
Integrative Analysis of Hepatopancreas Transcriptome and Proteome in Female Eriocheir sinensis under Thermal Stress

Abstract
:1. Introduction
2. Results
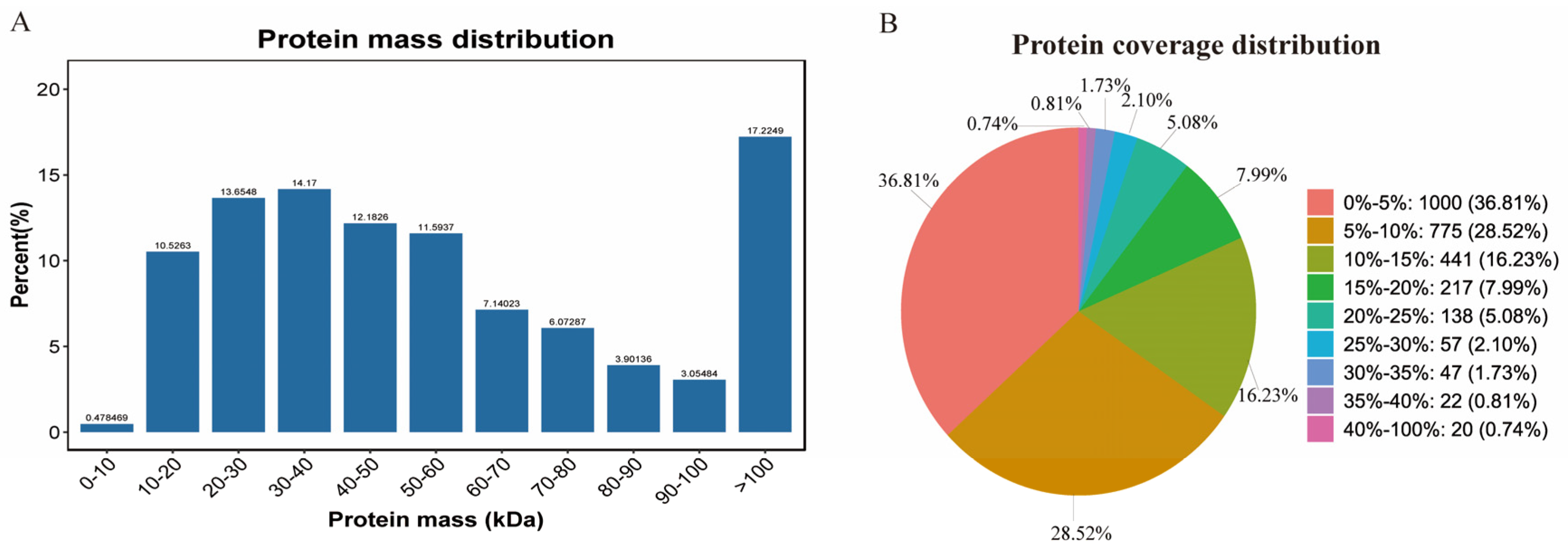
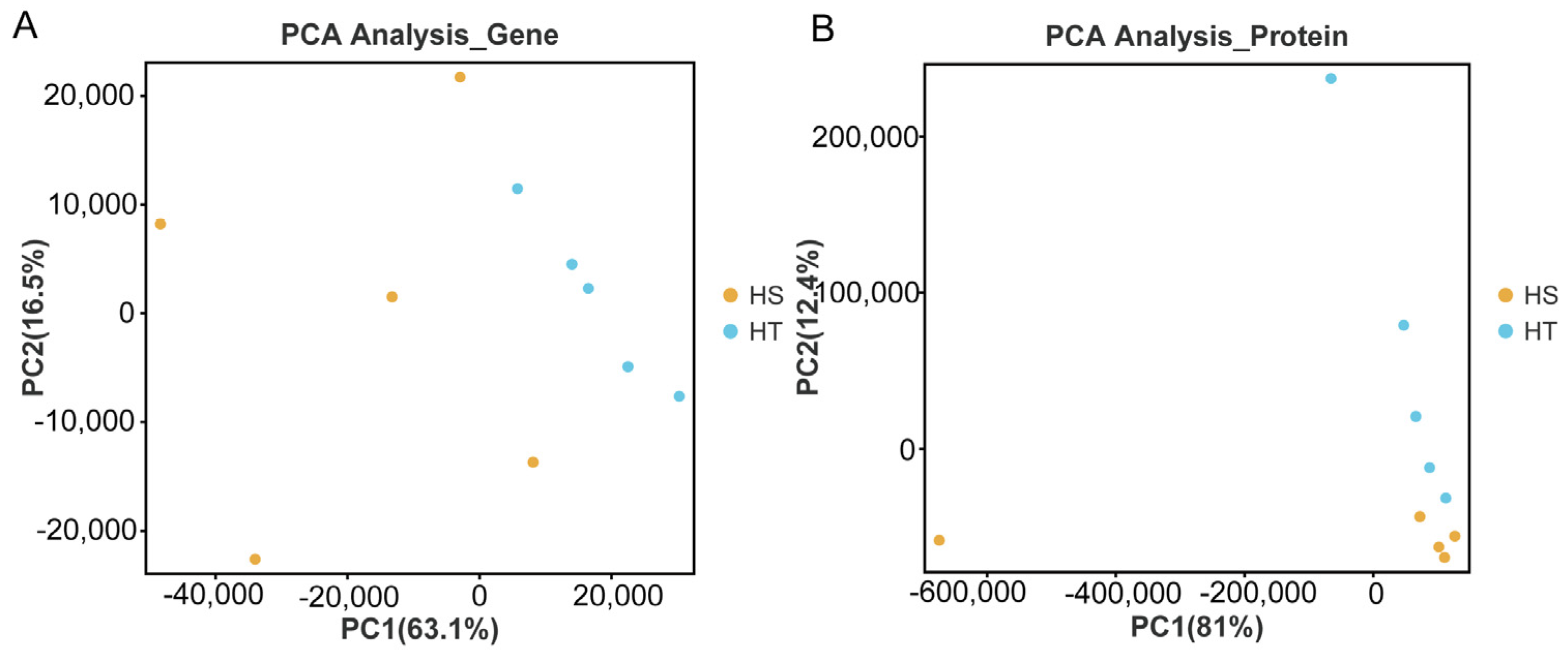
2.1. Transcriptome and Proteome Annotation
2.2. Identification of DEGs and DEPs
2.3. Functional Analysis of DEGs and DEPs
3. Discussion
3.1. Survival
3.2. Common DEGs and DEPs in Two Omics
3.3. Common GO Terms in Two Omics
4. Materials and Methods
4.1. Ethics Approval
4.2. Experimental Design
4.3. Transcriptome Analysis
4.4. Proteome Analysis
4.5. Integrated Analysis of the Transcriptome and Proteome
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sanda, T.; Shimizu, T.; Iwasaki, T.; Dan, S.; Hamasaki, K. Effect of temperature on survival, intermolt period, and growth of juveniles of two mud crab species, Scylla paramamosain and Scylla serrata (Decapoda: Brachyura: Portunidae), under laboratory conditions. Nauplius 2022, 30, e2022012. [Google Scholar] [CrossRef]
- Yang, Y.; Tian, J.; Du, X.; Huang, Y.; Li, Y.; Huang, Y.; Jiang, Q.; Zhao, Y. Effects of temperature on the growth parameters, hepatopancreas structures, antioxidant ability, and non-specific immunity of the crayfish, Cherax destructor. Aquac. Int. 2023, 31, 349–365. [Google Scholar] [CrossRef]
- Liu, J.; Shi, C.; Ye, Y.; Ma, Z.; Mu, C.; Ren, Z.; Wu, Q.; Wang, C. Effects of temperature on growth, molting, feed intake, and energy metabolism of individually cultured juvenile mud crab Scylla paramamosain in the recirculating aquaculture system. Water 2022, 14, 2988. [Google Scholar] [CrossRef]
- Wang, Z.; Luo, H.; Yang, S. Different mechanisms for the extremely hot central-eastern China in July–August 2022 from a Eurasian large-scale circulation perspective. Environ. Res. Lett. 2023, 18, 024023. [Google Scholar] [CrossRef]
- Zhang, G.; Zeng, G.; Yang, X.; Jiang, Z. Future changes in extreme high temperature over China at 1.5 C–5 C global warming based on CMIP6 simulations. Adv. Atmos. Sci. 2021, 38, 253–267. [Google Scholar] [CrossRef]
- Gotthard, K. Growth strategies of ectothermic animals in temperate environments. Environ. Anim. Dev. 2001, 98, 287–304. [Google Scholar]
- Schulte, P.M.; Healy, T.M.; Fangue, N.A. Thermal performance curves, phenotypic plasticity, and the time scales of temperature exposure. Integr. Comp. Biol. 2011, 51, 691–702. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.B.; Su, Y.Q.; Mao, Y.; You, X.X.; Ding, S.X.; Wang, J. Dietary supplementation with Bacillus can improve the growth and survival of the kuruma shrimp Marsupenaeus japonicus in high-temperature environments. Aquac. Int. 2013, 22, 607–617. [Google Scholar] [CrossRef]
