
Regulatory T Cell Dysfunction in Autoimmune Diseases

Abstract
:1. Introduction
2. Suppressive Function of Regulatory T Cells
- (1)
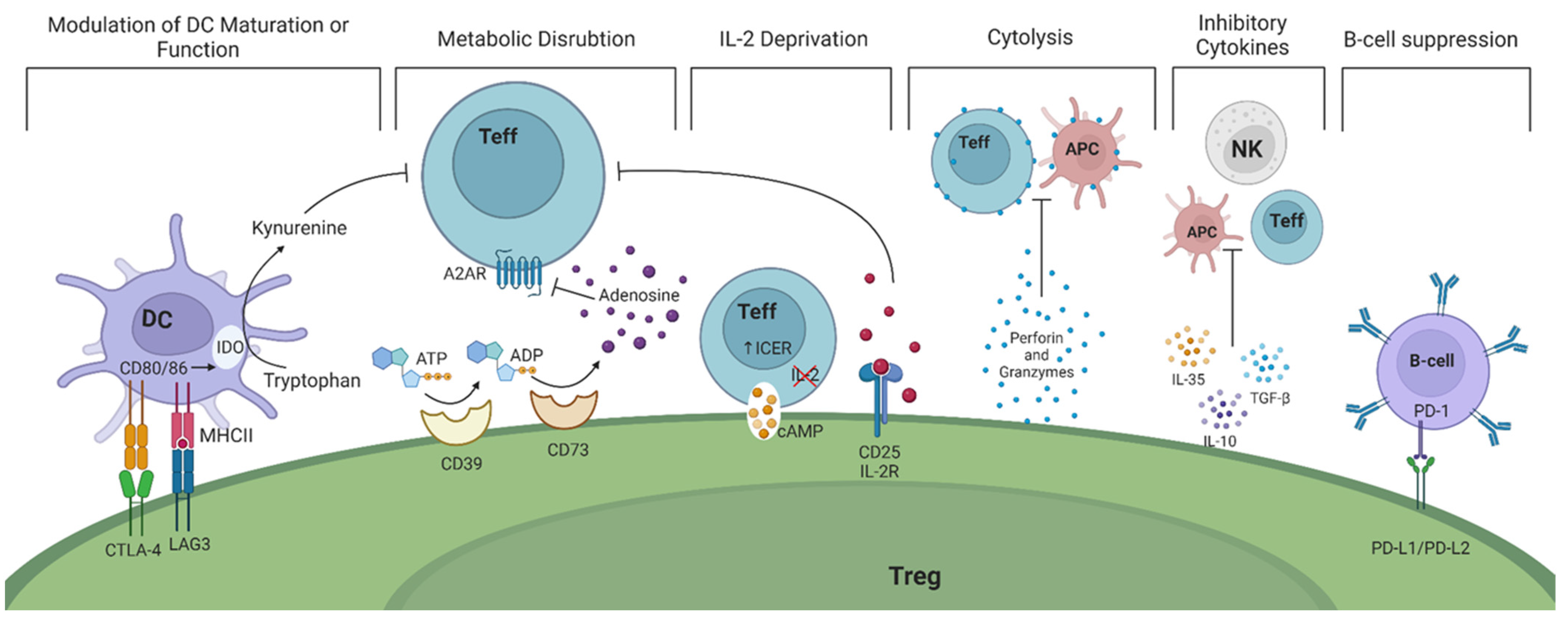
- Modulation of dendritic cells (DCs) in a cell—cell dependent manner ultimately prevents their maturation. This is accomplished through the expression of CTLA-4 and lymphocyte activation gene 3 (LAG-3) on the surface of Tregs. CTLA-4 and LAG-3 can interact with CD80/86 co-stimulatory molecules and major histocompatibility complex (MHC) class II on DCs, respectively [10,11]. As a result, the function and maturation of DCs are disrupted and they become unable to perform their effector function, namely activating Teffs. Studies in mice have shown that CTLA-4 is essential for the suppressive function of FoxP3+ Tregs while LAG-3 is required for maximal Treg activity in vitro and in vivo [12,13]. In humans, it is intriguing that only high levels of CTLA-4 expression are observed in CD45RO+FoxP3high effector Tregs (Table 1) [14]. Recent evidence suggest that Tregs may exert a dual suppressive action via trogocytosis of APC membranes by depleting CD80/86 (in a CTLA-4 dependent manner) and stable peptide-MHCII complexes to simultaneously deprive co-stimulation and antigenic stimulation of both naïve and activated (antigen-specific) Tconvs [15,16]. Whether the interaction of APCs with CTLA-4 is indispensable for the in vitro suppressive function of Tregs and whether TCR/MHC peptide engagement is necessary or enhances Treg suppressive function remain to be elucidated.
- (2)
- Tregs exert their suppressive function by disrupting metabolic pathways via the highly expressed ectoenzymes CD39/CD73 on their cell surface. These enzymes catalyze the hydrolysis of extracellular adenosine triphosphate (ATP) and adenosine diphosphate (ADP) to adenosine monophosphate (AMP) and subsequently adenosine, which binds to adenosine receptor 2A (A2AR), expressed on Teffs. The adenosine-A2AR signaling pathway ultimately suppresses Teff responses [36]. The expression of CD39 and CD73 have increasingly been used as markers of Tregs due to their potential contribution to the suppressive activity of Tregs (Table 2) [37,38].
- Upon activation, resting T cells rapidly upregulate their metabolic and biosynthetic machinery, including increased translational activity, prior to impending cell division. Recent data suggest that through mTORc signaling mediated by IL-10 and TGFβ, Tregs inhibit protein synthesis in effector T cells by preventing translation of mRNAs encoding components of protein synthesis machinery, thereby preventing proliferation of Teff cells before it has begun [55]. In addition, a functional mitochondrial complex-III in Tregs is needed to maintain immune regulatory gene expression and suppressive function of Tregs [56].
- (3)
- Tregs inhibit Teffs through the deprivation of interleukin-2 (IL-2), a cytokine that plays a critical role in T cell activation and proliferation. Tregs upregulate IL-2 receptor (CD25), allowing them to bind IL-2 and prevent neighboring cells from receiving pro-inflammatory cytokine signals [57]. IL-2 is also necessary for the suppressive activity of Tregs since it activates and phosphorylates the signal transducer and activator of transcription 5 (STAT5), leading to FoxP3 gene transcription [58]. Signaling through IL-2 receptors significantly upregulates CD25 expression, enhancing Tregs’ ability to bind IL-2 and further deprive surrounding Teffs [59,60]. Mice with deficiencies in either IL-2 or its receptor display enlarged peripheral lymphoid organs, impaired activation-induced cell death, and autoimmune disorders, all attributed to a diminished Treg generation [5].
- (4)
- Tregs also secrete various anti-inflammatory cytokines, including IL-10, IL-35, and transforming growth factor-β (TGF-β). These cytokines restrain the immune responses of T helper (Th)1 and Th17 cells and the production of interferon γ (IFN-γ) and IL-17, respectively [19].
- (5)
- The release of granzyme A, granzyme B, and perforin by Tregs directly kills Teffs and B cells by the induction of cytolysis [19].
- Extracellular vesicles (EVs) derived from Tregs is another mode deployed by Tregs to modulate immune responses. Treg-derived EV cargo includes immunomodulatory proteins and cytokines such as CD25 and IL-35, and miRNAs that reduce T-cell proliferation, IL-2 and IFNγ release, modify DC function, and promote IL-10 production by murine DCs [53,61,62].
- (6)
- Tregs have also been found to directly impact B cells through the interaction of programmed death-ligand 1 (PD-L1)/programmed cell death protein (PD-1) [63]. Tregs indirectly prevent B cell activation by inhibiting T follicular helper (Tfh) and Th-cells. This results in the inability of Tfh cells to activate B cells and Th cells to produce antibodies [63,64].
