
Inflammatory Response in the Pathogenesis and Treatment of Hepatocellular Carcinoma: A Double-Edged Weapon
 , , , ,
, , , ,  and
and
Abstract
1. Introduction
2. Viral Etiological Factors Associated with Inflammation and Hepatocellular Carcinoma
2.1. Hepatitis B Infection

{kind=link}
| Study | Molecular Pathway | Description |
|---|---|---|
| Zhao et al. [22] | NTCP receptor pathway | HBV entry into hepatocytes occurs via the NTCP receptor, which is specific to hepatocytes and bile acids. |
| Zhao et al. [22] | Integration of cccDNA | HBV integrates covalently closed circular DNA (cccDNA) into the hepatocyte genome, affecting cancer-related genes like CCNA2, TERT, MLL4, TP53, CTNNB1, KMT2B, and CCNE1. |
| Tu et al. [23] | Cis-mediated insertional mutagenesis | HBV integration into the genome causes insertional mutagenesis, chromosomal instability, and expression of aberrant viral proteins, increasing the risk of liver cancer. |
| Wangensteen et al. [25] | Immune response pathways | Chronic HBV infections trigger persistent cellular damage by activating innate and adaptive immune responses, leading to T-cell exhaustion and immune inhibitory receptor expression (e.g., PD-1, CTLA-4, TIM-3). |
| Rehermann et al. [26] | Innate immune system pathways | Components like dendritic cells, Kupffer cells, and NK cells perpetuate inflammation and hepatocellular injury in chronic HBV infections. |
| Revill et al. [27] | Host immune response | Host immune response, rather than the virus itself, is responsible for hepatocellular damage, necessitating continuous lifelong treatment. |
| Lim et al. [28] | Immunosuppressive tumor microenvironment | HBV–HCC creates an immunosuppressive environment within the tumor to mitigate host damage during prolonged viral infection. |
| He et al. [29] | Cytokine secretion by NK cells | NK cells secrete cytokines like IL-10, TGFβ, IL-4, and IL-13, facilitating fibrogenesis and tumorigenesis. |
| Tian et al. [30] Chen et al. [31] | NK cell cytotoxicity and survival | HBV impairs NK cell cytotoxicity and survival, promoting chronic hepatocellular injury and epithelial-to-mesenchymal transition. |
| Borgia et al. [32] | Factors affecting NK cell dysfunction | Factors like severe hypoxia, tumor cell metabolites, inhibitory receptors, and molecules like TGFβ, PGE2, IDO1 contribute to NK cell dysfunction in HBV–HCC. |
| Wang. et al. [33] | Role of Kupffer cells and monocytes | Kupffer cells and inflammatory monocytes promote the transition from chronic hepatitis to liver cancer by releasing proinflammatory cytokines such as TNFα, IL-6, and MCP-1, and inducing CD8+ T-cell exhaustion in collaboration with myeloid-derived suppressor cells (MDSCs). |
| Bertoletti et al. [34] Hao, X et al. [35] | CD8+ T-cell antiviral activity | HBV-specific CD8+ T cells produce proinflammatory cytokines and cytotoxic molecules to eliminate infected cells but contribute to sustained hepatic inflammation and HCC development in chronic HBV infection. |
| Zong et al. [36] Fisicaro et al. [37] | CD8+ T-cell dysfunction | In chronic HBV infection, CD8+ T cells become dysfunctional, expressing exhaustion markers like PD-1, CTLA-4, LAG3, TIM-3, and TIGIT, contributing to defective anti-tumoral surveillance and chronic hepatic inflammation. |
| Heim et al. [38] Liu. et al. [39] | Cytokine production by exhausted CD8+ T cells | Exhausted CD8+ T cells maintain chronic hepatic inflammation by producing inflammatory cytokines (IFN-α, TNFα, IL-17A, IL-22) in response to continuous HBV antigen stimulation. |
| Yan. et al. [40] Fu et al. [41] | CD4+ T-cell response | Chronic HBV infection leads to a shift from Th1 to Th17 CD4+ T cells and a reduction in effector cytotoxic CD4+ T cells, contributing to ongoing hepatocellular injury and diminished anti-tumoral surveillance. |
| Li et al. [42] | Expansion of Treg cells | Treg cells expand during chronic HBV infection, stimulated by cytokines like TGFβ, IL-10, and CCL22, creating an immunosuppressive microenvironment for HCC initiation and progression. |
| Schollmeier et al. [44] | HBx protein and hepatocarcinogenesis | HBx protein disrupts hepatocellular cycle regulation, affecting cell proliferation, death, transcription, and DNA repair pathways, and interacts with cellular signaling pathways like CREB, Ras/Raf, MAPK, and JAK-STAT, contributing to genetic instability and neoplastic transformation. |
| Datfar et al. [45] Levrero et al. [46] Ling et al. [47] | HBx protein and cellular signaling pathways | HBx protein modulates CREB-dependent transcription and engages cellular signaling pathways such as Ras/Raf, MAPK, and JAK-STAT. |
| Popa et al. [48] | HBx protein and apoptotic effects | HBx protein affects proteasomes, mitochondrial proteins, p53, and DDB1, leading to apoptotic effects and contributing to HCC progression and metastasis. |
| Yang et al. [49] | HBx protein and HCC progression | Studies demonstrate HBx protein’s involvement in HCC progression and metastasis. |
| Liu et al. [51] | HBc protein and Src/PI3K/Akt pathway | HBc protein promotes hepatocarcinogenesis by enhancing apoptosis resistance and stimulating hepatoma cell proliferation via the Src/PI3K/Akt pathway. |
| Capasso et al. [52] | HBc and HBx protein synergy | HBc protein synergizes with HBx protein to exacerbate liver cancer progression by suppressing p53 promoter activity. |
2.2. Hepatitis C Infection
| Study | Molecular Pathway | Description |
|---|---|---|
| Rosen et al. [57] | Pattern recognition receptors (PRRs) and cytokine production | HCV replication detected by PRRs as PAMPs triggers cytokine and chemokine production (e.g., IFN-λ1, IL-1β, MIP-1), initiating innate and adaptive immune responses. |
| Ghouri et al. [59] | Nonstructural proteins and MAPK pathway | HCV nonstructural proteins (e.g., NS2, NS3, NS4A, NS4B, NS5A, NS5B) disrupt the MAPK cellular signaling pathway, inhibiting apoptosis and causing cellular changes in infected liver cells. |
| Zeng et al. [60] | Compromised immune function | HCV compromises antigen presentation, increases T-cell depletion markers, and enhances regulatory T-cell activity, leading to immune inefficiency and compromised antiviral function. |
