceuticals 2023, 16, 1157. [Google Scholar] [CrossRef] [PubMed]
- Manley, P.W.; Cowan-Jacob, S.W.; Fendrich, G.; Jahnke, W.; Fabbro, D. Nilotinib, in Comparison to Both Dasatinib and Imatinib, Possesses a Greatly Prolonged Residence Time When Bound to the BCR-ABL Kinase SH1 Domain. Blood 2011, 118, 1674. [Google Scholar] [CrossRef]
- Kitagawa, D.; Gouda, M.; Kirii, Y. Quick evaluation of kinase inhibitors by surface plasmon resonance using single-site specifically biotinylated kinases. J. Biomol. Screen. 2014, 19, 453–461. [Google Scholar] [CrossRef]
- Ojha, A.A.; Srivastava, A.; Votapka, L.W.; Amaro, R.E. Selectivity and Ranking of Tight-Binding JAK-STAT Inhibitors Using Markovian Milestoning with Voronoi Tessellations. J. Chem. Inf. Model. 2023, 63, 2469–2482. [Google Scholar] [CrossRef]
- Shamsudin, Y.; Kazemi, M.; Gutierrez-de-Teran, H.; Aqvist, J. Origin of the Enigmatic Stepwise Tight-Binding Inhibition of Cyclooxygenase-1. Biochemistry 2015, 54, 7283–7291. [Google Scholar] [CrossRef]
- Carroll, M.J.; Mauldin, R.V.; Gromova, A.V.; Singleton, S.F.; Collins, E.J.; Lee, A.L. Evidence for dynamics in proteins as a mechanism for ligand dissociation. Nat. Chem. Biol. 2012, 8, 246–252. [Google Scholar] [CrossRef]
- Rawat, R.; Whitty, A.; Tonge, P.J. The isoniazid-NAD adduct is a slow, tight-binding inhibitor of InhA, the Mycobacterium tuberculosis enoyl reductase: Adduct affinity and drug resistance. Proc. Natl. Acad. Sci. USA 2003, 100, 13881–13886. [Google Scholar] [CrossRef]
- Copeland, R.A.; Pompliano, D.L.; Meek, T.D. Drug-target residence time and its implications for lead optimization. Nat. Rev. Drug Discov. 2006, 5, 730–739. [Google Scholar] [CrossRef]