3. Molecular Signatures for Identifying Regulatory T Cells
4. Subsets of Regulatory T Cells
4.1. FoxP3+CD4+ Regulatory T Cells
4.2. Subpopulations of CD4+ Regulatory T Cells
4.3. Plasticity of Treg Phenotypes
5. Regulatory T Cells Dysfunction in Autoimmune Diseases
5.1. Type 1 Diabetes
5.2. Systemic Lupus Erythematosus
5.3. Rheumatoid Arthritis
5.4. Inflammatory Bowel Disease
6. Therapeutic Approaches to Restore Immune Tolerance
7. Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sakaguchi, S.; Yamaguchi, T.; Nomura, T.; Ono, M. Regulatory T Cells and Immune Tolerance. Cell 2008, 133, 775–787. [Google Scholar] [CrossRef] [PubMed]
- Hori, S.; Nomura, T.; Sakaguchi, S. Control of Regulatory T Cell Development by the Transcription Factor Foxp3. Science 2003, 299, 1057–1061. [Google Scholar] [CrossRef] [PubMed]
- Bennett, C.L.; Christie, J.; Ramsdell, F.; Brunkow, M.E.; Ferguson, P.J.; Whitesell, L.; Kelly, T.E.; Saulsbury, F.T.; Chance, P.F.; Ochs, H.D. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat. Genet. 2001, 27, 20–21. [Google Scholar] [CrossRef] [PubMed]
- Wildin, R.S.; Ramsdell, F.; Peake, J.; Faravelli, F.; Casanova, J.L.; Buist, N.; Levy-Lahad, E.; Mazzella, M.; Goulet, O.; Perroni, L.; et al. X-linked neonatal diabetes mellitus, enteropathy and endocrinopathy syndrome is the human equivalent of mouse scurfy. Nat. Genet. 2001, 27, 18–20. [Google Scholar] [CrossRef] [PubMed]
- Goudy, K.; Aydin, D.; Barzaghi, F.; Gambineri, E.; Vignoli, M.; Ciullini Mannurita, S.; Doglioni, C.; Ponzoni, M.; Cicalese, M.P.; Assanelli, A.; et al. Human IL2RA null mutation mediates immunodeficiency with lymphoproliferation and autoimmunity. Clin. Immunol. 2013, 146, 248–261. [Google Scholar] [CrossRef] [PubMed]
- Schubert, D.; Bode, C.; Kenefeck, R.; Hou, T.Z.; Wing, J.B.; Kennedy, A.; Bulashevska, A.; Petersen, B.S.; Schäffer, A.A.; Grüning, B.A.; et al. Autosomal dominant immune dysregulation syndrome in humans with CTLA4 mutations. Nat. Med. 2014, 20, 1410–1416. [Google Scholar] [CrossRef] [PubMed]
- Kucuk, Z.Y.; Charbonnier, L.M.; McMasters, R.L.; Chatila, T.; Bleesing, J.J. CTLA-4 haploinsufficiency in a patient with an autoimmune lymphoproliferative disorder. J. Allergy Clin. Immunol. 2017, 140, 862–864.e4. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, S.; Tsang, J.S.; Park, K. Systems immunology of regulatory T cells: Can one circuit explain it all? Trends Immunol. 2023, 44, 766–781. [Google Scholar] [CrossRef]
- Ohue, Y.; Nishikawa, H. Regulatory T (Treg) cells in cancer: Can Treg cells be a new therapeutic target? Cancer Sci. 2019, 110, 2080–2089. [Google Scholar] [CrossRef]
- Walker, L.S.; Sansom, D.M. The emerging role of CTLA4 as a cell-extrinsic regulator of T cell responses. Nat. Rev.Immunol. 2011, 11, 852–863. [Google Scholar] [CrossRef]
- Maruhashi, T.; Sugiura, D.; Okazaki, I.M.; Shimizu, K.; Maeda, T.K.; Ikubo, J.; Yoshikawa, H.; Maenaka, K.; Ishimaru, N.; Kosako, H.; et al. Binding of LAG-3 to stable peptide-MHC class II limits T cell function and suppresses autoimmunity and anti-cancer immunity. Immunity 2022, 55, 912–924.e8. [Google Scholar] [CrossRef] [PubMed]
- Wing, K.; Onishi, Y.; Prieto-Martin, P.; Yamaguchi, T.; Miyara, M.; Fehervari, Z.; Nomura, T.; Sakaguchi, S. CTLA-4 control over Foxp3+ regulatory T cell function. Science 2008, 322, 271–275. [Google Scholar] [CrossRef] [PubMed]
- Park, J.M.; Greten, F.R.; Wong, A.; Westrick, R.J.; Arthur, J.S.; Otsu, K.; Hoffmann, A.; Montminy, M.; Karin, M. Signaling pathways and genes that inhibit pathogen-induced macrophage apoptosis—CREB and NF-kappaB as key regulators. Immunity 2005, 23, 319–329. [Google Scholar] [CrossRef]
- Miyara, M.; Yoshioka, Y.; Kitoh, A.; Shima, T.; Wing, K.; Niwa, A.; Parizot, C.; Taflin, C.; Heike, T.; Valeyre, D.; et al. Functional Delineation and Differentiation Dynamics of Human CD4+ T Cells Expressing the FoxP3 Transcription Factor. Immunity 2009, 30, 899–911. [Google Scholar] [CrossRef] [PubMed]
- Tekguc, M.; Wing, J.B.; Osaki, M.; Long, J.; Sakaguchi, S. Treg-expressed CTLA-4 depletes CD80/CD86 by trogocytosis, releasing free PD-L1 on antigen-presenting cells. Proc. Natl. Acad. Sci. USA 2021, 118, e2023739118. [Google Scholar] [CrossRef] [PubMed]
- Akkaya, B.; Oya, Y.; Akkaya, M.; Al Souz, J.; Holstein, A.H.; Kamenyeva, O.; Kabat, J.; Matsumura, R.; Dorward, D.W.; Glass, D.D.; et al. Regulatory T cells mediate specific suppression by depleting peptide–MHC class II from dendritic cells. Nat. Immunol. 2019, 20, 218–231. [Google Scholar] [CrossRef] [PubMed]
- Seddiki, N.; Santner-Nanan, B.; Tangye, S.G.; Alexander, S.I.; Solomon, M.; Lee, S.; Nanan, R.; Fazekas de Saint Groth, B. Persistence of naive CD45RA+ regulatory T cells in adult life. Blood 2006, 107, 2830–2838. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.R.; Yang, W.C.; Chen, H.W. The fate of regulatory T cells: Survival or apoptosis. Cell. Mol. Immunol. 2014, 11, 11–13. [Google Scholar] [CrossRef] [PubMed]
- Goswami, T.K.; Singh, M.; Dhawan, M.; Mitra, S.; Emran, T.B.; Rabaan, A.A.; Mutair, A.A.; Alawi, Z.A.; Alhumaid, S.; Dhama, K. Regulatory T cells (Tregs) and their therapeutic potential against autoimmune disorders—Advances and challenges. Hum. Vaccin. Immunother. 2022, 18, 2035117. [Google Scholar] [CrossRef] [PubMed]
- Jordan, M.S.; Boesteanu, A.; Reed, A.J.; Petrone, A.L.; Holenbeck, A.E.; Lerman, M.A.; Naji, A.; Caton, A.J. Thymic selection of CD4+CD25+ regulatory T cells induced by an agonist self-peptide. Nat. Immunol. 2001, 2, 301–306. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Hanabuchi, S.; Wang, Y.H.; Park, W.R.; Arima, K.; Bover, L.; Qin, F.X.; Gilliet, M.; Liu, Y.J. Two functional subsets of FOXP3+ regulatory T cells in human thymus and periphery. Immunity 2008, 28, 870–880. [Google Scholar] [CrossRef] [PubMed]
- Feger, U.; Tolosa, E.; Huang, Y.H.; Waschbisch, A.; Biedermann, T.; Melms, A.; Wiendl, H. HLA-G expression defines a novel regulatory T-cell subset present in human peripheral blood and sites of inflammation. Blood 2007, 110, 568–577. [Google Scholar] [CrossRef] [PubMed]