- Phuong, N.T.; Ha, N.T.K.; Nguyen, T.E.; Do Thi, T.H. Effects of different temperatures on the growth, survival and digestive enzyme activities of mud crab Scylla paramamosain at juvenile stage. Aquac. Aquar. Conserv. Legis. 2021, 14, 2741–2750. [Google Scholar]
- Matozzo, V.; Gallo, C.; Marin, M.G. Effects of temperature on cellular and biochemical parameters in the crab Carcinusaestuarii (Crustacea, Decapoda). Mar. Environ. Res. 2011, 71, 351–356. [Google Scholar] [CrossRef]
- Chen, D.-W.; Zhang, M.; Shrestha, S. Compositional characteristics and nutritional quality of Chinese mitten crab (Eriocheir sinensis). Food Chem. 2007, 103, 1343–1349. [Google Scholar] [CrossRef]
- ChinaFisheryBureau. China Fisheries Yearbook; Bureau, C.F., Ed.; Chinese Agriculture Express: Beijing, China, 2023; p. 24. [Google Scholar]
- Yuan, Q.; Wang, Q.; Zhang, T.; Li, Z.; Liu, J. Effects of water temperature on growth, feeding and molting of juvenile Chinese mitten crab Eriocheir sinensis. Aquaculture 2017, 468, 169–174. [Google Scholar] [CrossRef]
- Bao, J.; Wang, X.; Feng, C.; Li, X.; Jiang, H. Trehalose metabolism in the Chinese mitten crab Eriocheir sinensis: Molecular cloning of trehalase and its expression during temperature stress. Aquac. Rep. 2021, 20, 100770. [Google Scholar] [CrossRef]
- Li, Z.; Zhao, Z.; Luo, L.; Wang, S.; Zhang, R.; Guo, K.; Yang, Y. Immune and intestinal microbiota responses to heat stress in Chinese mitten crab (Eriocheir sinensis). Aquaculture 2023, 563, 738965. [Google Scholar] [CrossRef]
- Dong, Y.; Jiang, H.W.; Yu, Y.Q.; Zhou, S.X.; Sun, B. Water-temperature characteristics of rice-field-crab “Panshan mode”. Agric. Sci. Technol. Hunan 2010, 11, 152–155. [Google Scholar]
- Pan, T.; Li, T.; Yang, M.; Jiang, H.; Ling, J.; Gao, Q. Cardiac Transcriptome and Histology of the Heart of the Male Chinese Mitten Crab (Eriocheir sinensis) under High-Temperature Stress. Fishes 2024, 9, 92. [Google Scholar] [CrossRef]
- Zhang, T.; Li, T. Ecological observations on molting of juveniles of the Chinese mitten crab, Eriocheir sinensis. J. Lake Sci. 1999, 11, 333–337. [Google Scholar]
- Peng, J.; Zhao, Y.; Xu, Z.; Liu, B.; Duan, C.; Tang, Y. Effect of temperature stress on the survival of juvenile Chinese mitten crab (Eriocheir sinensis). Iran. J. Fish. Sci. 2019, 18, 763–774. [Google Scholar]