- Capdeville, R.; Buchdunger, E.; Zimmermann, J.; Matter, A. Glivec (STI571, imatinib), a rationally developed, targeted anticancer drug. Nat. Rev. Drug Discov. 2002, 1, 493–502. [Google Scholar] [CrossRef] [PubMed]
- Manley, P.W.; Cowan-Jacob, S.W.; Buchdunger, E.; Fabbro, D.; Fendrich, G.; Furet, P.; Meyer, T.; Zimmermann, J. Imatinib: A selective tyrosine kinase inhibitor. Eur. J. Cancer 2002, 38 (Suppl. S5), S19–S27. [Google Scholar] [CrossRef] [PubMed]
- Wood, E.R.; Truesdale, A.T.; McDonald, O.B.; Yuan, D.; Hassell, A.; Dickerson, S.H.; Ellis, B.; Pennisi, C.; Horne, E.; Lackey, K.; et al. A unique structure for epidermal growth factor receptor bound to GW572016 (Lapatinib): Relationships among protein conformation, inhibitor off-rate, and receptor activity in tumor cells. Cancer Res. 2004, 64, 6652–6659. [Google Scholar] [CrossRef] [PubMed]
- Tanramluk, D.; Schreyer, A.; Pitt, W.R.; Blundell, T.L. On the origins of enzyme inhibitor selectivity and promiscuity: A case study of protein kinase binding to staurosporine. Chem. Biol. Drug Des. 2009, 74, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Kersten, C.; Fleischer, E.; Kehrein, J.; Borek, C.; Jaenicke, E.; Sotriffer, C.; Brenk, R. How To Design Selective Ligands for Highly Conserved Binding Sites: A Case Study Using N-Myristoyltransferases as a Model System. J. Med. Chem. 2020, 63, 2095–2113. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.H.; Shiao, H.Y.; Tu, C.H.; Liu, P.M.; Hsu, J.T.; Amancha, P.K.; Wu, J.S.; Coumar, M.S.; Chen, C.H.; Wang, S.Y.; et al. Protein kinase inhibitor design by targeting the Asp-Phe-Gly (DFG) motif: The role of the DFG motif in the design of epidermal growth factor receptor inhibitors. J. Med. Chem. 2013, 56, 3889–3903. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Hernandez, D.; Vijayan, K.; Zigweid, R.; Fenwick, M.K.; Sankaran, B.; Roobsoong, W.; Sattabongkot, J.; Glennon, E.K.K.; Myler, P.J.; Sunnerhagen, P.; et al. Identification of potent and selective N-myristoyltransferase inhibitors of Plasmodium vivax liver stage hypnozoites and schizonts. Nat. Commun. 2023, 14, 5408. [Google Scholar] [CrossRef] [PubMed]
- Schindler, T.; Bornmann, W.; Pellicena, P.; Miller, W.T.; Clarkson, B.; Kuriyan, J. Structural mechanism for STI-571 inhibition of abelson tyrosine kinase. Science 2000, 289, 1938–1942. [Google Scholar] [CrossRef] [PubMed]
- Leung, C.S.; Leung, S.S.; Tirado-Rives, J.; Jorgensen, W.L. Methyl effects on protein-ligand binding. J. Med. Chem. 2012, 55, 4489–4500. [Google Scholar] [CrossRef] [PubMed]
- Ferreira de Freitas, R.; Schapira, M. A systematic analysis of atomic protein-ligand interactions in the PDB. MedChemComm 2017, 8, 1970–1981. [Google Scholar] [CrossRef]
- Spassov, D.S.; Atanasova, M.; Doytchinova, I. A role of salt bridges in mediating drug potency: A lesson from the N-myristoyltransferase inhibitors. Front. Mol. Biosci. 2023, 9, 1066029. [Google Scholar] [CrossRef]
- Schiebel, J.; Gaspari, R.; Wulsdorf, T.; Ngo, K.; Sohn, C.; Schrader, T.E.; Cavalli, A.; Ostermann, A.; Heine, A.; Klebe, G. Intriguing role of water in protein-ligand binding studied by neutron crystallography on trypsin complexes. Nat. Commun. 2018, 9, 3559. [Google Scholar] [CrossRef] [PubMed]