- Pankratz, S.; Bittner, S.; Herrmann, A.M.; Schuhmann, M.K.; Ruck, T.; Meuth, S.G.; Wiendl, H. Human CD4+ HLA-G+ regulatory T cells are potent suppressors of graft-versus-host disease in vivo. Faseb J. 2014, 28, 3435–3445. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.-H.; Zozulya, A.L.; Weidenfeller, C.; Schwab, N.; Wiendl, H. T cell suppression by naturally occurring HLA-G-expressing regulatory CD4+ T cells is IL-10-dependent and reversible. J. Leukoc. Biol. 2009, 86, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Baecher-Allan, C.; Wolf, E.; Hafler, D.A. MHC Class II Expression Identifies Functionally Distinct Human Regulatory T Cells1. J. Immunol. 2006, 176, 4622–4631. [Google Scholar] [CrossRef] [PubMed]
- Thornton, A.M.; Korty, P.E.; Tran, D.Q.; Wohlfert, E.A.; Murray, P.E.; Belkaid, Y.; Shevach, E.M. Expression of Helios, an Ikaros transcription factor family member, differentiates thymic-derived from peripherally induced Foxp3+ T regulatory cells. J. Immunol. 2010, 184, 3433–3441. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Chen, M.; Liu, Y.; Guo, Z.; He, X.; Brand, D.; Zheng, S.G. Advances in distinguishing natural from induced Foxp3(+) regulatory T cells. Int. J. Clin. Exp. Pathol. 2013, 6, 116–123. [Google Scholar] [PubMed]
- Zeng, H.; Zhang, R.; Jin, B.; Chen, L. Type 1 regulatory T cells: A new mechanism of peripheral immune tolerance. Cell. Mol. Immunol. 2015, 12, 566–571. [Google Scholar] [CrossRef] [PubMed]
- Groux, H.; O’Garra, A.; Bigler, M.; Rouleau, M.; Antonenko, S.; de Vries, J.E.; Roncarolo, M.G. A CD4+T-cell subset inhibits antigen-specific T-cell responses and prevents colitis. Nature 1997, 389, 737–742. [Google Scholar] [CrossRef] [PubMed]
- Levings, M.K.; Sangregorio, R.; Galbiati, F.; Squadrone, S.; de Waal Malefyt, R.; Roncarolo, M.G. IFN-alpha and IL-10 induce the differentiation of human type 1 T regulatory cells. J. Immunol. 2001, 166, 5530–5539. [Google Scholar] [CrossRef] [PubMed]
- Weiner, H.L. Induction and mechanism of action of transforming growth factor-beta-secreting Th3 regulatory cells. Immunol. Rev. 2001, 182, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Weiner, H.L. The mucosal milieu creates tolerogenic dendritic cells and TR1 and TH3 regulatory cells. Nat. Immunol. 2001, 2, 671–672. [Google Scholar] [CrossRef] [PubMed]
- Collison, L.W.; Chaturvedi, V.; Henderson, A.L.; Giacomin, P.R.; Guy, C.; Bankoti, J.; Finkelstein, D.; Forbes, K.; Workman, C.J.; Brown, S.A.; et al. IL-35-mediated induction of a potent regulatory T cell population. Nat. Immunol. 2010, 11, 1093–1101. [Google Scholar] [CrossRef] [PubMed]
- Hsu, L.H.; Li, K.P.; Chu, K.H.; Chiang, B.L. A B-1a cell subset induces Foxp3(−) T cells with regulatory activity through an IL-10-independent pathway. Cell. Mol. Immunol. 2015, 12, 354–365. [Google Scholar] [CrossRef] [PubMed]
- Chien, C.H.; Chiang, B.L. Regulatory T cells induced by B cells: A novel subpopulation of regulatory T cells. J. Biomed. Sci. 2017, 24, 86. [Google Scholar] [CrossRef] [PubMed]
- Vignali, D.A.; Collison, L.W.; Workman, C.J. How regulatory T cells work. Nat. Rev. Immunol. 2008, 8, 523–532. [Google Scholar] [CrossRef] [PubMed]
- Antonioli, L.; Pacher, P.; Vizi, E.S.; Haskó, G. CD39 and CD73 in immunity and inflammation. Trends Mol. Med. 2013, 19, 355–367. [Google Scholar] [CrossRef] [PubMed]
- Schuler, P.J.; Harasymczuk, M.; Schilling, B.; Lang, S.; Whiteside, T.L. Separation of human CD4+CD39+ T cells by magnetic beads reveals two phenotypically and functionally different subsets. J. Immunol. Methods 2011, 369, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Haribhai, D.; Relland, L.M.; Truong, N.; Carlson, M.R.; Williams, C.B.; Chatila, T.A. Regulatory T cell development in the absence of functional Foxp3. Nat. Immunol. 2007, 8, 359–368. [Google Scholar] [CrossRef]
- Fontenot, J.D.; Rasmussen, J.P.; Williams, L.M.; Dooley, J.L.; Farr, A.G.; Rudensky, A.Y. Regulatory T cell lineage specification by the forkhead transcription factor foxp3. Immunity 2005, 22, 329–341. [Google Scholar] [CrossRef]
- Sakaguchi, S.; Sakaguchi, N.; Asano, M.; Itoh, M.; Toda, M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J. Immunol. 1995, 155, 1151–1164. [Google Scholar] [CrossRef] [PubMed]
- Baecher-Allan, C.; Brown, J.A.; Freeman, G.J.; Hafler, D.A. CD4+CD25high regulatory cells in human peripheral blood. J. Immunol. 2001, 167, 1245–1253. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Putnam, A.L.; Xu-Yu, Z.; Szot, G.L.; Lee, M.R.; Zhu, S.; Gottlieb, P.A.; Kapranov, P.; Gingeras, T.R.; Fazekas de St Groth, B.; et al. CD127 expression inversely correlates with FoxP3 and suppressive function of human CD4+ T reg cells. J. Exp. Med. 2006, 203, 1701–1711. [Google Scholar] [CrossRef] [PubMed]
- Seddiki, N.; Santner-Nanan, B.; Martinson, J.; Zaunders, J.; Sasson, S.; Landay, A.; Solomon, M.; Selby, W.; Alexander, S.I.; Nanan, R.; et al. Expression of interleukin (IL)-2 and IL-7 receptors discriminates between human regulatory and activated T cells. J. Exp. Med. 2006, 203, 1693–1700. [Google Scholar] [CrossRef] [PubMed]
- Aerts, N.E.; Dombrecht, E.J.; Ebo, D.G.; Bridts, C.H.; Stevens, W.J.; De Clerck, L.S. Activated T cells complicate the identification of regulatory T cells in rheumatoid arthritis. Cell. Immunol. 2008, 251, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Ni, X.; Pan, X.; Lu, H.; Lu, Y.; Zhao, J.; Guo Zheng, S.; Hippen, K.L.; Wang, X.; Lu, L. Human CD39(hi) regulatory T cells present stronger stability and function under inflammatory conditions. Cell. Mol.Immunol. 2017, 14, 521–528. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, J.; Yamazaki, S.; Takahashi, T.; Ishida, Y.; Sakaguchi, S. Stimulation of CD25(+)CD4(+) regulatory T cells through GITR breaks immunological self-tolerance. Nat. Immunol. 2002, 3, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Ronchetti, S.; Ricci, E.; Petrillo, M.G.; Cari, L.; Migliorati, G.; Nocentini, G.; Riccardi, C. Glucocorticoid-induced tumour necrosis factor receptor-related protein: A key marker of functional regulatory T cells. J. Immunol. Res. 2015, 2015, 171520. [Google Scholar] [CrossRef] [PubMed]
- Tai, X.; Van Laethem, F.; Pobezinsky, L.; Guinter, T.; Sharrow, S.O.; Adams, A.; Granger, L.; Kruhlak, M.; Lindsten, T.; Thompson, C.B.; et al. Basis of CTLA-4 function in regulatory and conventional CD4(+) T cells. Blood 2012, 119, 5155–5163. [Google Scholar] [CrossRef]