- Chen, Q.; Ma, Q.; Shen, Z.; Li, E.; Chen, L. Effects of dietary methionine, bile acid, taurine supplementation in low fishmeal diet on growth performance, feed utilization and antioxidant capacity of juvenile Eriocheir sinensis. Mar. Fish. 2018, 40, 65–75. [Google Scholar]
- Ren, X.; Yu, Z.; Xu, Y.; Zhang, Y.; Mu, C.; Liu, P.; Li, J. Integrated transcriptomic and metabolomic responses in the hepatopancreas of kuruma shrimp (Marsupenaeus japonicus) under cold stress. Ecotoxicol. Environ. Saf. 2020, 206, 111360. [Google Scholar] [CrossRef]
- Duan, P.; Tian, Y.; Li, Z.; Chen, S.; Li, L.; Wang, X.; Wang, L.; Liu, Y.; Zhai, J.; Li, W. Comparative transcriptome analysis of hybrid Jinhu grouper (Epinephelusfuscoguttatus♀×Epinephelustukula♂) and Epinephelusfuscoguttatus under temperature stress. Aquaculture 2024, 578, 740037. [Google Scholar] [CrossRef]
- Pan, T.; Yang, M.; Jiang, H.; Li, T.; Duan, G.; Ling, J.; Gao, Q. Effect of Astragalus membranaceus on Transcriptome and Survival of Hybrid Yellow Catfish (Pseudobagrusvachellii♂×Tachysurusfulvidraco♀) in Response to Aeromonas hydrophila Challenge. Fishes 2023, 8, 454. [Google Scholar] [CrossRef]
- Liu, F.; Zhang, T.; He, Y.; Zhan, W.; Xie, Q.; Lou, B. Integration of transcriptome and proteome analyses reveals the regulation mechanisms of Larimichthyspolyactis liver exposed to heat stress. Fish Shellfish Immunol. 2023, 135, 108704. [Google Scholar] [CrossRef] [PubMed]
- Corteel, M.; Dantas-Lima, J.; Wille, M.; Alday-Sanz, V.; Pensaert, M.; Sorgeloos, P.; Nauwynck, H. Molt stage and cuticle damage influence white spot syndrome virus immersion infection in penaeid shrimp. Vet. Microbiol. 2009, 137, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Ni, H.; Zhang, X.; Sun, Q.; Wu, X.; He, J. Comparative transcriptomics reveals the immune dynamics during the molting cycle of swimming crab Portunustrituberculatus. Front. Immunol. 2022, 13, 1037739. [Google Scholar]
- Xu, Z.; Liu, A.; Li, S.; Wang, G.; Ye, H. Hepatopancreas immune response during molt cycle in the mud crab, Scylla paramamosain. Sci. Rep. 2020, 10, 13102. [Google Scholar] [CrossRef] [PubMed]
- Lou, F.; Wang, Y.; Han, Z.; Shui, B. Comparative transcriptome reveals the molecular regulation mechanism of Charybdis japonica to high-and low-temperature stresses. Front. Mar. Sci. 2022, 9, 849485. [Google Scholar] [CrossRef]