- Segala, E.; Guo, D.; Cheng, R.K.; Bortolato, A.; Deflorian, F.; Dore, A.S.; Errey, J.C.; Heitman, L.H.; AP, I.J.; Marshall, F.H.; et al. Controlling the Dissociation of Ligands from the Adenosine A2A Receptor through Modulation of Salt Bridge Strength. J. Med. Chem. 2016, 59, 6470–6479. [Google Scholar] [CrossRef]
- Takano, K.; Tsuchimori, K.; Yamagata, Y.; Yutani, K. Contribution of salt bridges near the surface of a protein to the conformational stability. Biochemistry 2000, 39, 12375–12381. [Google Scholar] [CrossRef]
- Pylaeva, S.; Brehm, M.; Sebastiani, D. Salt Bridge in Aqueous Solution: Strong Structural Motifs but Weak Enthalpic Effect. Sci. Rep. 2018, 8, 13626. [Google Scholar] [CrossRef]
- Huang, S.Y.; Zou, X. Inclusion of solvation and entropy in the knowledge-based scoring function for protein-ligand interactions. J. Chem. Inf. Model. 2010, 50, 262–273. [Google Scholar] [CrossRef]
- Pantsar, T.; Kaiser, P.D.; Kudolo, M.; Forster, M.; Rothbauer, U.; Laufer, S.A. Decisive role of water and protein dynamics in residence time of p38alpha MAP kinase inhibitors. Nat. Commun. 2022, 13, 569. [Google Scholar] [CrossRef] [PubMed]
- Seo, M.H.; Park, J.; Kim, E.; Hohng, S.; Kim, H.S. Protein conformational dynamics dictate the binding affinity for a ligand. Nat. Commun. 2014, 5, 3724. [Google Scholar] [CrossRef] [PubMed]
- Vajpai, N.; Strauss, A.; Fendrich, G.; Cowan-Jacob, S.W.; Manley, P.W.; Grzesiek, S.; Jahnke, W. Solution conformations and dynamics of ABL kinase-inhibitor complexes determined by NMR substantiate the different binding modes of imatinib/nilotinib and dasatinib. J. Biol. Chem. 2008, 283, 18292–18302. [Google Scholar] [CrossRef] [PubMed]