- Yokosuka, T.; Kobayashi, W.; Takamatsu, M.; Sakata-Sogawa, K.; Zeng, H.; Hashimoto-Tane, A.; Yagita, H.; Tokunaga, M.; Saito, T. Spatiotemporal Basis of CTLA-4 Costimulatory Molecule-Mediated Negative Regulation of T Cell Activation. Immunity 2010, 33, 326–339. [Google Scholar] [CrossRef]
- Huang, C.T.; Workman, C.J.; Flies, D.; Pan, X.; Marson, A.L.; Zhou, G.; Hipkiss, E.L.; Ravi, S.; Kowalski, J.; Levitsky, H.I.; et al. Role of LAG-3 in regulatory T cells. Immunity 2004, 21, 503–513. [Google Scholar] [CrossRef] [PubMed]
- Mandapathil, M.; Lang, S.; Gorelik, E.; Whiteside, T.L. Isolation of functional human regulatory T cells (Treg) from the peripheral blood based on the CD39 expression. J. Immunol. Methods 2009, 346, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Grover, P.; Goel, P.N.; Greene, M.I. Regulatory T Cells: Regulation of Identity and Function. Front. Immunol. 2021, 12, 750542. [Google Scholar] [CrossRef] [PubMed]
- Akkaya, B.; Shevach, E.M. Regulatory T cells: Master thieves of the immune system. Cell. Immunol. 2020, 355, 104160. [Google Scholar] [CrossRef] [PubMed]
- So, L.; Obata-Ninomiya, K.; Hu, A.; Muir, V.S.; Takamori, A.; Song, J.; Buckner, J.H.; Savan, R.; Ziegler, S.F. Regulatory T cells suppress CD4+ effector T cell activation by controlling protein synthesis. J. Exp. Med. 2023, 220, e20221676. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, S.E.; Singer, B.D.; Steinert, E.M.; Martinez, C.A.; Mehta, M.M.; Martínez-Reyes, I.; Gao, P.; Helmin, K.A.; Abdala-Valencia, H.; Sena, L.A.; et al. Mitochondrial complex III is essential for suppressive function of regulatory T cells. Nature 2019, 565, 495–499. [Google Scholar] [CrossRef]
- Fan, M.Y.; Low, J.S.; Tanimine, N.; Finn, K.K.; Priyadharshini, B.; Germana, S.K.; Kaech, S.M.; Turka, L.A. Differential Roles of IL-2 Signaling in Developing versus Mature Tregs. Cell Rep. 2018, 25, 1204–1213.e4. [Google Scholar] [CrossRef] [PubMed]
- Zorn, E.; Nelson, E.A.; Mohseni, M.; Porcheray, F.; Kim, H.; Litsa, D.; Bellucci, R.; Raderschall, E.; Canning, C.; Soiffer, R.J.; et al. IL-2 regulates FOXP3 expression in human CD4+CD25+ regulatory T cells through a STAT-dependent mechanism and induces the expansion of these cells in vivo. Blood 2006, 108, 1571–1579. [Google Scholar] [CrossRef] [PubMed]
- Malek, T.R.; Castro, I. Interleukin-2 receptor signaling: At the interface between tolerance and immunity. Immunity 2010, 33, 153–165. [Google Scholar] [CrossRef]
- Malek, T.R. The biology of interleukin-2. Annu. Rev. Immunol. 2008, 26, 453–479. [Google Scholar] [CrossRef]
- Aloi, N.; Drago, G.; Ruggieri, S.; Cibella, F.; Colombo, P.; Longo, V. Extracellular Vesicles and Immunity: At the Crossroads of Cell Communication. Int. J. Mol. Sci. 2024, 25, 1205. [Google Scholar] [CrossRef]
- Lin, C.; Guo, J.; Jia, R. Roles of Regulatory T Cell-Derived Extracellular Vesicles in Human Diseases. Int. J. Mol. Sci. 2022, 23, 1206. [Google Scholar] [CrossRef]
- Gotot, J.; Gottschalk, C.; Leopold, S.; Knolle, P.A.; Yagita, H.; Kurts, C.; Ludwig-Portugall, I. Regulatory T cells use programmed death 1 ligands to directly suppress autoreactive B cells in vivo. Proc. Natl. Acad. Sci. USA 2012, 109, 10468–10473. [Google Scholar] [CrossRef]
- Wang, P.; Zheng, S.G. Regulatory T cells and B cells: Implication on autoimmune diseases. Int. J. Clin. Exp. Pathol. 2013, 6, 2668–2674. [Google Scholar]
- Dikiy, S.; Rudensky, A.Y. Principles of regulatory T cell function. Immunity 2023, 56, 240–255. [Google Scholar] [CrossRef]
- Yang, S.; Fujikado, N.; Kolodin, D.; Benoist, C.; Mathis, D. Regulatory T cells generated early in life play a distinct role in maintaining self-tolerance. Science 2015, 348, 589–594. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Zhao, W.; Wang, Y.; Jin, L.; Jin, G.; Sun, X.; Wang, W.; Wang, K.; Xu, X.; Hao, J.; et al. A wave of Foxp3+ regulatory T cell accumulation in the neonatal liver plays unique roles in maintaining self-tolerance. Cell. Mol. Immunol. 2020, 17, 507–518. [Google Scholar] [CrossRef] [PubMed]
- Cipolletta, D.; Feuerer, M.; Li, A.; Kamei, N.; Lee, J.; Shoelson, S.E.; Benoist, C.; Mathis, D. PPAR-γ is a major driver of the accumulation and phenotype of adipose tissue Treg cells. Nature 2012, 486, 549–553. [Google Scholar] [CrossRef] [PubMed]
- Hirata, Y.; Furuhashi, K.; Ishii, H.; Li, H.W.; Pinho, S.; Ding, L.; Robson, S.C.; Frenette, P.S.; Fujisaki, J. CD150(high) Bone Marrow Tregs Maintain Hematopoietic Stem Cell Quiescence and Immune Privilege via Adenosine. Cell Stem Cell 2018, 22, 445–453.e5. [Google Scholar] [CrossRef] [PubMed]
- Ito, M.; Komai, K.; Mise-Omata, S.; Iizuka-Koga, M.; Noguchi, Y.; Kondo, T.; Sakai, R.; Matsuo, K.; Nakayama, T.; Yoshie, O.; et al. Brain regulatory T cells suppress astrogliosis and potentiate neurological recovery. Nature 2019, 565, 246–250. [Google Scholar] [CrossRef] [PubMed]
- Ali, N.; Zirak, B.; Rodriguez, R.S.; Pauli, M.L.; Truong, H.A.; Lai, K.; Ahn, R.; Corbin, K.; Lowe, M.M.; Scharschmidt, T.C.; et al. Regulatory T Cells in Skin Facilitate Epithelial Stem Cell Differentiation. Cell 2017, 169, 1119–1129.e11. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Hu, X.; Liang, Y.; Yu, J.; Li, H.; Shokhirev, M.N.; Zheng, Y. Glucocorticoid signaling and regulatory T cells cooperate to maintain the hair-follicle stem-cell niche. Nat. Immunol. 2022, 23, 1086–1097. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.N.; Gouirand, V.; Macon, C.E.; Lowe, M.M.; Boothby, I.C.; Moreau, J.M.; Gratz, I.K.; Stoecklinger, A.; Weaver, C.T.; Sharpe, A.H.; et al. Regulatory T cells in skin mediate immune privilege of the hair follicle stem cell niche. Sci. Immunol. 2024, 9, eadh0152. [Google Scholar] [CrossRef] [PubMed]
- Dieckmann, D.; Plottner, H.; Berchtold, S.; Berger, T.; Schuler, G. Ex vivo isolation and characterization of CD4(+)CD25(+) T cells with regulatory properties from human blood. J. Exp. Med. 2001, 193, 1303–1310. [Google Scholar] [CrossRef] [PubMed]
- Taylor, P.A.; Noelle, R.J.; Blazar, B.R. CD4(+)CD25(+) immune regulatory cells are required for induction of tolerance to alloantigen via costimulatory blockade. J. Exp. Med. 2001, 193, 1311–1318. [Google Scholar] [CrossRef] [PubMed]
- Fontenot, J.D.; Gavin, M.A.; Rudensky, A.Y. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat. Immunol. 2003, 4, 330–336. [Google Scholar] [CrossRef] [PubMed]