- Liu, C.; Chen, Z.; Chen, J.; Wang, S.; Li, J.; Mao, X. Transcriptome analysis reveals the potential mechanism of carotenoids change in hepatopancreas under low-temperature storage from swimming crab (Portunustrituberculatus). Food Chem. 2023, 408, 135241. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.; Feng, G.; Zhao, F.; Huang, X.; Wang, M.; Wang, H. Integration of Transcriptomics and Proteomics Analysis Reveals the Molecular Mechanism of Eriocheir sinensis Gills Exposed to Heat Stress. Antioxidants 2023, 12, 2020. [Google Scholar] [CrossRef]
- Moritz, C.P.; Mühlhaus, T.; Tenzer, S.; Schulenborg, T.; Friauf, E. Poor transcript-protein correlation in the brain: Negatively correlating gene products reveal neuronal polarity as a potential cause. J. Neurochem. 2019, 149, 582–604. [Google Scholar] [CrossRef]
- Bukau, B.; Weissman, J.; Horwich, A. Molecular chaperones and protein quality control. Cell 2006, 125, 443–451. [Google Scholar] [CrossRef] [PubMed]
- Hartl, F.U.; Bracher, A.; Hayer-Hartl, M. Molecular chaperones in protein folding and proteostasis. Nature 2011, 475, 324–332. [Google Scholar] [CrossRef] [PubMed]
- Saibil, H. Chaperone machines for protein folding, unfolding and disaggregation. Nat. Rev. Mol. Cell Biol. 2013, 14, 630–642. [Google Scholar] [CrossRef] [PubMed]
- Mohindra, V.; Tripathi, R.K.; Yadav, P.; Singh, R.K.; Lal, K.K. Hypoxia induced altered expression of heat shock protein genes (Hsc71, Hsp90α and Hsp10) in Indian Catfish, Clarias batrachus (Linnaeus, 1758) under oxidative stress. Mol. Biol. Rep. 2015, 42, 1197–1209. [Google Scholar] [CrossRef] [PubMed]
- Niu, D.; Zhao, Y.; Gong, X.; Yang, R.; Hu, L.; Zhang, W. Stress response and silencing verification of heat shock proteins in Dermatophagoidesfarinae under temperature stress. Int. J. Biol. Macromol. 2020, 144, 351–361. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Ashraf, S.; Goud, T.S.; Grewal, A.; Singh, S.; Yadav, B.; Upadhyay, R. Expression profiling of major heat shock protein genes during different seasons in cattle (Bos indicus) and buffalo (Bubalus bubalis) under tropical climatic condition. J. Therm. Biol. 2015, 51, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Al-Thuwaini, T.M.; Al-Shuhaib, M.B.S.; Hussein, Z.M. A novel T177P missense variant in the HSPA8 gene associated with the low tolerance of Awassi sheep to heat stress. Trop. Anim. Health Prod. 2020, 52, 2405–2416. [Google Scholar] [CrossRef] [PubMed]