| Selimovic et al. [61] | Structural proteins and oncogenesis | HCV structural proteins, especially the core protein, manipulate intracellular pathways (e.g., NF-κB pathway) and enhance expression of proteins like IL-6 and STAT3, contributing to transformative changes in hepatocytes and disrupted immune responses. |
| Li et al. [62] | NF-κB pathway activation | HCV core protein activates the NF-κB pathway. |
| Wangensteen et al. [25] | Immune evasion in inflamed liver microenvironment | Disrupted immune responses within the inflamed liver microenvironment undermine anti-tumor immunity and facilitate tumor immune evasion. |
| Irshad et al. [63] | Hepatic stellate cells (HSCs) activation | HCV and inflammatory cytokines promote the differentiation of HSCs into myofibroblasts, contributing to liver cirrhosis. |
| Sahin et al. [64] | Cytokine and growth factor overexpression | Viral proteins induce overexpression of PDGF, EGF, VEGF, CXCL5, and CXCL9, enhancing both innate and adaptive immune responses. |
| Dolina et al. [66] | T-cell dysfunction and exhaustion | Chronic HCV impedes IFN synthesis in T cells and induces exhaustion markers (e.g., TIM-3), promoting HCC in an immunosuppressive environment. |
| Datfar et al. [45] | M2-macrophage polarization and NK cell dysfunction | Chronic HCV induces TGFβ-mediated M2-macrophage polarization, IL-10-mediated Treg expansion, and NK cell depletion and dysfunction, attenuating antiviral and anticancer responses. |
| Gurzu et al. [67] | Epithelial–mesenchymal transition (EMT) | HCV promotes EMT through pathways like IL-6/STAT3/lncTCF7, IL-6/STAT3/Snail–Smad3/TGFβ1, and NS5A-mediated Wntβ catenin pathway dysregulation. |
| Heredia-Torres et al. [68] | Oxidative stress, insulin resistance, and steatosis | HCV-induced oxidative stress, insulin resistance, and steatosis contribute to HCC development by disrupting lipid metabolism and insulin signaling pathways. |
| Smirnova et al. [69] | NS5A and core protein effects on ROS and mitochondrial function | NS5A protein elevates ROS by increasing Ca2+ influx into mitochondria, activating NF-κB and STAT3, while core protein disrupts mitochondrial respiratory chain complex I, increasing ROS levels. |
| Zhao et al. [70] | TGFβ and hepatic stellate cell activation | TGFβ recognition triggers hepatic stellate cell activation and ECM production, leading to liver fibrosis, while EMT facilitates hepatocyte transition to myofibroblasts. |
2.3. Hepatitis Delta Infection
3. Metabolic Etiological Factors Associated with Inflammation and Hepatocellular Carcinoma
3.1. Metabolic Dysfunction-Associated Steatotic Liver Disease (MASLD)
3.2. Obesity
4. Alcohol Etiological Factors Associated with Inflammation and Hepatocellular Carcinoma
5. The Contribution of the Gut–Liver Axis to Hepatocarcinogenesis through Inflammation
| Study | Molecular Pathway | Description |
|---|---|---|
| Luo et al. [113] | LPS/TLR4 signaling pathway | Activation in Kupffer cells leads to TNFα- and IL-6/STAT3-dependent hepatocyte compensatory proliferation. In hepatic stellate cells, TLR4 signaling upregulates NF-κB/p65-dependent epiregulin, inhibiting apoptosis. |
| Kang et al. [114] | Inflammatory MAPK4 (MKK4)/c-Jun N-terminal kinase (JNK) pathway | Activation stimulates production of MMP2, MMP9, IL-6, IL-23, IL-17A, and TNFα, fostering EMT and HCC invasiveness. |
| Shi et al. [116] | Bile acid metabolism and FXR pathway | Altered bile acid metabolism leads to toxic bile acid accumulation, damaging the gut lining and disrupting the microbiota. Bile acids interact with FXR, which regulates inflammation and gut barrier integrity. |
| Luo et al. [113] | p38/MAPK-dependent activation of p53 and NF-κB | Bile acid metabolism alterations cause hepatocyte apoptosis, mitochondrial and ER stress, ROS generation, and the overproduction of proinflammatory cytokines (e.g., TNFα, IL-1β, IL-6). |
| Huang et al. [117] Wei et al. [118] | FXR pathway and oncosuppression | Downregulation of FXR by toxic bile acid overload and proinflammatory cytokines in chronic liver diseases is key in hepatocarcinogenesis. FXR normally suppresses local inflammation. |
| Chen et al. [119] Jia et al. [120] | Deoxycholic acid and SCFA in HCC progression | Deoxycholic acid overproduction promotes HCC by activating HSCs to secrete proinflammatory cytokines, creating an immunosuppressive milieu. SCFAs favor HBx-mediated carcinogenesis. |
| Ren et al. [121] Huang et al. [122] Zheng et al. [123] | Gut microbiota shift in liver diseases | Shift from beneficial bacteria to proinflammatory LPS- and bile acid-producing genera (e.g., Bacteroides, Lachnospiraceae incertae sedis, Clostridium XIVa) is observed during cirrhosis and early HCC. |
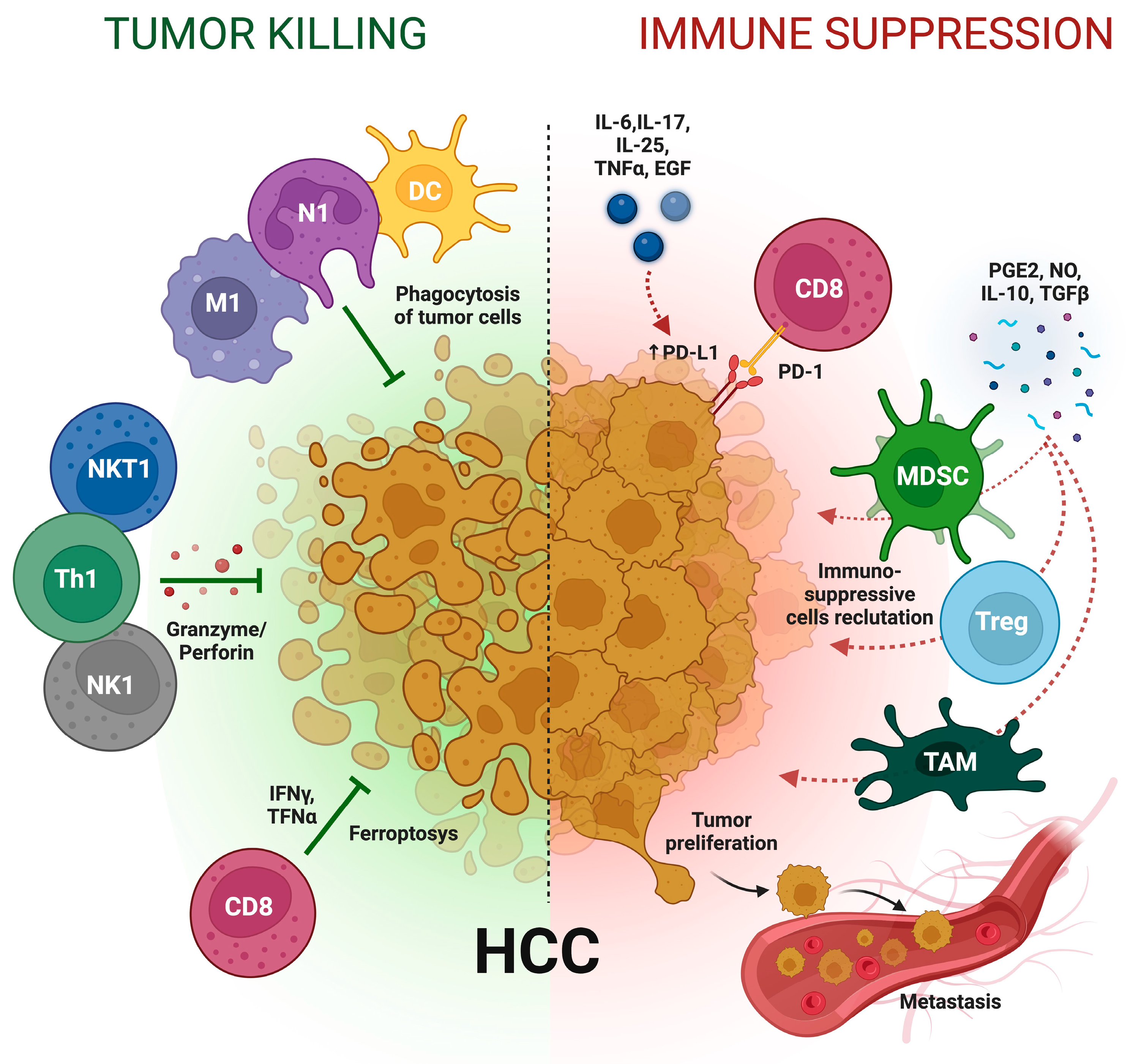
6. Inflammation and Treatment of Hepatocellular Carcinoma: A Double-Edged Weapon
7. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Wong, G.; Anstee, Q.M.; Henry, L. The Global Burden of Liver Disease. Clin. Gastroenterol. Hepatol. 2023, 21, 1978–1991. [Google Scholar] [CrossRef] [PubMed]
- Rumgay, H.; Arnold, M.; Ferlay, J.; Lesi, O.; Cabasag, C.J.; Vignat, J.; Laversanne, M.; McGlynn, K.A.; Soerjomataram, I. Global burden of primary liver cancer in 2020 and predictions to 2040. J. Hepatol. 2022, 77, 1598–1606. [Google Scholar] [CrossRef] [PubMed]
- Samant, H.; Amiri, H.S.; Zibari, G.B. Addressing the worldwide hepatocellular carcinoma: Epidemiology, prevention and management. J. Gastrointest. Oncol. 2021, 12, S361–S373. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.D.; Hainaut, P.; Gores, G.J.; Amadou, A.; Plymoth, A.; Roberts, L.R. A global view of hepatocellular carcinoma: Trends, risk, prevention and management. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 589–604. [Google Scholar] [CrossRef] [PubMed]
- Janevska, D.; Chaloska-Ivanova, V.; Janevski, V. Hepatocellular Carcinoma: Risk Factors, Diagnosis and Treatment. Open Access Maced. J. Med. Sci. 2015, 3, 732–736. [Google Scholar] [CrossRef] [PubMed]