- Roncador, G.; Brown, P.J.; Maestre, L.; Hue, S.; Martínez-Torrecuadrada, J.L.; Ling, K.L.; Pratap, S.; Toms, C.; Fox, B.C.; Cerundolo, V.; et al. Analysis of FOXP3 protein expression in human CD4+CD25+ regulatory T cells at the single-cell level. Eur. J. Immunol. 2005, 35, 1681–1691. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Ioan-Facsinay, A.; van der Voort, E.I.; Huizinga, T.W.; Toes, R.E. Transient expression of FOXP3 in human activated nonregulatory CD4+ T cells. Eur. J. Immunol. 2007, 37, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Janson, P.C.; Winerdal, M.E.; Marits, P.; Thörn, M.; Ohlsson, R.; Winqvist, O. FOXP3 promoter demethylation reveals the committed Treg population in humans. PLoS ONE 2008, 3, e1612. [Google Scholar] [CrossRef]
- Klein, S.; Kretz, C.C.; Krammer, P.H.; Kuhn, A. CD127(low/-) and FoxP3(+) expression levels characterize different regulatory T-cell populations in human peripheral blood. J. Investig. Dermatol. 2010, 130, 492–499. [Google Scholar] [CrossRef]
- Akimova, T.; Beier, U.H.; Wang, L.; Levine, M.H.; Hancock, W.W. Helios expression is a marker of T cell activation and proliferation. PLoS ONE 2011, 6, e24226. [Google Scholar] [CrossRef] [PubMed]
- Matos, T.R.; Hirakawa, M.; Alho, A.C.; Neleman, L.; Graca, L.; Ritz, J. Maturation and Phenotypic Heterogeneity of Human CD4+ Regulatory T Cells From Birth to Adulthood and After Allogeneic Stem Cell Transplantation. Front. Immunol. 2020, 11, 570550. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, K.; Kim, H.T.; McDonough, S.; Bascug, G.; Warshauer, B.; Koreth, J.; Cutler, C.; Ho, V.T.; Alyea, E.P.; Antin, J.H.; et al. Altered regulatory T cell homeostasis in patients with CD4+ lymphopenia following allogeneic hematopoietic stem cell transplantation. J. Clin. Investig. 2010, 120, 1479–1493. [Google Scholar] [CrossRef] [PubMed]
- Yadav, M.; Louvet, C.; Davini, D.; Gardner, J.M.; Martinez-Llordella, M.; Bailey-Bucktrout, S.; Anthony, B.A.; Sverdrup, F.M.; Head, R.; Kuster, D.J.; et al. Neuropilin-1 distinguishes natural and inducible regulatory T cells among regulatory T cell subsets in vivo. J. Exp. Med. 2012, 209, 1713–1722. [Google Scholar] [CrossRef] [PubMed]
- Workman, C.J.; Szymczak-Workman, A.L.; Collison, L.W.; Pillai, M.R.; Vignali, D.A. The development and function of regulatory T cells. Cell. Mol. Life Sci. 2009, 66, 2603–2622. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Josefowicz, S.; Chaudhry, A.; Peng, X.P.; Forbush, K.; Rudensky, A.Y. Role of conserved non-coding DNA elements in the Foxp3 gene in regulatory T-cell fate. Nature 2010, 463, 808–812. [Google Scholar] [CrossRef] [PubMed]
- Morikawa, H.; Sakaguchi, S. Genetic and epigenetic basis of Treg cell development and function: From a FoxP3-centered view to an epigenome-defined view of natural Treg cells. Immunol. Rev. 2014, 259, 192–205. [Google Scholar] [CrossRef] [PubMed]
- Gavin, M.A.; Torgerson, T.R.; Houston, E.; DeRoos, P.; Ho, W.Y.; Stray-Pedersen, A.; Ocheltree, E.L.; Greenberg, P.D.; Ochs, H.D.; Rudensky, A.Y. Single-cell analysis of normal and FOXP3-mutant human T cells: FOXP3 expression without regulatory T cell development. Proc. Natl. Acad. Sci. USA 2006, 103, 6659–6664. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Lopes, J.E.; Chong, M.M.; Ivanov, I.I.; Min, R.; Victora, G.D.; Shen, Y.; Du, J.; Rubtsov, Y.P.; Rudensky, A.Y.; et al. TGF-beta-induced Foxp3 inhibits T(H)17 cell differentiation by antagonizing RORgammat function. Nature 2008, 453, 236–240. [Google Scholar] [CrossRef]
- Malek, T.R.; Yu, A.; Zhu, L.; Matsutani, T.; Adeegbe, D.; Bayer, A.L. IL-2 family of cytokines in T regulatory cell development and homeostasis. J. Clin. Immunol. 2008, 28, 635–639. [Google Scholar] [CrossRef]
- Komatsu, N.; Okamoto, K.; Sawa, S.; Nakashima, T.; Oh-hora, M.; Kodama, T.; Tanaka, S.; Bluestone, J.A.; Takayanagi, H. Pathogenic conversion of Foxp3+ T cells into TH17 cells in autoimmune arthritis. Nat. Med. 2014, 20, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Jonuleit, H.; Schmitt, E. The regulatory T cell family: Distinct subsets and their interrelations. J. Immunol. 2003, 171, 6323–6327. [Google Scholar] [CrossRef] [PubMed]
- Hoeppli, R.E.; MacDonald, K.G.; Levings, M.K.; Cook, L. How antigen specificity directs regulatory T-cell function: Self, foreign and engineered specificity. Hla 2016, 88, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Paiva, R.S.; Lino, A.C.; Bergman, M.L.; Caramalho, I.; Sousa, A.E.; Zelenay, S.; Demengeot, J. Recent thymic emigrants are the preferential precursors of regulatory T cells differentiated in the periphery. Proc. Natl. Acad. Sci. USA 2013, 110, 6494–6499. [Google Scholar] [CrossRef] [PubMed]
- Long, S.A.; Rieck, M.; Tatum, M.; Bollyky, P.L.; Wu, R.P.; Muller, I.; Ho, J.-C.; Shilling, H.G.; Buckner, J.H. Low-dose antigen promotes induction of FOXP3 in human CD4+ T cells. J. Immunol. 2011, 187, 3511–3520. [Google Scholar] [CrossRef] [PubMed]
- Chien, C.H.; Yu, H.H.; Chiang, B.L. Single allergen-induced oral tolerance inhibits airway inflammation in conjugated allergen immunized mice. J. Allergy Clin. Immunol. 2015, 136, 1110–1113.e4. [Google Scholar] [CrossRef] [PubMed]
- Shao, T.Y.; Hsu, L.H.; Chien, C.H.; Chiang, B.L. Novel Foxp3(−) IL-10(−) Regulatory T-cells Induced by B-Cells Alleviate Intestinal Inflammation in Vivo. Sci. Rep. 2016, 6, 32415. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.Y.; Hsu, W.T.; Chen, Y.L.; Chien, C.H.; Chiang, B.L. Lymphocyte-activation gene 3(+) (LAG3(+)) forkhead box protein 3(−) (FOXP3(−)) regulatory T cells induced by B cells alleviates joint inflammation in collagen-induced arthritis. J. Autoimmun. 2016, 68, 75–85. [Google Scholar] [CrossRef]
- Lee, J.; Kim, D.; Min, B. Tissue Resident Foxp3(+) Regulatory T Cells: Sentinels and Saboteurs in Health and Disease. Front. Immunol. 2022, 13, 865593. [Google Scholar] [CrossRef]
- Feuerer, M.; Herrero, L.; Cipolletta, D.; Naaz, A.; Wong, J.; Nayer, A.; Lee, J.; Goldfine, A.B.; Benoist, C.; Shoelson, S.; et al. Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat. Med. 2009, 15, 930–939. [Google Scholar] [CrossRef]
- Spanier, J.A.; Fung, V.; Wardell, C.M.; Alkhatib, M.H.; Chen, Y.; Swanson, L.A.; Dwyer, A.J.; Weno, M.E.; Silva, N.; Mitchell, J.S.; et al. Tregs with an MHC class II peptide-specific chimeric antigen receptor prevent autoimmune diabetes in mice. J. Clin. Investig. 2023, 133, e168601. [Google Scholar] [CrossRef] [PubMed]