- Füzéry, A.K.; Oh, J.J.; Ta, D.T.; Vickery, L.E.; Markley, J.L. Three hydrophobic amino acids in Escherichia coli HscB make the greatest contribution to the stability of the HscB-IscU complex. BMC Biochem. 2011, 12, 3. [Google Scholar] [CrossRef]
- Duncan, R.F.; Cavener, D.R.; Qu, S. Heat shock effects on phosphorylation of protein synthesis initiation factor proteins eIF-4E and eIF-2. alpha. in Drosophila. Biochemistry 1995, 34, 2985–2997. [Google Scholar] [CrossRef]
- Cao, L.; Wang, G.; Fahim, A.M.; Pang, Y.; Zhang, Q.; Zhang, X.; Wang, Z.; Lu, X. Comprehensive Analysis of the DnaJ/HSP40 Gene Family in Maize (Zea mays L.) Reveals that ZmDnaJ96 Enhances Abiotic Stress Tolerance. J. Plant Growth Regul. 2024, 43, 1548–1569. [Google Scholar] [CrossRef]
- Cebula, M.; Morgenstern, R. Enzymology of reactive intermediate protection: Kinetic analysis and temperature dependence of the mesophilic membrane protein catalyst MGST1. FEBS J. 2023, 290, 3448–3460. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. bioRxiv 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Wei, F.; Liang, J.; Tian, W.; Yu, L.; Feng, Z.; Hua, Q. Transcriptomic and proteomic analyses provide insights into the adaptive responses to the combined impact of salinity and alkalinity in Gymnocyprisprzewalskii. Bioresour. Bioprocess. 2022, 9, 104. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reads Summary | HS | NS |
|---|---|---|
| Raw reads | 200,718,242 | 191,482,932 |
| Clean reads | 199,113,626 | 189,706,812 |
| Raw data | 30,107,736,300 | 28,722,439,800 |
| Clean data | 29,761,887,212 | 28,345,746,874 |
| Valid ratio (reads)/% | 99.20 | 99.07 |
| GC content % | 50.04 | 52.60 |
| Mapping reads (ratio) | 174,600,253 (88.30%) | 169,753,006 (90.19%) |
| Unique mapped reads (ratio) | 164,765,803 (83.33%) | 159,186,593 (84.58%) |
| Expression Pattern | Transcriptome | Proteome | Number of Genes | In Total |
|---|---|---|---|---|
| Significance of differential expression | Significant | Non-significant | 536 | 2641 |
| Non-significant | Significant | 119 | ||
| Significant | Significant | 25 | ||
| Non-significant | Non-significant | 1961 | ||
| Regulated model | Upregulated | Downregulated | 4 | 25 |
| Downregulated | Upregulated | 10 | ||
| Upregulated | Upregulated | 6 | ||
| Downregulated | Downregulated | 5 |
| GO ID | GO Term | GO Function | p Value | |
|---|---|---|---|---|
| Transcriptome | Proteome | |||
| GO:0051087 | Chaperone Binding | Molecular function | 0.018406 | 8.07 × 10−7 |