- Stamos, J.; Sliwkowski, M.X.; Eigenbrot, C. Structure of the epidermal growth factor receptor kinase domain alone and in complex with a 4-anilinoquinazoline inhibitor. J. Biol. Chem. 2002, 277, 46265–46272. [Google Scholar] [CrossRef] [PubMed]
- Hajduk, P.J.; Sheppard, G.; Nettesheim, D.G.; Olejniczak, E.T.; Shuker, S.B.; Meadows, R.P.; Steinman, D.H.; Carrera, G.M.; Marcotte, P.A.; Severin, J.; et al. Discovery of Potent Nonpeptide Inhibitors of Stromelysin Using SAR by NMR. J. Am. Chem. Soc. 1997, 119, 5818–5827. [Google Scholar] [CrossRef]
- Kirsch, P.; Hartman, A.M.; Hirsch, A.K.H.; Empting, M. Concepts and Core Principles of Fragment-Based Drug Design. Molecules 2019, 24, 4309. [Google Scholar] [CrossRef] [PubMed]
- Petros, A.M.; Dinges, J.; Augeri, D.J.; Baumeister, S.A.; Betebenner, D.A.; Bures, M.G.; Elmore, S.W.; Hajduk, P.J.; Joseph, M.K.; Landis, S.K.; et al. Discovery of a potent inhibitor of the antiapoptotic protein Bcl-xL from NMR and parallel synthesis. J. Med. Chem. 2006, 49, 656–663. [Google Scholar] [CrossRef]
- Mortenson, P.N.; Berdini, V.; O’Reilly, M. Fragment-based approaches to the discovery of kinase inhibitors. Methods Enzymol. 2014, 548, 69–92. [Google Scholar] [CrossRef]
- McCullagh, M.; Zeczycki, T.N.; Kariyawasam, C.S.; Durie, C.L.; Halkidis, K.; Fitzkee, N.C.; Holt, J.M.; Fenton, A.W. What is allosteric regulation? Exploring the exceptions that prove the rule! J. Biol. Chem. 2024, 300, 105672. [Google Scholar] [CrossRef]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef] [PubMed]
- Zacharias, M. Accounting for conformational changes during protein-protein docking. Curr. Opin. Struct. Biol. 2010, 20, 180–186. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Bradley, P.; Baker, D. Protein-protein docking with backbone flexibility. J. Mol. Biol. 2007, 373, 503–519. [Google Scholar] [CrossRef] [PubMed]