- Kitz, A.; Dominguez-Villar, M. Molecular mechanisms underlying Th1-like Treg generation and function. Cell. Mol. Life Sci. 2017, 74, 4059–4075. [Google Scholar] [CrossRef] [PubMed]
- Blatner, N.R.; Mulcahy, M.F.; Dennis, K.L.; Scholtens, D.; Bentrem, D.J.; Phillips, J.D.; Ham, S.; Sandall, B.P.; Khan, M.W.; Mahvi, D.M.; et al. Expression of RORγt marks a pathogenic regulatory T cell subset in human colon cancer. Sci. Transl. Med. 2012, 4, 164ra159. [Google Scholar] [CrossRef] [PubMed]
- Beriou, G.; Costantino, C.M.; Ashley, C.W.; Yang, L.; Kuchroo, V.K.; Baecher-Allan, C.; Hafler, D.A. IL-17-producing human peripheral regulatory T cells retain suppressive function. Blood 2009, 113, 4240–4249. [Google Scholar] [CrossRef] [PubMed]
- Ueno, A.; Jeffery, L.; Kobayashi, T.; Hibi, T.; Ghosh, S.; Jijon, H. Th17 plasticity and its relevance to inflammatory bowel disease. J. Autoimmun. 2018, 87, 38–49. [Google Scholar] [CrossRef] [PubMed]
- Eastaff-Leung, N.; Mabarrack, N.; Barbour, A.; Cummins, A.; Barry, S. Foxp3+ Regulatory T Cells, Th17 Effector Cells, and Cytokine Environment in Inflammatory Bowel Disease. J. Clin. Immunol. 2010, 30, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Kryczek, I.; Wu, K.; Zhao, E.; Wei, S.; Vatan, L.; Szeliga, W.; Huang, E.; Greenson, J.; Chang, A.; Roliński, J.; et al. IL-17+ regulatory T cells in the microenvironments of chronic inflammation and cancer. J. Immunol. 2011, 186, 4388–4395. [Google Scholar] [CrossRef] [PubMed]
- Halim, L.; Romano, M.; McGregor, R.; Correa, I.; Pavlidis, P.; Grageda, N.; Hoong, S.J.; Yuksel, M.; Jassem, W.; Hannen, R.F.; et al. An Atlas of Human Regulatory T Helper-like Cells Reveals Features of Th2-like Tregs that Support a Tumorigenic Environment. Cell Rep. 2017, 20, 757–770. [Google Scholar] [CrossRef] [PubMed]
- Wing, J.B.; Lim, E.L.; Sakaguchi, S. Control of foreign Ag-specific Ab responses by Treg and Tfr. Immunol. Rev. 2020, 296, 104–119. [Google Scholar] [CrossRef]
- Zhou, X.; Bailey-Bucktrout, S.L.; Jeker, L.T.; Penaranda, C.; Martínez-Llordella, M.; Ashby, M.; Nakayama, M.; Rosenthal, W.; Bluestone, J.A. Instability of the transcription factor Foxp3 leads to the generation of pathogenic memory T cells in vivo. Nat. Immunol. 2009, 10, 1000–1007. [Google Scholar] [CrossRef]
- McRitchie, B.R.; Akkaya, B. Exhaust the exhausters: Targeting regulatory T cells in the tumor microenvironment. Front. Immunol. 2022, 13, 940052. [Google Scholar] [CrossRef] [PubMed]
- Wong, H.S.; Park, K.; Gola, A.; Baptista, A.P.; Miller, C.H.; Deep, D.; Lou, M.; Boyd, L.F.; Rudensky, A.Y.; Savage, P.A.; et al. A local regulatory T cell feedback circuit maintains immune homeostasis by pruning self-activated T cells. Cell 2021, 184, 3981–3997.e22. [Google Scholar] [CrossRef] [PubMed]
- Becker, M.; Joseph, S.S.; Garcia-Carrizo, F.; Tom, R.Z.; Opaleva, D.; Serr, I.; Tschöp, M.H.; Schulz, T.J.; Hofmann, S.M.; Daniel, C. Regulatory T cells require IL6 receptor alpha signaling to control skeletal muscle function and regeneration. Cell Metab. 2023, 35, 1736–1751.e7. [Google Scholar] [CrossRef]
- Kukreja, A.; Cost, G.; Marker, J.; Zhang, C.; Sun, Z.; Lin-Su, K.; Ten, S.; Sanz, M.; Exley, M.; Wilson, B.; et al. Multiple immuno-regulatory defects in type-1 diabetes. J. Clin. Investig. 2002, 109, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Lindley, S.; Dayan, C.M.; Bishop, A.; Roep, B.O.; Peakman, M.; Tree, T.I. Defective suppressor function in CD4(+)CD25(+) T-cells from patients with type 1 diabetes. Diabetes 2005, 54, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Brusko, T.M.; Wasserfall, C.H.; Clare-Salzler, M.J.; Schatz, D.A.; Atkinson, M.A. Functional defects and the influence of age on the frequency of CD4+ CD25+ T-cells in type 1 diabetes. Diabetes 2005, 54, 1407–1414. [Google Scholar] [CrossRef] [PubMed]
- Brusko, T.; Wasserfall, C.; McGrail, K.; Schatz, R.; Viener, H.L.; Schatz, D.; Haller, M.; Rockell, J.; Gottlieb, P.; Clare-Salzler, M.; et al. No alterations in the frequency of FOXP3+ regulatory T-cells in type 1 diabetes. Diabetes 2007, 56, 604–612. [Google Scholar] [CrossRef] [PubMed]
- Putnam, A.L.; Vendrame, F.; Dotta, F.; Gottlieb, P.A. CD4+CD25high regulatory T cells in human autoimmune diabetes. J Autoimmun. 2005, 24, 55–62. [Google Scholar] [CrossRef]
- Mason, G.M.; Lowe, K.; Melchiotti, R.; Ellis, R.; de Rinaldis, E.; Peakman, M.; Heck, S.; Lombardi, G.; Tree, T.I. Phenotypic Complexity of the Human Regulatory T Cell Compartment Revealed by Mass Cytometry. J. Immunol. 2015, 195, 2030–2037. [Google Scholar] [CrossRef]
- Okubo, Y.; Torrey, H.; Butterworth, J.; Zheng, H.; Faustman, D.L. Treg activation defect in type 1 diabetes: Correction with TNFR2 agonism. Clin. Transl. Immunol. 2016, 5, e56. [Google Scholar] [CrossRef]
- Mills, K.H. Regulatory T cells: Friend or foe in immunity to infection? Nat. Rev. Immunol. 2004, 4, 841–855. [Google Scholar] [CrossRef] [PubMed]
- Bender, C.; Wiedeman, A.E.; Hu, A.; Ylescupidez, A.; Sietsema, W.K.; Herold, K.C.; Griffin, K.J.; Gitelman, S.E.; Long, S.A.; on behalf of the T-Rex Study Group. A phase 2 randomized trial with autologous polyclonal expanded regulatory T cells in children with new-onset type 1 diabetes. Sci. Transl. Med. 2024, 16, eadn2404. [Google Scholar] [CrossRef] [PubMed]
- Ward, N.C.; Yu, A.; Moro, A.; Ban, Y.; Chen, X.; Hsiung, S.; Keegan, J.; Arbanas, J.M.; Loubeau, M.; Thankappan, A.; et al. IL-2/CD25: A Long-Acting Fusion Protein That Promotes Immune Tolerance by Selectively Targeting the IL-2 Receptor on Regulatory T Cells. J. Immunol. 2018, 201, 2579–2592. [Google Scholar] [CrossRef] [PubMed]
- Ward, N.C.; Lui, J.B.; Hernandez, R.; Yu, L.; Struthers, M.; Xie, J.; Santos Savio, A.; Dwyer, C.J.; Hsiung, S.; Yu, A.; et al. Persistent IL-2 Receptor Signaling by IL-2/CD25 Fusion Protein Controls Diabetes in NOD Mice by Multiple Mechanisms. Diabetes 2020, 69, 2400–2413. [Google Scholar] [CrossRef] [PubMed]
- Bonelli, M.; Savitskaya, A.; Steiner, C.W.; Rath, E.; Smolen, J.S.; Scheinecker, C. Phenotypic and functional analysis of CD4+ CD25- Foxp3+ T cells in patients with systemic lupus erythematosus. J. Immunol. 2009, 182, 1689–1695. [Google Scholar] [CrossRef] [PubMed]