- Sankar, K.; Gong, J.; Osipov, A.; Miles, S.A.; Kosari, K.; Nissen, N.N.; Hendifar, A.E.; Koltsova, E.K.; Yang, J.D. Recent advances in the management of hepatocellular carcinoma. Clin. Mol. Hepatol. 2024, 30, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.Q.; Singal, A.G.; Kono, Y.; Tan, D.J.H.; El-Serag, H.B.; Loomba, R. Changing global epidemiology of liver cancer from 2010 to 2019: NASH is the fastest growing cause of liver cancer. Cell Metab. 2022, 34, 969–977. [Google Scholar] [CrossRef] [PubMed]
- Motta, B.M.; Masarone, M.; Torre, P.; Persico, M. From Non-Alcoholic Steatohepatitis (NASH) to Hepatocellular Carcinoma (HCC): Epidemiology, Incidence, Predictions, Risk Factors, and Prevention. Cancers 2023, 15, 5458. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.Y. Changing etiology and epidemiology of hepatocellular carcinoma: Asia and worldwide. J. Liver Cancer 2024, 24, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Phoolchund, A.G.S.; Khakoo, S.I. MASLD and the Development of HCC: Pathogenesis and Therapeutic Challenges. Cancers 2024, 16, 259. [Google Scholar] [CrossRef] [PubMed]
- Soto, A.; Spongberg, C.; Martinino, A.; Giovinazzo, F. Exploring the Multifaceted Landscape of MASLD: A Comprehensive Synthesis of Recent Studies, from Pathophysiology to Organoids and Beyond. Biomedicines 2024, 12, 397. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.X.; Ling, Y.; Wang, H.Y. Role of nonresolving inflammation in hepatocellular carcinoma development and progression. npj Precis. Oncol. 2018, 2, 6. [Google Scholar] [CrossRef] [PubMed]
- Żeromski, J.; Kierepa, A.; Brzezicha, B.; Kowala-Piaskowska, A.; Mozer-Lisewska, I. Pattern Recognition Receptors: Significance of Expression in the Liver. Arch. Immunol. Ther. Exp. (Warsz) 2020, 68, 29. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Patidar, A.; Selvaraj, S.; Sarode, A.; Chauhan, P.; Chattopadhyay, D.; Saha, B. DAMP-TLR-cytokine axis dictates the fate of tumor. Cytokine 2018, 104, 114–123. [Google Scholar] [CrossRef] [PubMed]
- Calvillo-Rodríguez, K.M.; Lorenzo-Anota, H.Y.; Rodríguez-Padilla, C.; Martínez-Torres, A.C.; Scott-Algara, D. Immunotherapies inducing immunogenic cell death in cancer: Insight of the innate immune system. Front. Immunol. 2023, 14, 1294434. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Galasso, L.; Cerrito, L.; Maccauro, V.; Termite, F.; Ainora, M.E.; Gasbarrini, A.; Zocco, M.A. Hepatocellular Carcinoma and the Multifaceted Relationship with Its Microenvironment: Attacking the Hepatocellular Carcinoma Defensive Fortress. Cancers 2024, 16, 1837. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Fu, Y.; Chung, F.L. Oxidative stress and hepatocarcinogenesis. Hepatoma Res. 2018, 4, 39. [Google Scholar] [CrossRef] [PubMed]
- Su, L.J.; Zhang, J.H.; Gomez, H.; Murugan, R.; Hong, X.; Xu, D.; Jiang, F.; Peng, Z.Y. Reactive Oxygen Species-Induced Lipid Peroxidation in Apoptosis, Autophagy, and Ferroptosis. Oxidative Med. Cell. Longev. 2019, 2019, 5080843. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Tan, H.Y.; Wang, N.; Zhang, Z.J.; Lao, L.; Wong, C.W.; Feng, Y. The Role of Oxidative Stress and Antioxidants in Liver Diseases. Int. J. Mol. Sci. 2015, 16, 26087–26124. [Google Scholar] [CrossRef] [PubMed]
- Boulahtouf, Z.; Virzì, A.; Baumert, T.F.; Verrier, E.R.; Lupberger, J. Signaling Induced by Chronic Viral Hepatitis: Dependence and Consequences. Int. J. Mol. Sci. 2022, 23, 2787. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.H.; Liu, X.; Yan, H.X.; Li, W.Y.; Zeng, X.; Yang, Y.; Zhao, J.; Liu, S.P.; Zhuang, X.H.; Lin, C.; et al. Genomic and oncogenic preference of HBV integration in hepatocellular carcinoma. Nat. Commun. 2016, 7, 12992. [Google Scholar] [CrossRef] [PubMed]
- Tu, T.; Budzinska, M.A.; Shackel, N.A.; Urban, S. HBV DNA Integration: Molecular Mechanisms and Clinical Implications. Viruses 2017, 9, 75. [Google Scholar] [CrossRef] [PubMed]
- European Association for the Study of the Liver. EASL 2017 Clinical Practice Guidelines on the management of hepatitis B virus infection. J. Hepatol. 2017, 67, 370–398. [Google Scholar] [CrossRef] [PubMed]
- Wangensteen, K.J.; Chang, K.M. Multiple Roles for Hepatitis B and C Viruses and the Host in the Development of Hepatocellular Carcinoma. Hepatology 2021, 73 (Suppl. 1), 27–37. [Google Scholar] [CrossRef] [PubMed]
- Rehermann, B.; Thimme, R. Insights from Antiviral Therapy into Immune Responses to Hepatitis B and C Virus Infection. Gastroenterology 2019, 156, 369–383. [Google Scholar] [CrossRef] [PubMed]
- Revill, P.A.; Tu, T.; Netter, H.J.; Yuen, L.K.W.; Locarnini, S.A.; Littlejohn, M. The evolution and clinical impact of hepatitis B virus genome diversity. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 618–634. [Google Scholar] [CrossRef] [PubMed]
- Lim, C.J.; Lee, Y.H.; Pan, L.; Lai, L.; Chua, C.; Wasser, M.; Lim, T.K.H.; Yeong, J.; Toh, H.C.; Lee, S.Y.; et al. Multidimensional analyses reveal distinct immune microenvironment in hepatitis B virus-related hepatocellular carcinoma. Gut 2019, 68, 916–927. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Tian, Z. NK cell education via nonclassical MHC and non-MHC ligands. Cell Mol. Immunol. 2017, 14, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Tian, Z.; Chen, Y.; Gao, B. Natural killer cells in liver disease. Hepatology 2013, 57, 1654–1662. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Hao, X.; Sun, R.; Wei, H.; Tian, Z. Natural Killer Cell-Derived Interferon-Gamma Promotes Hepatocellular Carcinoma Through the Epithelial Cell Adhesion Molecule-Epithelial-to-Mesenchymal Transition Axis in Hepatitis B Virus Transgenic Mice. Hepatology 2019, 69, 1735–1750. [Google Scholar] [CrossRef] [PubMed]
- Borgia, M.; Dal Bo, M.; Toffoli, G. Role of Virus-Related Chronic Inflammation and Mechanisms of Cancer Immune-Suppression in Pathogenesis and Progression of Hepatocellular Carcinoma. Cancers 2021, 13, 4387. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhao, W.; Cheng, L.; Guo, M.; Li, D.; Li, X.; Tan, Y.; Ma, S.; Li, S.; Yang, Y.; et al. CD137-mediated pathogenesis from chronic hepatitis to hepatocellular carcinoma in hepatitis B virus-transgenic mice. J. Immunol. 2010, 185, 7654–7662. [Google Scholar] [CrossRef] [PubMed]