- Ayaz, P.; Lyczek, A.; Paung, Y.; Mingione, V.R.; Iacob, R.E.; de Waal, P.W.; Engen, J.R.; Seeliger, M.A.; Shan, Y.; Shaw, D.E. Structural mechanism of a drug-binding process involving a large conformational change of the protein target. Nat. Commun. 2023, 14, 1885. [Google Scholar] [CrossRef] [PubMed]
- Amaro, R.E.; Baudry, J.; Chodera, J.; Demir, O.; McCammon, J.A.; Miao, Y.; Smith, J.C. Ensemble Docking in Drug Discovery. Biophys. J. 2018, 114, 2271–2278. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- You, W.; Tang, Z.; Chang, C.A. Potential Mean Force from Umbrella Sampling Simulations: What Can We Learn and What Is Missed? J. Chem. Theory Comput. 2019, 15, 2433–2443. [Google Scholar] [CrossRef] [PubMed]
- Zazeri, G.; Povinelli, A.P.R.; Lima, M.F.; Cornelio, M.L. Detailed Characterization of the Cooperative Binding of Piperine with Heat Shock Protein 70 by Molecular Biophysical Approaches. Biomedicines 2020, 8, 629. [Google Scholar] [CrossRef]
- Zazeri, G.; Povinelli, A.P.R.; de Freitas Lima, M.; Cornelio, M.L. The Cytokine IL-1beta and Piperine Complex Surveyed by Experimental and Computational Molecular Biophysics. Biomolecules 2020, 10, 1337. [Google Scholar] [CrossRef]

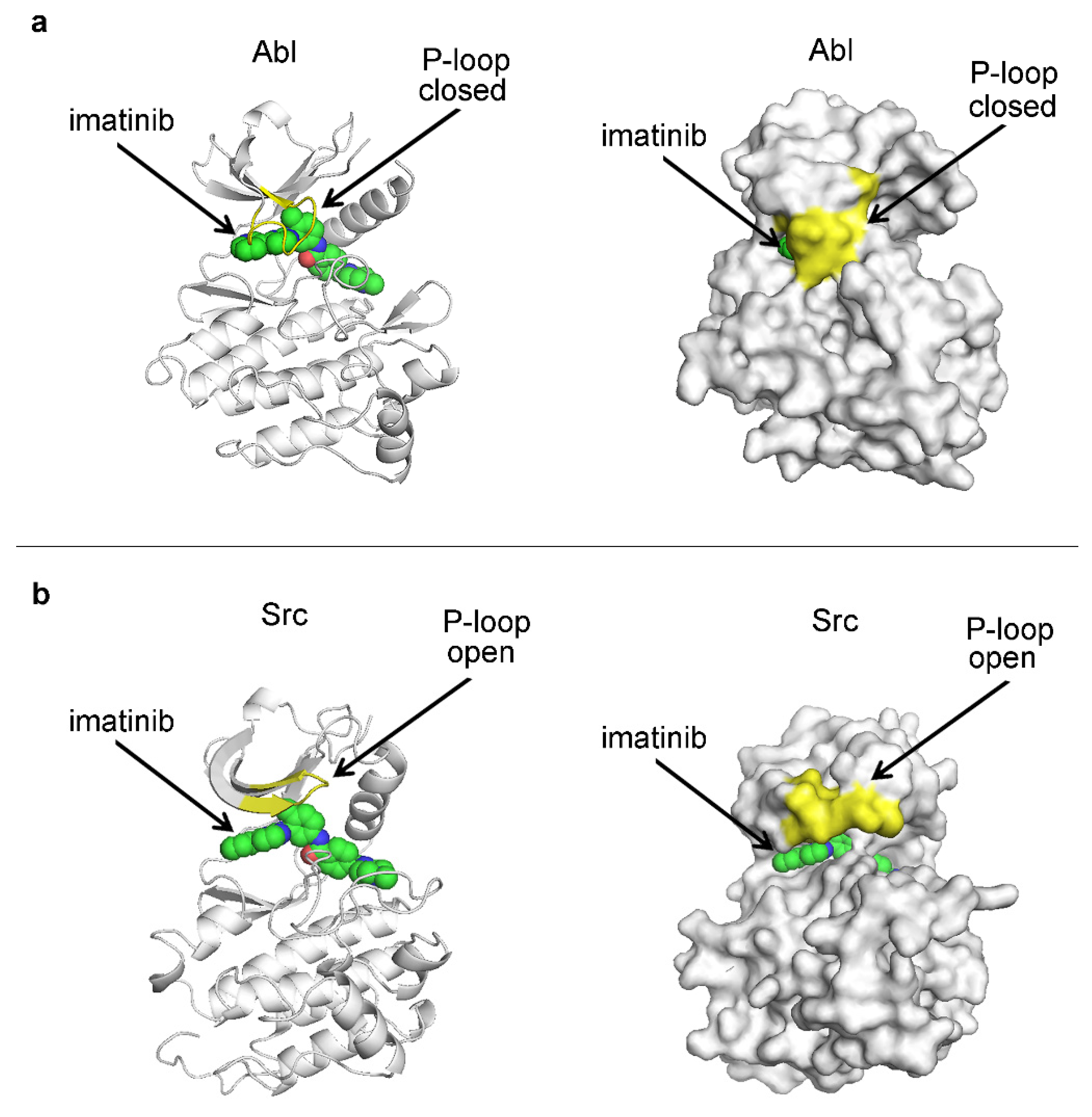

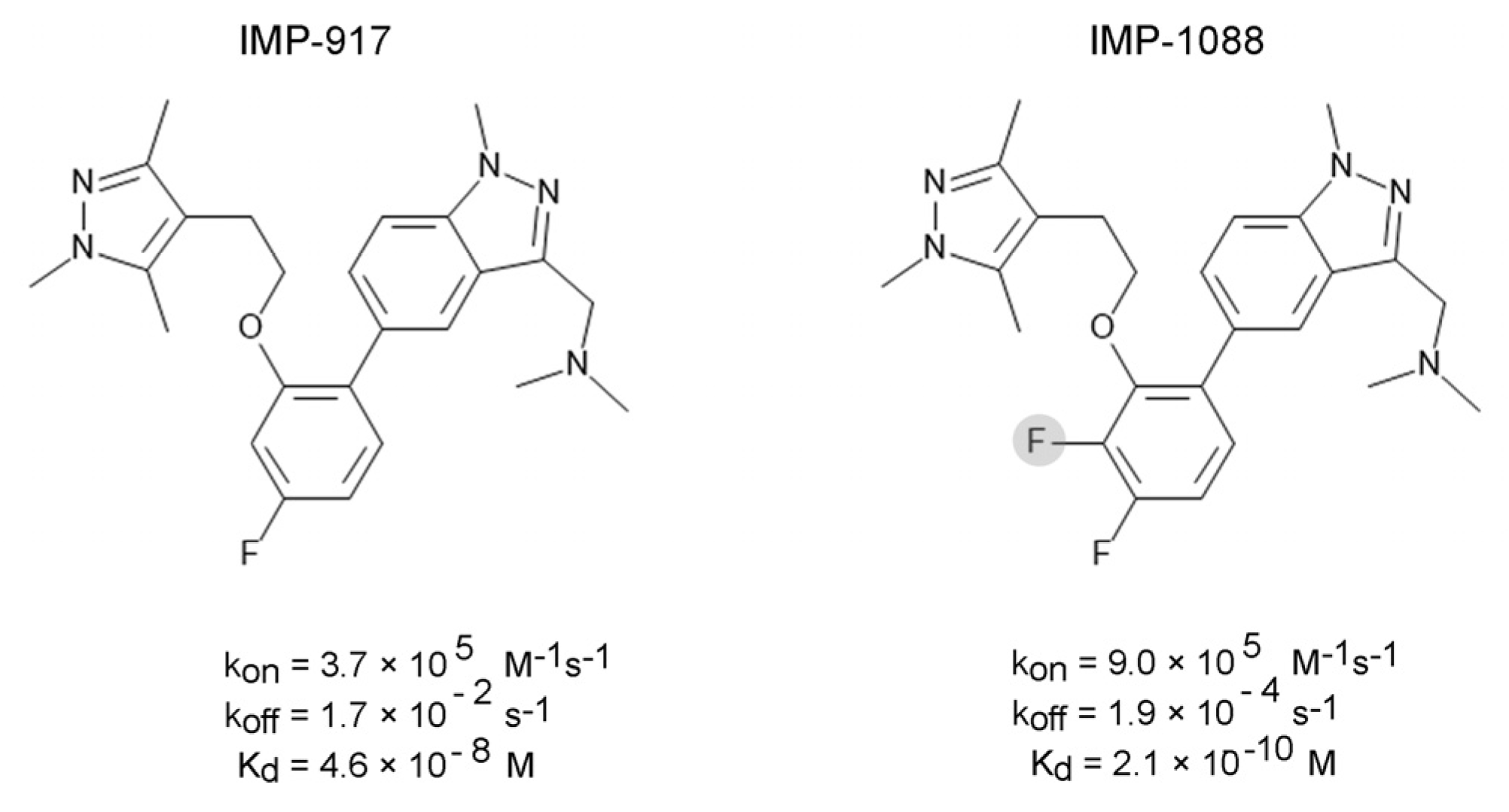
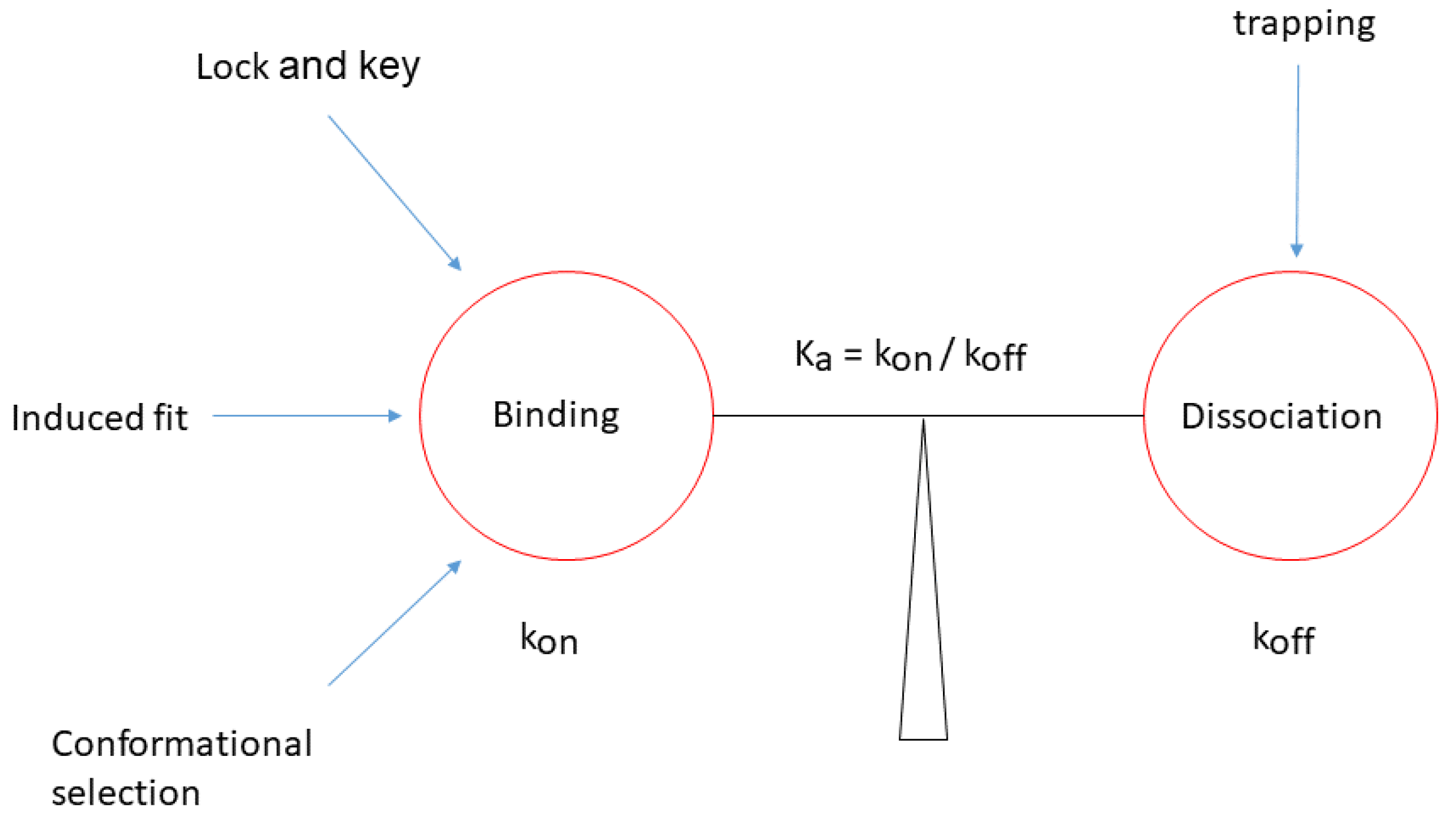

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligand | Protein | Kd, M | kon, M−1s−1, | koff, s−1 | Res Time *, s | Ref | |
|---|---|---|---|---|---|---|---|
| 1 | biotin | streptavidin | 5.0 × 10−15 | 5.40 × 107 | 2.70 × 10−7 | 3,703,704 | [66] |