- Kleczynska, W.; Jakiela, B.; Plutecka, H.; Milewski, M.; Sanak, M.; Musial, J. Imbalance between Th17 and regulatory T-cells in systemic lupus erythematosus. Folia Histochem. Cytobiol. 2011, 49, 646–653. [Google Scholar] [CrossRef]
- Zhu, Y.; Huang, Y.; Ming, B.; Wu, X.; Chen, Y.; Dong, L. Regulatory T-cell levels in systemic lupus erythematosus patients: A meta-analysis. Lupus 2019, 28, 445–454. [Google Scholar] [CrossRef] [PubMed]
- Mizui, M.; Tsokos, G.C. Targeting Regulatory T Cells to Treat Patients With Systemic Lupus Erythematosus. Front. Immunol. 2018, 9, 786. [Google Scholar] [CrossRef] [PubMed]
- Cao, D.; van Vollenhoven, R.; Klareskog, L.; Trollmo, C.; Malmström, V. CD25brightCD4+ regulatory T cells are enriched in inflamed joints of patients with chronic rheumatic disease. Arthritis Res. Ther. 2004, 6, R335–R346. [Google Scholar] [CrossRef]
- Moradi, B.; Schnatzer, P.; Hagmann, S.; Rosshirt, N.; Gotterbarm, T.; Kretzer, J.P.; Thomsen, M.; Lorenz, H.M.; Zeifang, F.; Tretter, T. CD4⁺CD25⁺/highCD127low/⁻ regulatory T cells are enriched in rheumatoid arthritis and osteoarthritis joints—Analysis of frequency and phenotype in synovial membrane, synovial fluid and peripheral blood. Arthritis Res. Ther. 2014, 16, R97. [Google Scholar] [CrossRef]
- Samson, M.; Audia, S.; Janikashvili, N.; Ciudad, M.; Trad, M.; Fraszczak, J.; Ornetti, P.; Maillefert, J.F.; Miossec, P.; Bonnotte, B. Brief report: Inhibition of interleukin-6 function corrects Th17/Treg cell imbalance in patients with rheumatoid arthritis. Arthritis Rheum. 2012, 64, 2499–2503. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, X.; Zhuang, L.; Xu, C.; Li, T.; Zhang, G.; Liu, Y. Decreased regulatory T-cell frequency and interleukin-35 levels in patients with rheumatoid arthritis. Exp. Ther. Med. 2018, 16, 5366–5372. [Google Scholar] [CrossRef] [PubMed]
- Walter, G.J.; Evans, H.G.; Menon, B.; Gullick, N.J.; Kirkham, B.W.; Cope, A.P.; Geissmann, F.; Taams, L.S. Interaction with activated monocytes enhances cytokine expression and suppressive activity of human CD4+CD45ro+CD25+CD127(low) regulatory T cells. Arthritis Rheum. 2013, 65, 627–638. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, C.; Jia, X.; Yu, J. Circulating Exosomal miR-17 Inhibits the Induction of Regulatory T Cells via Suppressing TGFBR II Expression in Rheumatoid Arthritis. Cell. Physiol. Biochem. 2018, 50, 1754–1763. [Google Scholar] [CrossRef] [PubMed]
- Morita, T.; Shima, Y.; Wing, J.B.; Sakaguchi, S.; Ogata, A.; Kumanogoh, A. The Proportion of Regulatory T Cells in Patients with Rheumatoid Arthritis: A Meta-Analysis. PLoS ONE 2016, 11, e0162306. [Google Scholar] [CrossRef] [PubMed]
- Bending, D.; Pesenacker, A.M.; Ursu, S.; Wu, Q.; Lom, H.; Thirugnanabalan, B.; Wedderburn, L.R. Hypomethylation at the regulatory T cell-specific demethylated region in CD25hi T cells is decoupled from FOXP3 expression at the inflamed site in childhood arthritis. J. Immunol. 2014, 193, 2699–2708. [Google Scholar] [CrossRef] [PubMed]
- Duurland, C.L.; Brown, C.C.; O’Shaughnessy, R.F.; Wedderburn, L.R. CD161(+) Tconv and CD161(+) Treg Share a Transcriptional and Functional Phenotype despite Limited Overlap in TCRβ Repertoire. Front. Immunol. 2017, 8, 103. [Google Scholar] [CrossRef] [PubMed]
- Pesenacker, A.M.; Bending, D.; Ursu, S.; Wu, Q.; Nistala, K.; Wedderburn, L.R. CD161 defines the subset of FoxP3+ T cells capable of producing proinflammatory cytokines. Blood 2013, 121, 2647–2658. [Google Scholar] [CrossRef] [PubMed]
- Morgan, M.E.; Flierman, R.; van Duivenvoorde, L.M.; Witteveen, H.J.; van Ewijk, W.; van Laar, J.M.; de Vries, R.R.; Toes, R.E. Effective treatment of collagen-induced arthritis by adoptive transfer of CD25+ regulatory T cells. Arthritis Rheum. 2005, 52, 2212–2221. [Google Scholar] [CrossRef]
- Pedros, C.; Duguet, F.; Saoudi, A.; Chabod, M. Disrupted regulatory T cell homeostasis in inflammatory bowel diseases. World J. Gastroenterol. 2016, 22, 974–995. [Google Scholar] [CrossRef]
- Maul, J.; Loddenkemper, C.; Mundt, P.; Berg, E.; Giese, T.; Stallmach, A.; Zeitz, M.; Duchmann, R. Peripheral and intestinal regulatory CD4+ CD25(high) T cells in inflammatory bowel disease. Gastroenterology 2005, 128, 1868–1878. [Google Scholar] [CrossRef] [PubMed]
- Negi, S.; Saini, S.; Tandel, N.; Sahu, K.; Mishra, R.P.N.; Tyagi, R.K. Translating Treg Therapy for Inflammatory Bowel Disease in Humanized Mice. Cells 2021, 10, 1847. [Google Scholar] [CrossRef] [PubMed]
- Mohammadnia-Afrouzi, M.; Zavaran Hosseini, A.; Khalili, A.; Abediankenari, S.; Hosseini, V.; Maleki, I. Decrease of CD4(+) CD25(+) CD127(low) FoxP3(+) regulatory T cells with impaired suppressive function in untreated ulcerative colitis patients. Autoimmunity 2015, 48, 556–561. [Google Scholar] [CrossRef] [PubMed]
- Mizoguchi, A. Animal models of inflammatory bowel disease. Prog. Mol. Biol. Transl. Sci. 2012, 105, 263–320. [Google Scholar] [CrossRef] [PubMed]
- Desreumaux, P.; Foussat, A.; Allez, M.; Beaugerie, L.; Hébuterne, X.; Bouhnik, Y.; Nachury, M.; Brun, V.; Bastian, H.; Belmonte, N.; et al. Safety and efficacy of antigen-specific regulatory T-cell therapy for patients with refractory Crohn’s disease. Gastroenterology 2012, 143, 1207–1217.e2. [Google Scholar] [CrossRef] [PubMed]
- Ho, P.; Cahir-McFarland, E.; Fontenot, J.D.; Lodie, T.; Nada, A.; Tang, Q.; Turka, L.A.; Bluestone, J.A. Harnessing regulatory T cells to establish immune tolerance. Sci. Transl. Med. 2024, 16, eadm8859. [Google Scholar] [CrossRef] [PubMed]
- Bittner, S.; Hehlgans, T.; Feuerer, M. Engineered Treg cells as putative therapeutics against inflammatory diseases and beyond. Trends Immunol. 2023, 44, 468–483. [Google Scholar] [CrossRef] [PubMed]
- Bluestone, J.A.; McKenzie, B.S.; Beilke, J.; Ramsdell, F. Opportunities for Treg cell therapy for the treatment of human disease. Front. Immunol. 2023, 14, 1166135. [Google Scholar] [CrossRef] [PubMed]
- Shao, Q.; Gu, J.; Zhou, J.; Wang, Q.; Li, X.; Deng, Z.; Lu, L. Tissue Tregs and Maintenance of Tissue Homeostasis. Front. Cell Dev. Biol. 2021, 9, 717903. [Google Scholar] [CrossRef]
- Eggenhuizen, P.J.; Ng, B.H.; Ooi, J.D. Treg Enhancing Therapies to Treat Autoimmune Diseases. Int. J. Mol. Sci. 2020, 21, 7015. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Subtype | Markers | Origin | Activation/Differentiation Factor | Main Suppressive Mechanism | Ref. |
|---|---|---|---|---|---|