- Bertoletti, A.; Ferrari, C. Adaptive immunity in HBV infection. J. Hepatol. 2016, 64 (Suppl. 1), S71–S83. [Google Scholar] [CrossRef] [PubMed]
- Hao, X.; Chen, Y.; Bai, L.; Wei, H.; Sun, R.; Tian, Z. HBsAg-specific CD8+ T cells as an indispensable trigger to induce murine hepatocellular carcinoma. Cell Mol. Immunol. 2021, 18, 128–137. [Google Scholar] [CrossRef] [PubMed]
- Zong, L.; Peng, H.; Sun, C.; Li, F.; Zheng, M.; Chen, Y.; Wei, H.; Sun, R.; Tian, Z. Breakdown of adaptive immunotolerance induces hepatocellular carcinoma in HBsAg-tg mice. Nat. Commun. 2019, 10, 221. [Google Scholar] [CrossRef] [PubMed]
- Fisicaro, P.; Barili, V.; Rossi, M.; Montali, I.; Vecchi, A.; Acerbi, G.; Laccabue, D.; Zecca, A.; Penna, A.; Missale, G.; et al. Pathogenetic Mechanisms of T Cell Dysfunction in Chronic HBV Infection and Related Therapeutic Approaches. Front. Immunol. 2020, 11, 849. [Google Scholar] [CrossRef] [PubMed]
- Heim, K.; Binder, B.; Sagar; Wieland, D.; Hensel, N.; Llewellyn-Lacey, S.; Gostick, E.; Price, D.A.; Emmerich, F.; Vingerhoet, H.; et al. TOX defines the degree of CD8+ T cell dysfunction in distinct phases of chronic HBV infection. Gut 2020, 70, 1550–1560. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Li, M.; Wang, X.; Dang, Z.; Jiang, Y.; Wang, X.; Kong, Y.; Yang, Z. PD-1+ TIGIT+ CD8+ T cells are associated with pathogenesis and progression of patients with hepatitis B virus-related hepatocellular carcinoma. Cancer Immunol. Immunother. 2019, 68, 2041–2054. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Liu, X.L.; Xiao, G.; Li, N.L.; Deng, Y.N.; Han, L.Z.; Yin, L.C.; Ling, L.J.; Liu, L.X. Prevalence and clinical relevance of T-helper cells, Th17 and Th1, in hepatitis B virus-related hepatocellular carcinoma. PLoS ONE 2014, 9, e96080. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Zhang, Z.; Zhou, L.; Qi, Z.; Xing, S.; Lv, J.; Shi, J.; Fu, B.; Liu, Z.; Zhang, J.Y.; et al. Impairment of CD4+ cytotoxic T cells predicts poor survival and high recurrence rates in patients with hepatocellular carcinoma. Hepatology 2013, 58, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Jiang, P.; Wei, S.; Xu, X.; Wang, J. Regulatory T cells in tumor microenvironment: New mechanisms, potential therapeutic strategies and future prospects. Mol. Cancer 2020, 19, 116. [Google Scholar] [CrossRef] [PubMed]
- Sivasudhan, E.; Blake, N.; Lu, Z.; Meng, J.; Rong, R. Hepatitis B Viral Protein HBx and the Molecular Mechanisms Modulating the Hallmarks of Hepatocellular Carcinoma: A Comprehensive Review. Cells 2022, 11, 741. [Google Scholar] [CrossRef] [PubMed]
- Schollmeier, A.; Basic, M.; Glitscher, M.; Hildt, E. The impact of HBx protein on mitochondrial dynamics and associated signaling pathways strongly depends on the hepatitis B virus genotype. J. Virol. 2024, 98, e0042424. [Google Scholar] [CrossRef] [PubMed]
- Datfar, T.; Doulberis, M.; Papaefthymiou, A.; Hines, I.N.; Manzini, G. Viral Hepatitis and Hepatocellular Carcinoma: State of the Art. Pathogens 2021, 10, 1366. [Google Scholar] [CrossRef] [PubMed]
- Levrero, M.; Zucman-Rossi, J. Mechanisms of HBV-induced hepatocellular carcinoma. J. Hepatol. 2016, 64 (Suppl. 1), S84–S101. [Google Scholar] [CrossRef] [PubMed]
- Ling, L.R.; Zheng, D.H.; Zhang, Z.Y.; Xie, W.H.; Huang, Y.H.; Chen, Z.X.; Wang, X.Z.; Li, D. Effect of HBx on inflammation and mitochondrial oxidative stress in mouse hepatocytes. Oncol. Lett. 2020, 19, 2861–2869. [Google Scholar] [CrossRef] [PubMed]
- Popa, G.L.; Popa, M.I. Oxidative Stress in Chronic Hepatitis B-An Update. Microorganisms 2022, 10, 1265. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Liu, Y.; Feng, X.; Wang, X.; Wu, M.; Gong, L.; Shu, B.; Lu, Q.; Dong, J. HBx acts as an oncogene and promotes the invasion and metastasis of hepatocellular carcinoma both in vivo and vitro. Dig. Liver Dis. 2021, 53, 360–366. [Google Scholar] [CrossRef] [PubMed]
- Kwon, J.A.; Rho, H.M. Transcriptional repression of the human p53 gene by hepatitis B viral core protein (HBc) in human liver cells. Biol. Chem. 2003, 384, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Guo, T.F.; Jing, Z.T.; Yang, Z.; Liu, L.; Yang, Y.P.; Lin, X.; Tong, Q.Y. Hepatitis B virus core protein promotes hepatocarcinogenesis by enhancing Src expression and activating the Src/PI3K/Akt pathway. FASEB J. 2018, 32, 3033–3046. [Google Scholar] [CrossRef] [PubMed]
- Capasso, M.; Cossiga, V.; Guarino, M.; Ranieri, L.; Morisco, F. The Role of Hepatitis Viruses as Drivers of Hepatocancerogenesis. Cancers 2024, 16, 1505. [Google Scholar] [CrossRef] [PubMed]
- Ali, H.; Vikash, F.; Moond, V.; Khalid, F.; Jamil, A.R.; Dahiya, D.S.; Sohail, A.H.; Gangwani, M.K.; Patel, P.; Satapathy, S.K. Global trends in hepatitis C-related hepatocellular carcinoma mortality: A public database analysis (1999–2019). World J. Virol. 2024, 13, 89469. [Google Scholar] [CrossRef]
- Matsuzaki, K.; Murata, M.; Yoshida, K.; Sekimoto, G.; Uemura, Y.; Sakaida, N.; Kaibori, M.; Kamiyama, Y.; Nishizawa, M.; Fujisawa, J.; et al. Chronic inflammation associated with hepatitis C virus infection perturbs hepatic transforming growth factor beta signaling, promoting cirrhosis and hepatocellular carcinoma. Hepatology 2007, 46, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Reddy, K.R.; McLerran, D.; Marsh, T.; Parikh, N.; Roberts, L.R.; Schwartz, M.; Nguyen, M.H.; Befeler, A.; Page-Lester, S.; Tang, R.; et al. Incidence and Risk Factors for Hepatocellular Carcinoma in Cirrhosis: The Multicenter Hepatocellular Carcinoma Early Detection Strategy (HEDS) Study. Gastroenterology 2023, 165, 1053–1063. [Google Scholar] [CrossRef] [PubMed]
- Farooq, H.Z.; James, M.; Abbott, J.; Oyibo, P.; Divall, P.; Choudhry, N.; Foster, G.R. Risk factors for hepatocellular carcinoma associated with hepatitis C genotype 3 infection: A systematic review. World J. Gastrointest. Oncol. 2024, 16, 1596–1612. [Google Scholar] [CrossRef] [PubMed]
- Rosen, H.R. Emerging concepts in immunity to hepatitis C virus infection. J. Clin. Investig. 2013, 123, 4121–4130. [Google Scholar] [CrossRef] [PubMed]