| GO:0016887 | ATP hydrolysis activity | Molecular function | 6.31 × 10−5 | 3.07 × 10−6 |
| GO:0016462 | Pyrophosphatase activity | Molecular function | 0.000202 | 3.23 × 10−6 |
| GO:0016818 | hydrolase activity, and acting on acid Anhydrides and phosphorus-containing anhydrides | Molecular function | 0.000222 | 3.36 × 10−6 |
| GO:0016817 | Hydrolase activity and acting on acid anhydrides | Molecular function | 0.000312 | 3.36 × 10−6 |
| GO:0017111 | Nucleoside-triphosphatase activity | Molecular function | 0.000318 | 1.61 × 10−6 |
| GO:0043232 | Intracellular non-membrane-bound organelle | Cellular component | 0.006763 | 8.3954 × 10−5 |
| GO:0043228 | Non-membrane-bound organelle | Cellular component | 0.006787 | 9.38 × 10−5 |
| GO:0009408 | Response to heat | Biological process | 0.026299 | 8.92 × 10−8 |
| GO:0009266 | Response to temperature stimulus | Biological process | 0.003881 | 9.02 × 10−7 |
| Pathway | Pathway Name | p Value | Number of Overlapped Genes | |
|---|---|---|---|---|
| Transcriptome | Proteome | |||
| ko05418 | Fluid shear stress and atherosclerosis | 0.039719 | 5.77 × 10−7 | 3 |
| ko04670 | Leukocyte transendothelial migration | 0.011702 | 1.48 × 10−5 | 2 |
| ko04918 | Thyroid hormone synthesis | 0.000732 | 0.012478 | 0 |
| Gene | Description | Transcription | Proteome | ||
|---|---|---|---|---|---|
| Log2FC | Regulation | Log2FC | Regulation | ||
| MGST1 | Microsomal glutathione S-transferase 1 | −1.83 | Down | 4.32 | Up |
| Act5C | Actin-4 | −1.01 | Down | 1.43 | Up |
| HSP90AB1 | Heat shock protein 90 | −1.38 | Down | 1.22 | Up |
| mys | Integrin | 2.458 | Up | 0.73 | Up |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pan, T.; Li, T.; Yang, M.; Jiang, H.; Ling, J. Integrative Analysis of Hepatopancreas Transcriptome and Proteome in Female Eriocheir sinensis under Thermal Stress. Int. J. Mol. Sci. 2024, 25, 7249. https://doi.org/10.3390/ijms25137249
Pan T, Li T, Yang M, Jiang H, Ling J. Integrative Analysis of Hepatopancreas Transcriptome and Proteome in Female Eriocheir sinensis under Thermal Stress. International Journal of Molecular Sciences. 2024; 25(13):7249. https://doi.org/10.3390/ijms25137249
Chicago/Turabian StylePan, Tingshuang, Tong Li, Min Yang, He Jiang, and Jun Ling. 2024. "Integrative Analysis of Hepatopancreas Transcriptome and Proteome in Female Eriocheir sinensis under Thermal Stress" International Journal of Molecular Sciences 25, no. 13: 7249. https://doi.org/10.3390/ijms25137249