| Naive Treg | CD25low/−FoxP3low/−CTLA-4low/−CD95− CD62L+CD45RA+CD45ROlow | Thymus | Antigen-dependent, TCR/CD28, TGF-β | Lack of suppressive activity until they are activated | [17] |
| Resting Treg | CD25highFoxP3lowKi-67−CD45RA+ | Thymus/Periphery | TCR stimulation | Not anergic and are able to proliferate upon TCR stimulation | [14] |
| Activated Treg | CD25high FoxP3high CD95+CD45RA− CTLA-4highCD45ROhigh | Thymus/Periphery | TCR/CD28, cytokines: IL-2, IL-10, TGF-β | Cell contact, IL-10 and TGF-β, modulation of the metabolism and activation of other immune cells | [14] |
| Memory Treg | CD25high CTLA-4high CD45RAlowCD45ROhighHelios+ | Thymus/Periphery | Previous antigen stimulation | Lack of suppressive activity until they are reactivated | [14] |
| Apoptotic Treg | CD95high Bcl-2low Bcl-XLlow | Thymus/Periphery | Loss of survival signals, increased pro-apoptotic signals, and reduced anti- apoptotic signals | None | [18] |
| nTreg | CD25+CD127−CTLA-4+GITR+Nrp-1+ Helios+ | Thymus | TCR/CD28, affinity-dependent, IL-2 | Cell contact | [19,20] |
| ICOS+ | ICOS+CD25+FoxP3+ | Thymus | TCR/CD28, ICOSL, affinity-dependent, IL-2 | IL-10 to suppress dendritic cell function and TGF-β to suppress T cell function | [21] |
| ICOS− | ICOS−CD25+FoxP3+ | Thymus | TCR/CD28, affinity-dependent | TGF-β | [21] |
| HLA-G+ | HLA-G+CD25−FoxP3− | Thymus | TCR/CD28, affinity-dependent | Cell contact, IL-10 and IL-35, soluble HLA-G5 | [22,23,24] |
| HLA-DR+ | HLA-DR+CD25high | Thymus | TCR/CD28, affinity-dependent, IL-2 and TGF-β | Early contact-dependent suppression | [25] |
| HLA-DR− | HLA-DR−CD25high | Thymus | TCR/CD28 (HLA-DQ, HLA-DP), affinity-dependent, IL-2 or IL-33 | Early IL-4 and IL-10 secretion and a late contact-dependent suppression | [25] |
| iTreg | CD25+CTLA-4+GITR+Nrp-1−/+?Helios −/+? FoxP3highLAG-3+ | Periphery | Antigen-dependent | IL-10, TGF-β | [26,27] |
| Tr1 | CD25−CTLA-4+GITR+FoxP3lowIL-10+ | Periphery | Antigen-dependent, IL-10, IFN-α | IL-10 | [28,29,30] |
| Th3 | CD25−/+CTLA-4lowGITR−FoxP3?TGF-β+ | Periphery | Antigen-dependent, TGF-β | TGF-β | [31,32] |
| iTr35 | CTLA-4+ FoxP3− IL-10− TGF-β− | Periphery | Antigen-dependent, IL-35 | IL-35 | [33] |
| Treg-of-B cell | CD25+CTLA-4+GITR+LAG+ICOS+OX40+ PD1+FoxP3−IL-10+TGF-β+ | Periphery | Cell-cell contact between B and T cells | Cell contact, IL-10 | [34,35] |
| Identification Markers | Human | Mice | Characteristics | Notes/Limitations | Ref. |
|---|---|---|---|---|---|
| FoxP3+ | X | X | FoxP3 expression act as a Treg cell lineage- specific marker and correlates with suppressor activity | Does not control all aspects of Treg biology as thymic CD25+Foxp3− Treg precursors are fate committed to the Treg lineage | [39,40] |
| CD25+ | X | CD25 expression is crucial for maintaining self-tolerance and dysfunction of this immunoregulation can underly autoimmune diseases | The use of CD25 expression as a marker is limited in human studies | [41] | |
| CD25highFoxP3+ | X | Exhibit strong in vitro regulatory function | No clear boundary between CD25+ and CD25high expression | [42] | |
| CD25+CD127- | X | X | Express the highest level of FoxP3, effectively suppress the proliferation of CD4+CD25− T cells, and no intracellular staining required | CD127 expression alone cannot accurately discriminate Treg cells from activate T cells | [43,44,45] |
| CD39high | X | Exhibit sustained FoxP3 levels, functional suppressive abilities even within an inflammatory environment, no intracellular staining required | - | [46] | |
| CD25+GITR+ | X | X | GITR expression is essential in immunological self-tolerance maintained by Tregs. | - | [47,48] |
| FoxP3+CTLA-4+ | X | X | CTLA-4 is a key inhibitory molecule that relies on FoxP3 that competes with CD28, leading to a shortened interaction between naïve T cells and APCs, and the ability to inhibit TCR signaling | - | [49,50] |
| CD25+LAG3+ | X | X | Tregs express LAG-3 upon activation, and Tregs from LAG-3− mice exhibit reduced regulatory activity | - | [51] |
| CD39+CD37+ | X | X | 90% of FoxP3+ Tregs are CD39+, CD39 and CD37 generate an immunosuppressed environment (increased adenosine levels) | Not specific for Tregs, expressed on endothelial cells and various immune cells | [37,52] |
| Subset | Markers | Differentiation Factor | Function | Ref. |
|---|---|---|---|---|
| Th1-like Tregs | T-bethigh CXCR3+ FoxP3+ | T-cell receptor stimulation in the presence of IFN-γ and IL-12 | Suppressing Th1-mediated responses, IFN-γ, IL-4 and IL-13 | [102] |
| Th17-like Tregs | FoxP3+CD45RA− ROR-γthigh CCR6+ | T-cell receptor stimulation in the presence of IL-6 and IL-1β | Suppressing Th17-mediated responses, IL-17 secretion | [103,104,107] |
| Th2-like Tregs | GATA3high ROR-γt-FoxP3+ | T-cell receptor stimulation in the presence IL-2 | Mostly found in tissues. Secreting IL-13, IL-4, and IL-5 chemotaxis to CCL17/22 | [108] |
| Tfh-like (Tfr) | PD-1highCXCR5high BCL6high | Antigen-dependent, vaccination, infection by influenza, CD28 | Suppression of Tfh function and antibody production, prevention of humoral autoimmunity, control of IgE | [109] |
| exFoxP3 Tregs | CD127high FoxP3− | Possible demethylation of FoxP3 triggered by proinflammatory cytokines, such as IL-6/deficiency of IL-2 signaling | Activated-memory T cell phenotype and produced inflammatory cytokines | [110] |
| Exhauster | PD-1+ FoxP3+ TIM-3+ LAG-3+ | Facilitated by chronic antigen and inflammation | Reduced suppressive function | [111] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Honing, D.Y.; Luiten, R.M.; Matos, T.R. Regulatory T Cell Dysfunction in Autoimmune Diseases. Int. J. Mol. Sci. 2024, 25, 7171. https://doi.org/10.3390/ijms25137171
Honing DY, Luiten RM, Matos TR. Regulatory T Cell Dysfunction in Autoimmune Diseases. International Journal of Molecular Sciences. 2024; 25(13):7171. https://doi.org/10.3390/ijms25137171
Chicago/Turabian StyleHoning, Dionne Y., Rosalie M. Luiten, and Tiago R. Matos. 2024. "Regulatory T Cell Dysfunction in Autoimmune Diseases" International Journal of Molecular Sciences 25, no. 13: 7171. https://doi.org/10.3390/ijms25137171
APA StyleHoning, D. Y., Luiten, R. M., & Matos, T. R. (2024). Regulatory T Cell Dysfunction in Autoimmune Diseases. International Journal of Molecular Sciences, 25(13), 7171. https://doi.org/10.3390/ijms25137171