- Shirvani-Dastgerdi, E.; Schwartz, R.E.; Ploss, A. Hepatocarcinogenesis associated with hepatitis B, delta and C viruses. Curr. Opin. Virol. 2016, 20, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Ghouri, Y.A.; Mian, I.; Rowe, J.H. Review of hepatocellular carcinoma: Epidemiology, etiology, and carcinogenesis. J. Carcinog. 2017, 16, 1. [Google Scholar] [CrossRef]
- Zeng, H.; Li, L.; Hou, Z.; Zhang, Y.; Tang, Z.; Liu, S. Direct-acting Antiviral in the Treatment of Chronic Hepatitis C: Bonuses and Challenges. Int. J. Med. Sci. 2020, 17, 892–902. [Google Scholar] [CrossRef]
- Selimovic, D.; El-Khattouti, A.; Ghozlan, H.; Haikel, Y.; Abdelkader, O.; Hassan, M. Hepatitis C virus-related hepatocellular carcinoma: An insight into molecular mechanisms and therapeutic strategies. World J. Hepatol. 2012, 4, 342–355. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.H.; Tang, Q.B.; Wang, J.; Zhou, L.; Huang, W.L.; Liu, R.Y.; Chen, R.F. Hepatitis C virus core protein induces malignant transformation of biliary epithelial cells by activating nuclear factor-kappaB pathway. J. Gastroenterol. Hepatol. 2010, 25, 1315–1320. [Google Scholar] [CrossRef] [PubMed]
- Irshad, M.; Gupta, P.; Irshad, K. Immunopathogenesis of Liver Injury During Hepatitis C Virus Infection. Viral Immunol. 2019, 32, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Sahin, H.; Borkham-Kamphorst, E.; Kuppe, C.; Zaldivar, M.M.; Grouls, C.; Al-samman, M.; Nellen, A.; Schmitz, P.; Heinrichs, D.; Berres, M.L.; et al. Chemokine Cxcl9 attenuates liver fibrosis-associated angiogenesis in mice. Hepatology 2012, 55, 1610–1619. [Google Scholar] [CrossRef] [PubMed]
- Robert, S.B., Jr. The Possible Association Between DAA Treatment for HCV Infection and HCC Recurrence. Gastroenterol. Hepatol. 2016, 12, 776–779. [Google Scholar]
- Dolina, J.S.; Braciale, T.J.; Hahn, Y.S. Liver-primed CD8+ T cells suppress antiviral adaptive immunity through galectin-9-independent T-cell immunoglobulin and mucin 3 engagement of high-mobility group box 1 in mice. Hepatology 2014, 59, 1351–1365. [Google Scholar] [CrossRef] [PubMed]
- Gurzu, S.; Kobori, L.; Fodor, D.; Jung, I. Epithelial Mesenchymal and Endothelial Mesenchymal Transitions in Hepatocellular Carcinoma: A Review. Biomed. Res. Int. 2019, 2019, 2962580. [Google Scholar] [CrossRef] [PubMed]
- Heredia-Torres, T.G.; Rincón-Sánchez, A.R.; Lozano-Sepúlveda, S.A.; Galan-Huerta, K.; Arellanos-Soto, D.; García-Hernández, M.; Garza-Juarez, A.J.; Rivas-Estilla, A.M. Unraveling the Molecular Mechanisms Involved in HCV-Induced Carcinogenesis. Viruses 2022, 14, 2762. [Google Scholar] [CrossRef] [PubMed]
- Smirnova, O.A.; Ivanova, O.N.; Bartosch, B.; Valuev-Elliston, V.T.; Mukhtarov, F.; Kochetkov, S.N.; Ivanov, A.V. Hepatitis C Virus NS5A Protein Triggers Oxidative Stress by Inducing NADPH Oxidases 1 and 4 and Cytochrome P450 2E1. Oxid. Med. Cell. Longev. 2016, 2016, 8341937. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Shen, M.; Wu, L.; Yang, H.; Yao, Y.; Yang, Q.; Du, J.; Liu, L.; Li, Y.; Bai, Y. Stromal cells in the tumor microenvironment: Accomplices of tumor progression? Cell Death Dis. 2023, 14, 587. [Google Scholar] [CrossRef] [PubMed]
- Baskiran, A.; Atay, A.; Baskiran, D.Y.; Akbulut, S. Hepatitis B/D-Related Hepatocellular Carcinoma. A Clinical Literature Review. J. Gastrointest. Cancer 2021, 52, 1192–1197. [Google Scholar] [CrossRef] [PubMed]
- Farci, P.; Niro, G.A.; Zamboni, F.; Diaz, G. Hepatitis D Virus and Hepatocellular Carcinoma. Viruses 2021, 13, 830. [Google Scholar] [CrossRef] [PubMed]
- Puigvehí, M.; Moctezuma-Velázquez, C.; Villanueva, A.; Llovet, J.M. The oncogenic role of hepatitis delta virus in hepatocellular carcinoma. JHEP Rep. 2019, 1, 120–130. [Google Scholar] [CrossRef] [PubMed]
- Kamal, H.; Fornes, R.; Simin, J.; Stål, P.; Duberg, A.S.; Brusselaers, N.; Aleman, S. Risk of hepatocellular carcinoma in hepatitis B and D virus co-infected patients: A systematic review and meta-analysis of longitudinal studies. J. Viral Hepat. 2021, 28, 1431–1442. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.H.; Jeong, S.H.; Hwang, S.B. Large hepatitis delta antigen modulates transforming growth factor-beta signaling cascades: Implication of hepatitis delta virus-induced liver fibrosis. Gastroenterology 2007, 132, 343–357. [Google Scholar] [CrossRef] [PubMed]
- Morita, M.; Yano, S.; Yamaguchi, T.; Sugimoto, T. Advanced glycation end products-induced reactive oxygen species generation is partly through NF-kappa B activation in human aortic endothelial cells. J. Diabetes Complicat. 2013, 27, 11–15. [Google Scholar] [CrossRef] [PubMed]
- Williams, V.; Brichler, S.; Khan, E.; Chami, M.; Dény, P.; Kremsdorf, D.; Gordien, E. Large hepatitis delta antigen activates STAT-3 and NF-κB via oxidative stress. J. Viral Hepat. 2012, 19, 744–753. [Google Scholar] [CrossRef] [PubMed]
- Chan, W.K.; Chuah, K.H.; Rajaram, R.B.; Lim, L.L.; Ratnasingam, J.; Vethakkan, S.R. Metabolic Dysfunction-Associated Steatotic Liver Disease (MASLD): A State-of-the-Art Review. J. Obes. Metab. Syndr. 2023, 32, 197–213. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global burden of NAFLD and NASH: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhang, L.; Dong, B. Molecular mechanisms in MASLD/MASH-related HCC. Hepatology 2024. online ahead of print. [Google Scholar] [CrossRef]
- Pinter, M.; Pinato, D.J.; Ramadori, P.; Heikenwalder, M. NASH and Hepatocellular Carcinoma: Immunology and Immunotherapy. Clin. Cancer Res. 2023, 29, 513–520. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.H.; Li, Y.L.; Li, D.; Wang, N.N.; Jing, L.; Huang, Y.H. The rs738409 (I148M) variant of the PNPLA3 gene and cirrhosis: A meta-analysis. J. Lipid Res. 2015, 56, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Meroni, M.; Longo, M.; Tria, G.; Dongiovanni, P. Genetics Is of the Essence to Face NAFLD. Biomedicines 2021, 9, 1359. [Google Scholar] [CrossRef] [PubMed]
- Lebeaupin, C.; Vallée, D.; Hazari, Y.; Hetz, C.; Chevet, E.; Bailly-Maitre, B. Endoplasmic reticulum stress signalling and the pathogenesis of non-alcoholic fatty liver disease. J. Hepatol. 2018, 69, 927–947. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.Z.; Xia, H.H.; Xin, Y.N.; Lin, Z.H.; Xuan, S.Y. TM6SF2 E167K Variant, a Novel Genetic Susceptibility Variant, Contributing to Nonalcoholic Fatty Liver Disease. J. Clin. Transl. Hepatol. 2015, 3, 265–270. [Google Scholar] [CrossRef] [PubMed]