| 2 | IMP-1088 | NMT | 2.1 × 10−10 | 9.00 × 105 | 1.90 × 10−4 | 5263 | [47] |
| 3 | imatinib | Abl | 3.83 × 10−9 | 1.53 × 105 | 5.86 × 10−4 | 1706 | [70] |
| 4 | nilotinib | Abl | 2.88 × 10−9 | 2.86 × 104 | 8.24 × 10−5 | 12,136 | [70] |
| 5 | dasatinib | Abl | 5.0 × 10−10 | 2.27 × 106 | 1.13 × 10−3 | 885 | [70] |
| 6 | dasatinib | EGFR | 1.28 × 10−8 | 7.81 × 105 | 1.00 × 10−2 | 100 | [71] |
| 7 | lapatinib | EGFR | 1.22 × 10−8 | 9.79 × 104 | 1.20 × 10−3 | 833 | [71] |
| 8 | staurosporine | EGFR | 6.00 × 10−8 | 4.93 × 105 | 3.00 × 10−2 | 33 | [71] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Spassov, D.S. Binding Affinity Determination in Drug Design: Insights from Lock and Key, Induced Fit, Conformational Selection, and Inhibitor Trapping Models. Int. J. Mol. Sci. 2024, 25, 7124. https://doi.org/10.3390/ijms25137124
Spassov DS. Binding Affinity Determination in Drug Design: Insights from Lock and Key, Induced Fit, Conformational Selection, and Inhibitor Trapping Models. International Journal of Molecular Sciences. 2024; 25(13):7124. https://doi.org/10.3390/ijms25137124
Chicago/Turabian StyleSpassov, Danislav S. 2024. "Binding Affinity Determination in Drug Design: Insights from Lock and Key, Induced Fit, Conformational Selection, and Inhibitor Trapping Models" International Journal of Molecular Sciences 25, no. 13: 7124. https://doi.org/10.3390/ijms25137124
APA StyleSpassov, D. S. (2024). Binding Affinity Determination in Drug Design: Insights from Lock and Key, Induced Fit, Conformational Selection, and Inhibitor Trapping Models. International Journal of Molecular Sciences, 25(13), 7124. https://doi.org/10.3390/ijms25137124