- Gil-Gómez, A.; Rojas, Á.; García-Lozano, M.R.; Muñoz-Hernández, R.; Gallego-Durán, R.; Maya-Miles, D.; Montero-Vallejo, R.; Gato, S.; Gallego, J.; Francés, R.; et al. Impact of a Loss-of-Function Variant in HSD17B13 on Hepatic Decompensation and Mortality in Cirrhotic Patients. Int. J. Mol. Sci. 2022, 23, 11840. [Google Scholar] [CrossRef] [PubMed]
- Dudek, M.; Pfister, D.; Donakonda, S.; Filpe, P.; Schneider, A.; Laschinger, M.; Hartmann, D.; Hüser, N.; Meiser, P.; Bayerl, F.; et al. Auto-aggressive CXCR6+ CD8 T cells cause liver immune pathology in NASH. Nature 2021, 592, 444–449. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Celton-Morizur, S.; Desdouets, C. NAFLD-Related HCC: Focus on the Latest Relevant Preclinical Models. Cancers 2023, 15, 3723. [Google Scholar] [CrossRef] [PubMed]
- Arvanitakis, K.; Papadakos, S.P.; Lekakis, V.; Koufakis, T.; Lempesis, I.G.; Papantoniou, E.; Kalopitas, G.; Georgakopoulou, V.E.; Stergiou, I.E.; Theocharis, S.; et al. Meeting at the Crossroad between Obesity and Hepatic Carcinogenesis: Unique Pathophysiological Pathways Raise Expectations for Innovative Therapeutic Approaches. Int. J. Mol. Sci. 2023, 24, 14704. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Wang, Y.; Xu, D. The roles of T cells in obese adipose tissue inflammation. Adipocyte 2021, 10, 435–445. [Google Scholar] [CrossRef]
- Yang, J.; He, J.; Feng, Y.; Xiang, M. Obesity contributes to hepatocellular carcinoma development via immunosuppressive microenvironment remodeling. Front. Immunol. 2023, 14, 1166440. [Google Scholar] [CrossRef] [PubMed]
- Herbel, C.; Patsoukis, N.; Bardhan, K.; Seth, P.; Weaver, J.D.; Boussiotis, V.A. Clinical significance of T cell metabolic reprogramming in cancer. Clin. Transl. Med. 2016, 5, 29. [Google Scholar] [CrossRef] [PubMed]
- Rajesh, Y.; Sarkar, D. Molecular Mechanisms Regulating Obesity-Associated Hepatocellular Carcinoma. Cancers 2020, 12, 1290. [Google Scholar] [CrossRef] [PubMed]
- Menendez, A.; Wanczyk, H.; Walker, J.; Zhou, B.; Santos, M.; Finck, C. Obesity and Adipose Tissue Dysfunction: From Pediatrics to Adults. Genes 2022, 13, 1866. [Google Scholar] [CrossRef] [PubMed]
- Christofides, A.; Konstantinidou, E.; Jani, C.; Boussiotis, V.A. The role of peroxisome proliferator-activated receptors (PPAR) in immune responses. Metabolism 2021, 114, 154338. [Google Scholar] [CrossRef] [PubMed]
- Eggert, T.; Greten, T.F. Tumor regulation of the tissue environment in the liver. Pharmacol. Ther. 2017, 173, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Taniai, M. Alcohol and hepatocarcinogenesis. Clin. Mol. Hepatol. 2020, 26, 736–741. [Google Scholar] [CrossRef] [PubMed]
- Welzel, T.M.; Graubard, B.I.; Quraishi, S.; Zeuzem, S.; Davila, J.A.; El-Serag, H.B.; McGlynn, K.A. Population-attributable fractions of risk factors for hepatocellular carcinoma in the United States. Am. J. Gastroenterol. 2013, 108, 1314–1321. [Google Scholar] [CrossRef] [PubMed]
- Malnick, S.D.H.; Alin, P.; Somin, M.; Neuman, M.G. Fatty Liver Disease-Alcoholic and Non-Alcoholic: Similar but Different. Int. J. Mol. Sci. 2022, 23, 16226. [Google Scholar] [CrossRef] [PubMed]
- Testino, G.; Leone, S.; Borro, P. Alcohol and hepatocellular carcinoma: A review and a point of view. World J. Gastroenterol. 2014, 20, 15943–15954. [Google Scholar] [CrossRef] [PubMed]
- Brandon-Warner, E.; Walling, T.L.; Schrum, L.W.; McKillop, I.H. Chronic ethanol feeding accelerates hepatocellular carcinoma progression in a sex-dependent manner in a mouse model of hepatocarcinogenesis. Alcohol. Clin. Exp. Res. 2012, 36, 641–653. [Google Scholar] [CrossRef] [PubMed]
- Salaspuro, M. Acetaldehyde: A cumulative carcinogen in humans. Addiction 2009, 104, 551–553. [Google Scholar] [CrossRef] [PubMed]
- Hagymási, K.; Blázovics, A.; Lengyel, G.; Kocsis, I.; Fehér, J. Oxidative damage in alcoholic liver disease. Eur. J. Gastroenterol. Hepatol. 2001, 13, 49–53. [Google Scholar] [CrossRef] [PubMed]
- Artru, F.; Bou Saleh, M.; Maggiotto, F.; Lassailly, G.; Ningarhari, M.; Demaret, J.; Ntandja-Wandji, L.C.; Pais de Barros, J.P.; Labreuche, J.; Drumez, E.; et al. IL-33/ST2 pathway regulates neutrophil migration and predicts outcome in patients with severe alcoholic hepatitis. J. Hepatol. 2021, 74, 1272–1273. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.Y.; Yamamoto, G.; Xu, J.; Liu, X.; Karin, D.; Kim, J.Y.; Alexandrov, L.B.; Koyama, Y.; Nishio, T.; Benner, C.; et al. IL-17 signaling in steatotic hepatocytes and macrophages promotes hepatocellular carcinoma in alcohol-related liver disease. J. Hepatol. 2020, 72, 946–959. [Google Scholar] [CrossRef] [PubMed]
- Strathearn, L.S.; Stepanov, A.I.; Font-Burgada, J. Inflammation in Primary and Metastatic Liver Tumorigenesis-Under the Influence of Alcohol and High-Fat Diets. Nutrients 2020, 12, 933. [Google Scholar] [CrossRef] [PubMed]
- Ramadori, P.; Cubero, F.J.; Liedtke, C.; Trautwein, C.; Nevzorova, Y.A. Alcohol and Hepatocellular Carcinoma: Adding Fuel to the Flame. Cancers 2017, 9, 130. [Google Scholar] [CrossRef] [PubMed]
- Ganne-Carrié, N.; Nahon, P. Hepatocellular carcinoma in the setting of alcohol-related liver disease. J. Hepatol. 2019, 70, 284–293. [Google Scholar] [CrossRef] [PubMed]
- Llopis, M.; Cassard, A.M.; Wrzosek, L.; Boschat, L.; Bruneau, A.; Ferrere, G.; Puchois, V.; Martin, J.C.; Lepage, P.; Le Roy, T.; et al. Intestinal microbiota contributes to individual susceptibility to alcoholic liver disease. Gut 2016, 65, 830–839. [Google Scholar] [CrossRef]
- Shasthry, S.M. Fecal microbiota transplantation in alcohol related liver diseases. Clin. Mol. Hepatol. 2020, 26, 294–301. [Google Scholar] [CrossRef] [PubMed]
- Heckley, G.A.; Jarl, J.; Asamoah, B.O.; G-Gerdtham, U. How the risk of liver cancer changes after alcohol cessation: A review and meta-analysis of the current literature. BMC Cancer 2011, 11, 446. [Google Scholar] [CrossRef] [PubMed]
- Di Tommaso, N.; Gasbarrini, A.; Ponziani, F.R. Intestinal Barrier in Human Health and Disease. Int. J. Environ. Res. Public Health 2021, 18, 12836. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.; Guo, S.; Zhou, Y.; Zhao, J.; Wang, M.; Sang, L.; Chang, B.; Wang, B. Hepatocellular Carcinoma: How the Gut Microbiota Contributes to Pathogenesis, Diagnosis, and Therapy. Front. Microbiol. 2022, 13, 873160. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.; Su, G.; Sun, J.; Zhang, Y. Activation of the TLR4/MyD88 signaling pathway contributes to the development of human hepatocellular carcinoma via upregulation of IL-23 and IL-17A. Oncol. Lett. 2018, 15, 9647–9654. [Google Scholar] [CrossRef] [PubMed]
- Sultan, S.; El-Mowafy, M.; Elgaml, A.; Ahmed, T.A.E.; Hassan, H.; Mottawea, W. Metabolic Influences of Gut Microbiota Dysbiosis on Inflammatory Bowel Disease. Front. Physiol. 2021, 12, 715506. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Jin, L.; Huang, W. Bile Acids, Intestinal Barrier Dysfunction, and Related Diseases. Cells 2023, 12, 1888. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Fan, M.; Huang, W. Pleiotropic roles of FXR in liver and colorectal cancers. Mol. Cell. Endocrinol. 2022, 543, 111543. [Google Scholar] [CrossRef]
- Wei, S.; Ma, X.; Zhao, Y. Mechanism of Hydrophobic Bile Acid-Induced Hepatocyte Injury and Drug Discovery. Front. Pharmacol. 2020, 11, 1084. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Ding, M.; Ji, L.; Yao, J.; Guo, Y.; Yan, W.; Yu, S.; Shen, Q.; Huang, M.; Zheng, Y.; et al. Bile acids promote the development of HCC by activating inflammasome. Hepatol. Commun. 2023, 7, e0217. [Google Scholar] [CrossRef] [PubMed]
- Jia, B.; Jeon, C.O. Promotion and induction of liver cancer by gut microbiome-mediated modulation of bile acids. PLoS Pathog. 2019, 15, e1007954. [Google Scholar] [CrossRef] [PubMed]
- Ren, Z.; Li, A.; Jiang, J.; Zhou, L.; Yu, Z.; Lu, H.; Xie, H.; Chen, X.; Shao, L.; Zhang, R.; et al. Gut microbiome analysis as a tool towards targeted non-invasive biomarkers for early hepatocellular carcinoma. Gut 2019, 68, 1014–1023. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Ren, Z.; Gao, X.; Hu, X.; Zhou, Y.; Jiang, J.; Lu, H.; Yin, S.; Ji, J.; Zhou, L.; et al. Integrated analysis of microbiome and host transcriptome reveals correlations between gut microbiota and clinical outcomes in HBV-related hepatocellular carcinoma. Genome Med. 2020, 12, 102. [Google Scholar] [CrossRef] [PubMed]
- Zheng, R.; Wang, G.; Pang, Z.; Ran, N.; Gu, Y.; Guan, X.; Yuan, Y.; Zuo, X.; Pan, H.; Zheng, J.; et al. Liver cirrhosis contributes to the disorder of gut microbiota in patients with hepatocellular carcinoma. Cancer Med. 2020, 9, 4232–4250. [Google Scholar] [CrossRef] [PubMed]
- Jinato, T.; Anuntakarun, S.; Satthawiwat, N.; Chuaypen, N.; Tangkijvanich, P. Distinct alterations of gut microbiota between viral- and non-viral-related hepatocellular carcinoma. Appl. Microbiol. Biotechnol. 2024, 108, 34. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ponziani, F.R.; Bhoori, S.; Castelli, C.; Putignani, L.; Rivoltini, L.; Del Chierico, F.; Sanguinetti, M.; Morelli, D.; Paroni Sterbini, F.; Petito, V.; et al. Hepatocellular Carcinoma Is Associated with Gut Microbiota Profile and Inflammation in Nonalcoholic Fatty Liver Disease. Hepatology 2019, 69, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Behary, J.; Amorim, N.; Jiang, X.-T.; Raposo, A.; Gong, L.; McGovern, E.; Ibrahim, R.; Chu, F.; Stephens, C.; Jebeili, H.; et al. Gut microbiota impact on the peripheral immune response in non-alcoholic fatty liver disease related hepatocellular carcinoma. Nat. Commun. 2021, 12, 187. [Google Scholar] [CrossRef] [PubMed]
- Gok Yavuz, B.; Datar, S.; Chamseddine, S.; Mohamed, Y.I.; LaPelusa, M.; Lee, S.S.; Hu, Z.I.; Koay, E.J.; Tran Cao, H.S.; Jalal, P.K.; et al. The Gut Microbiome as a Biomarker and Therapeutic Target in Hepatocellular Carcinoma. Cancers 2023, 15, 4875. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zhang, H.L.; Yu, L.X.; Yang, W.; Tang, L.; Lin, Y.; Wu, H.; Zhai, B.; Tan, Y.X.; Shan, L.; Liu, Q.; et al. Profound impact of gut homeostasis on chemically-induced pro-tumorigenic inflammation and hepatocarcinogenesis in rats. J. Hepatol. 2012, 57, 803–812. [Google Scholar] [CrossRef] [PubMed]
- Thilakarathna, W.P.D.W.; Rupasinghe, H.P.V.; Ridgway, N.D. Mechanisms by Which Probiotic Bacteria Attenuate the Risk of Hepatocellular Carcinoma. Int. J. Mol. Sci. 2021, 22, 2606. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Wang, T.; Tu, X.; Huang, Y.; Zhang, H.; Tan, D.; Jiang, W.; Cai, S.; Zhao, P.; Song, R.; et al. Gut microbiome affects the response to anti-PD-1 immunotherapy in patients with hepatocellular carcinoma. J. Immunother. Cancer 2019, 7, 193. [Google Scholar] [CrossRef] [PubMed]
- Zhou, A.; Tang, L.; Zeng, S.; Lei, Y.; Yang, S.; Tang, B. Gut microbiota: A new piece in understanding hepatocarcinogenesis. Cancer Lett. 2020, 474, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Ruan, W.Y.; Zhang, L.; Lei, S.; Zeng, Z.R.; Yang, Y.S.; Cao, W.P.; Hao, Q.Y.; Lu, M.; Tian, X.B.; Peng, P.L. An inflammation-associated ferroptosis signature optimizes the diagnosis, prognosis evaluation and immunotherapy options in hepatocellular carcinoma. J. Cell. Mol. Med. 2023, 27, 1820–1835. [Google Scholar] [CrossRef] [PubMed]
- Devan, A.R.; Nair, B.; Aryan, M.K.; Liju, V.B.; Koshy, J.J.; Mathew, B.; Valsan, A.; Kim, H.; Nath, L.R. Decoding Immune Signature to Detect the Risk for Early-Stage HCC Recurrence. Cancers 2023, 15, 2729. [Google Scholar] [CrossRef]
- Wang, T.C.; An, T.Z.; Li, J.X.; Pang, P.F. Systemic Inflammation Response Index is a Prognostic Risk Factor in Patients with Hepatocellular Carcinoma Undergoing TACE. Risk Manag. Healthc. Policy 2021, 14, 2589–2600. [Google Scholar] [CrossRef] [PubMed]
- Sanghera, C.; Teh, J.J.; Pinato, D.J. The systemic inflammatory response as a source of biomarkers and therapeutic targets in hepatocellular carcinoma. Liver Int. 2019, 39, 2008–2023. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Mezzadra, R.; Schumacher, T.N. Regulation and Function of the PD-L1 Checkpoint. Immunity 2018, 48, 434–452. [Google Scholar] [CrossRef] [PubMed]
- Sanceau, J.; Gougelet, A. Epigenetic mechanisms of liver tumor resistance to immunotherapy. World J. Hepatol. 2021, 13, 979–1002. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.Y.; Kudo, M.; Breder, V.; Merle, P.; Kaseb, A.O.; et al. IMbrave150 Investigators. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N. Engl. J. Med. 2020, 382, 1894–1905. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.L.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.Y.; Lim, H.Y.; Kudo, M.; Breder, V.; Merle, P.; et al. Updated efficacy and safety data from IMbrave150: Atezolizumab plus bevacizumab vs. sorafenib for unresectable hepatocellular carcinoma. J. Hepatol. 2022, 76, 862–873. [Google Scholar] [CrossRef] [PubMed]
- Kelley, R.K.; Rimassa, L.; Cheng, A.L.; Kaseb, A.; Qin, S.; Zhu, A.X.; Chan, S.L.; Melkadze, T.; Sukeepaisarnjaroen, W.; Breder, V.; et al. Cabozantinib plus atezolizumab versus sorafenib for advanced hepatocellular carcinoma (COSMIC-312): A multicentre, open-label, randomised, phase 3 trial. Lancet Oncol. 2022, 23, 995–1008. [Google Scholar] [CrossRef] [PubMed]
- Pfister, D.; Núñez, N.G.; Pinyol, R.; Govaere, O.; Pinter, M.; Szydlowska, M.; Gupta, R.; Qiu, M.; Deczkowska, A.; Weiner, A.; et al. NASH limits anti-tumour surveillance in immunotherapy-treated HCC. Nature 2021, 592, 450–456. [Google Scholar] [CrossRef] [PubMed]
- Ren, Z.; Xu, J.; Bai, Y.; Xu, A.; Cang, S.; Du, C.; Li, Q.; Lu, Y.; Chen, Y.; Guo, Y.; et al. ORIENT-32 study group. Sintilimab plus a bevacizumab biosimilar (IBI305) versus sorafenib in unresectable hepatocellular carcinoma (ORIENT-32): A randomised, open-label, phase 2-3 study. Lancet Oncol. 2021, 22, 977–990. [Google Scholar] [CrossRef] [PubMed]
- Abou-Alfa, G.K.; Lau, G.; Kudo, M.; Chan, S.L.; Kelley, R.K.; Furuse, J.; Sukeepaisarnjaroen, W.; Kang, Y.K.; Van Dao, T.; De Toni, E.N.; et al. Tremelimumab plus Durvalumab in Unresectable Hepatocellular Carcinoma. NEJM Evid. 2022, 1, EVIDoa2100070. [Google Scholar] [CrossRef] [PubMed]
- Yau, T.; Kang, Y.K.; Kim, T.Y.; El-Khoueiry, A.B.; Santoro, A.; Sangro, B.; Melero, I.; Kudo, M.; Hou, M.M.; Matilla, A.; et al. Efficacy and Safety of Nivolumab Plus Ipilimumab in Patients with Advanced Hepatocellular Carcinoma Previously Treated with Sorafenib: The CheckMate 040 Randomized Clinical Trial. JAMA Oncol. 2020, 6, e204564. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.S.; Yang, H.; Chon, H.J.; Kim, C. Combination of anti-angiogenic therapy and immune checkpoint blockade normalizes vascular-immune crosstalk to potentiate cancer immunity. Exp. Mol. Med. 2020, 52, 1475–1485. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.F.; Zou, T.; Tuo, Q.Z.; Xu, S.; Li, H.; Belaidi, A.A.; Lei, P. Ferroptosis: Mechanisms and links with diseases. Signal Transduct. Target. Ther. 2021, 6, 49. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Zhou, X.; Xie, F.; Zhang, L.; Yan, H.; Huang, J.; Zhang, C.; Zhou, F.; Chen, J.; Zhang, L. Ferroptosis in cancer and cancer immunotherapy. Cancer Commun. 2022, 42, 88–116. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.J.; Li, S.R.; Huang, Y. An inflammation-related gene landscape predicts prognosis and response to immunotherapy in virus-associated hepatocellular carcinoma. Front. Oncol. 2023, 13, 1118152. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Pang, Y.; Xu, L.; Xu, X. Efficacy and safety of sorafenib combined with TACE in the treatment of advanced hepatocellular carcinoma: A meta-analysis. J. BUON 2021, 26, 1355–1364. [Google Scholar] [PubMed]
- Qasim, W.; Brunetto, M.; Gehring, A.J.; Xue, S.A.; Schurich, A.; Khakpoor, A.; Zhan, H.; Ciccorossi, P.; Gilmour, K.; Cavallone, D.; et al. Immunotherapy of HCC metastases with autologous T cell receptor redirected T cells, targeting HBsAg in a liver transplant patient. J. Hepatol. 2015, 62, 486–491. [Google Scholar] [CrossRef] [PubMed]
- Lakshmaiah, K.C.; Jacob, L.A.; Aparna, S.; Lokanatha, D.; Saldanha, S.C. Epigenetic therapy of cancer with histone deacetylase inhibitors. J. Cancer Res. Ther. 2014, 10, 469–478. [Google Scholar] [CrossRef] [PubMed]
- Bitzer, M.; Horger, M.; Giannini, E.G.; Ganten, T.M.; Wörns, M.A.; Siveke, J.T.; Dollinger, M.M.; Gerken, G.; Scheulen, M.E.; Wege, H.; et al. Resminostat plus sorafenib as second-line therapy of advanced hepatocellular carcinoma—The SHELTER study. J. Hepatol. 2016, 65, 280–288. [Google Scholar] [CrossRef] [PubMed]
- Straś, W.A.; Wasiak, D.; Łągiewska, B.; Tronina, O.; Hreńczuk, M.; Gotlib, J.; Lisik, W.; Małkowski, P. Recurrence of Hepatocellular Carcinoma After Liver Transplantation: Risk Factors and Predictive Models. Ann. Transplant. 2022, 27, e934924. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Galasso, L.; Cerrito, L.; Maccauro, V.; Termite, F.; Mignini, I.; Esposto, G.; Borriello, R.; Ainora, M.E.; Gasbarrini, A.; Zocco, M.A. Inflammatory Response in the Pathogenesis and Treatment of Hepatocellular Carcinoma: A Double-Edged Weapon. Int. J. Mol. Sci. 2024, 25, 7191. https://doi.org/10.3390/ijms25137191
Galasso L, Cerrito L, Maccauro V, Termite F, Mignini I, Esposto G, Borriello R, Ainora ME, Gasbarrini A, Zocco MA. Inflammatory Response in the Pathogenesis and Treatment of Hepatocellular Carcinoma: A Double-Edged Weapon. International Journal of Molecular Sciences. 2024; 25(13):7191. https://doi.org/10.3390/ijms25137191
Chicago/Turabian StyleGalasso, Linda, Lucia Cerrito, Valeria Maccauro, Fabrizio Termite, Irene Mignini, Giorgio Esposto, Raffaele Borriello, Maria Elena Ainora, Antonio Gasbarrini, and Maria Assunta Zocco. 2024. "Inflammatory Response in the Pathogenesis and Treatment of Hepatocellular Carcinoma: A Double-Edged Weapon" International Journal of Molecular Sciences 25, no. 13: 7191. https://doi.org/10.3390/ijms25137191
APA StyleGalasso, L., Cerrito, L., Maccauro, V., Termite, F., Mignini, I., Esposto, G., Borriello, R., Ainora, M. E., Gasbarrini, A., & Zocco, M. A. (2024). Inflammatory Response in the Pathogenesis and Treatment of Hepatocellular Carcinoma: A Double-Edged Weapon. International Journal of Molecular Sciences, 25(13), 7191. https://doi.org/10.3390/ijms25137191